Abstract
Small molecules with the potential to initiate different types of programmed cell death could be useful ‘adjunct therapy’ where current anticancer modalities fail to generate significant activity due to a defective apoptotic machinery or resistance of cancer cells to the specific death mechanism induced by that treatment. The current study identified silibinin, for the first time, as one such natural agent, having dual efficacy against colorectal cancer (CRC) cells. First, silibinin rapidly induced oxidative stress in CRC SW480 cells due to reactive oxygen species (ROS) generation with a concomitant dissipation of mitchondrial potential (ΔΨm) and cytochrome c release leading to mild apoptosis as a biological effect. However, with increased exposure to silibinin, cytoplasmic vacuolization intensified within the cells followed by sequestration of the organelles, which inhibits the further release of cytochrome c. Interestingly, this decrease in apoptotic response correlated with increased autophagic events as evidenced by tracking the dynamics of LC3-II within the cells. Mechanistic studies revealed that silibinin strongly inhibited PIK3CA-AKT–MTOR but activated MAP2K1/2-MAPK1/3 pathways for its biological effects. Corroborating these effects, endoplasmic reticulum stress was generated and glucose uptake inhibition as well as energy restriction were induced by silibinin, thus, mimicking starvation-like conditions. Further, the cellular damage to tumor cells by silibinin was severe and irreparable due to sustained interference in essential cellular processes such as mitochondrial metabolism, phospholipid and protein synthesis, suggesting that silibinin harbors a deadly ‘double-edged sword’ against CRC cells thereby further advocating its clinical effectiveness against this malignancy.
Introduction
Colorectal cancer (CRC) is the second leading cause (both genders combined) of cancer-related deaths in US; statistical estimates for 2012 indicate ~103,170 new CRC cases and 51,690 associated deaths in US alone.Citation1 The current anticancer therapy fails to generate significant antitumor effect due to a defective apoptotic machinery or resistance of the CRC cells to the specific death mechanisms induced by the drugs.Citation2-Citation7 Thus, new and alternate approaches need to be explored which can help overcome this anomaly. In this regard, it has been appreciated that molecules which are capable of inducing both autophagic cell death and apoptosis have the potential to be the ‘golden bullets’ since they will be capable of triggering both caspase-dependent and autophagic caspase-independent cell death.Citation3,Citation8,Citation9 In a broader scenario, the use of these molecules can be extended as ‘adjunct’ to such treatment conditions where the prescribed therapy fails to augment the anticancer effect.Citation2,Citation4
Metabolomic approaches in recent times have been used to establish the metabolic phenotypes of tumors and to assess/monitor the efficacy of an anticancer drug treatment.Citation10,Citation11 These studies have shown that the metabolic profile of various cancerous /noncancerous tissues can be correlated with cell growth and specific type of cell death, specific tumor type as well as the pathological stage of tumor.Citation11-Citation13 In this regard, the metabolomics of tissues from CRC patients using spectroscopy has also helped identify and establish the metabolic profiles specific to CRC malignancy.Citation14,Citation15 We recently reported the strong preventive and therapeutic efficacy of silibinin (flavonolignan isolated from the seeds of milk thistle), in different CRC preclinical models and subsequently analyzed the protein molecules possibly involved with silibinin efficacy.Citation16-Citation23 From a broader viewpoint, the limitation of these studies was that it did not discuss the efficacy of the silibinin treatment in terms of the metabolic profile and energy state of the colonic tumors. To answer these specific questions we have performed a metabolomics study utilizing quantitative high-resolution nuclear magnetic resonance spectroscopy (1H-, 13C- and 31P-NMR) to assess the metabolic profile and energy state of the silibinin-treated CRC cells so as to complement the proteomics data as well as the programmed cell-death type II generated in response to energy restrictions induced by silibinin. This study, inarguably, not only provides us with a complete scenario of the anticancer effect of silibinin against CRC growth but in terms of practical and translational aspects, further validates the clinical usefulness of this drug. Our results showed that silibinin possesses dual efficacy in terms of inducing cell death in CRC cells, where first it initiates an apoptotic response triggered by ROS generation and then imposes energy restrictions within the cells, thereby activating autophagic machinery to induce autophagic death.
Results
Silibinin causes intense cytoplasmic vacuolization
First we assessed the efficacy of silibinin against the mitogenic effects of EGF and IGF1 in SW480 CRC cells. However, during the course of study, in Trypan blue assay, treatment of SW480 cells with pharmacologically achievable concentration (100 μM) of silibininCitation24 resulted in a significant (p < 0.001) death of serum-starved control cells quite early (2–12 h); even mitogens failed to rescue silibinin-induced cell death (). These results were corroborated by MTT assay, where treatment of serum-starved SW480 cells with different doses of silibinin (25–100 μM) for 6 to 12 h also decreased the cell viability while cotreatment with IGF1 failed to show any protective effect (). To address whether these effects of silibinin are specific to SW480 cells, two additional CRC cell lines LoVo and HT-29 were also assessed; similar treatments in these cells failed to cause any significant death by 12 h (), though cellular death was observed after 24 h (data not shown). Next, we assessed whether a similar effect of silibinin could be seen under serum conditions in SW480 cells (). Results indicated that under serum conditions, silibinin exhibited delayed cytotoxicity () compared with when the treatment was carried under serum-starved conditions; with 100 μM dose decreasing viability by ~30% at 72 h.Citation17 Furthermore, silibinin (50–100 μM dose) treatment arrested SW480 cells at ‘S’ phase followed by a significant nonapoptotic cell death at 72 h, proceeding to absolute cellular death at later time points (data not shown). These results were corroborated by clonogenic assay showing that silibinin causes a dose-dependent inhibition of colony formation; however, with doses of 100 μM and beyond, there were no viable/live cells present to colonize ().
Figure 1. Comparative effect of silibinin on viability of human CRC cells. (A) Effect of mitogens on silibinin-induced cell death in different human CRC cells under serum-starved (SS) conditions. (B) Effect of silibinin on viability, apoptotic death, cell cycle distribution and clonogenic potential of SW480 cells cultured under serum conditions. (C) Representative photomicrographs of SW480 cells cultured under different serum conditions, depicting intense cytoplasmic vacuolization due to silibinin treatment. All experimental procedures and statistical analysis were performed as detailed in Materials and Methods. #p < 0.01; *p < 0.001.
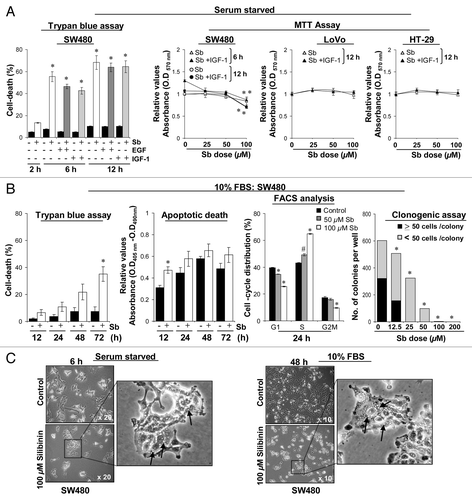
To ascertain this differential effect of silibinin on SW480 cells cultured under serum and serum-starved conditions, we compared their cellular morphology (). Microscopic investigations revealed that silibinin induces massive vacuolization in the cytoplasm of the cells cultured under serum-starved conditions. Vacuoles were initially pearly type and started to be visible from 2 h onwards, which became large, asymmetrical, and their number/cell increased with treatment time (peaked by 12 h), the proportion of dead cells also increased dramatically thereafter; essentially all cells were dead after continued silibinin treatment. Interestingly, under serum conditions, the vacuoles started appearing slowly (24 h onwards) and became massive with continued silibinin exposure. Thus, it was inferred that the same silibinin effect was being mimicked under serum/serum-starved conditions, however, under serum-starved conditions, the effect was immediate (within 2 h) with appearance of vacuolization as a much earlier phenomenon (). To understand in detail the mechanism of silibinin effect, we further chose working under serum conditions as this had the advantage of a broader time frame.
Silibinin causes ROS generation
We used cationic dye DiOC6(3) as an indicator of dissipation of mitochondrial potential (ΔΨm) together with dihydroethidium (DHE), which, on oxidization to ethidium by superoxide radical, yields bright red fluorescence and intercalates with nuclear DNA.Citation25-Citation28 In control cells, DiOC6(3)-fluorescence was bright green and identified viable/normal thread like mitochondrial network, spread uniformly within cytoplasm; but red nuclear ethidium-fluorescence was almost absent (, left panel). Conversely, in silibinin-treated cells, green DiOC6(3)-fluorescence appeared weak and of punctate type, and was either quite diminished or altogether absent in some cells. Silibinin-treated cells, however, showed bright red nuclear ethidium-fluorescence indicative of ROS superoxide radical generation (, left panel). FACS analysis revealed a time-dependent decay of DiOC6(3)-fluorescence (indicating a decline in ΔΨm) and a concomitant increase in ethidium-fluorescence intensity in silibinin-treated cells (, middle and right panels). Also, there was a significant time- and dose-dependent increase in ethidium-fluorescence positive cells indicating silibinin potential to encompass the entire cell population with oxidative stress (, left and middle panels). To confirm that DHE was specifically being oxidized by ROS only, cells were pretreated with ROS scavengers N-acetyl cysteine (NAC), ascorbic acid (AA); metalloporphyrins: Fe (III) meso-Tetra (4-carboxyphenyl) porphine chloride (FeTCP) and Mn(III)tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP)Citation29,Citation30 and ethidium-fluorescence was determined after silibinin treatment. All four agents decreased ethidium-fluorescence positive cells in the silibinin-treated set, reconfirming ROS generation by silibinin (, right panel). To determine whether ROS generation was involved in the apoptotic response initiated after silibinin treatment, cells were pretreated with these antioxidants and the effect on apoptotic cell death by silibinin was determined. The results indicated that indeed ROS generation served as a trigger for apoptosis as indicated by significant reversal of silibinin caused apoptotic death by AA; of all the antioxidants used, AA was most effective in significantly decreasing both basal and silibinin-induced ROS levels and this justified its role in causing the most significant reversal in apoptosis (, left panel).
Figure 2. Effect of silibinin on reactive oxygen species (ROS) generation in SW480 cells. (A and B) Time and dose-dependent effect of silibinin on intracellular superoxide radical generation and dissipation of mitochondrial potential (Δψm) as measured using dihydroethidium (DHE) and DiOC6(3) dyes, respectively. (B) Right panel, effect of cotreatment of antioxidants on ROS generation by silibinin. (C) Left panel, effect of cotreatment of antioxidants on apoptotic induction by silibinin; middle panel, effect of silibinin on expression levels of H2O2 quenching enzymes, CYCS release; right panel, mitochondrial network as traced by MitoTracker Red using confocal microscopy. All experimental procedures and statistical analysis were performed as detailed in Materials and Methods. *p < 0.001.
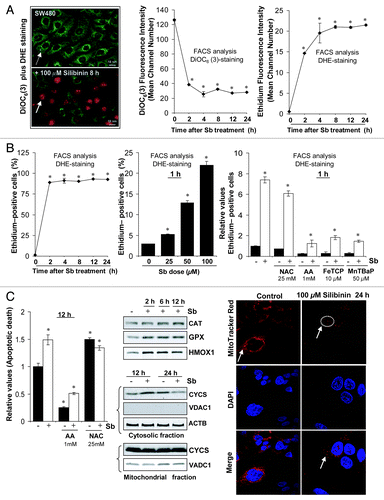
We also found that ROS was specifically the superoxide radical, due to a lack of H2O2 detection in DCFH2-DA dye experiments (data not shown), which could be attributed to increased levels of GPX and CAT enzymes, H2O2 quencher,Citation28 by silibinin (, middle panel). An increase in cellular stress associated with ROS generation by silibinin was also indicated by induced HMOX1 levels (, middle panel).
Our additional studies suggested that ROS generation in SW480 cells preceded changes in DiOC6(3) retention indicating that silibinin triggers oxidative stress prior to mitochondrial disruption and apoptosis. Dissipation of ΔΨm associated with mitochondrial disruption (a pre-requisite for apoptosis) was observed only at earlier time points (up to 12 h) as indicated by cytochrome c (CYCS) release in the cytoplasm of silibinin-treated cells (, middle panel). However, there was a significant decrease in CYCS release, compared with untreated cells, with an increase in silibinin exposure time (24 h data shown in , middle panel). This was also confirmed by confocal studies using MitoTracker Red which indicated that mitochondria were intact and thread-like in control cells, but looked condensed and fragmented and were sequestered in the cytoplasm due to intense cytoplasmic vacuolization following silibinin treatment (, right panel). These results were consistent with those in , where SW480 cells do undergo a slight apoptotic death until 12 h of silibinin treatment, but it was not quite high compared with total dead cell population.
Silibinin induces autophagic death
To confirm whether the depolarized/damaged mitochondria are being sequestered and subsequently removed by autophagosomes, preventing CYCS from forming a functional apoptosome in the cytoplasm,Citation6,Citation8,Citation31 experiments were next performed to identify and establish autophagy induction by silibinin in SW480 cells (). Silibinin indeed induced autophagy as indicated by a significant time-dependent increase in LC3-II/LC3-I ratio. Furthermore, LC3-II turnover also markedly increased by treatment of cells with a combination of lysosomal inhibitors E64d and pepstatin A, confirming both autophagy induction and maturation to completion (). We also observed a decrease in sequestosome 1 (SQSTM1) protein levels by silibinin (), even though the mRNA levels of SQSTM1 were increased by treatment (Table S1); SQSTM1 serves as a selective substrate of autophagy and its protein levels are decreased during starvation-induced autophagy.Citation32 An additional important observation for autophagy induction by silibinin was a significant accumulation of monodansylcadaverine (MDC), a specific autolysosome/autolysosomal marker,Citation33,Citation34 in the form of spherical structures distributed in the cytoplasm and perinuclear region of treated cells (). Fluorometric measurement of intracellular MDC, corrected to cell number, also showed its significant accumulation in silibinin-treated cells (). Recent reports indicate that BCL2 at the endoplasmic reticulum (ER) can inhibit autophagy by physically interacting with BECN1 and can also prevent Ca2+ release from ER to block the activation of PRKAA2 which represses MTOR to activate autophagy.Citation6 We also observed a decrease () in the levels of BCL2 family members, BCL2 and MCL1 which are known to inhibit both apoptosis and autophagy.Citation6 Interestingly, while ATG5 levels were increased, silibinin treatment decreased the protein levels of BECN1 (). Further, silibinin treatment was also able to differentially modulate the gene expression levels of autophagy-associated molecules (Table S1), however, it is not still established whether such a modulation in mRNA levels of these molecules does necessarily precede autophagic events.
Figure 3. Effect of silibinin on autophagy induction in SW480 cells. (A) Effect of silibinin on the expression levels of LC3-I, LC3-II and SQSTM1 levels, and the turnover of LC3-II after cotreatment with lysosomal inhibitors (E64d and pepstatin A). (B) Effect of silibinin on MDC incorporation and relative MDC fluorescence as detected by fluorescence photometry. (C) Effect of silibinin on SQSTM1 expression in CRC tumor xenografts in nude mice. Archival xenograft tissues of SW480 cells in athymic (nu/nu) nude male mice, as described in Materials and Methods were used in the present study. Representative DAB-stained tissue specimens from control and silibinin-fed groups are shown. Quantification of SQSTM1-positive cells represented as immunoreactivity score is shown as mean and ± SEM (error bars) of each group. Densitometric analysis of band intensity for SQSTM1 protein in immunoblots was adjusted with ACTB (blots not shown), and is shown as mean ± SEM (error bars) of the three bands from individual tumor tissue in each group. All experimental procedures and statistical analysis were performed as detailed in Materials and Methods. $p < 0.05; *p < 0.001.
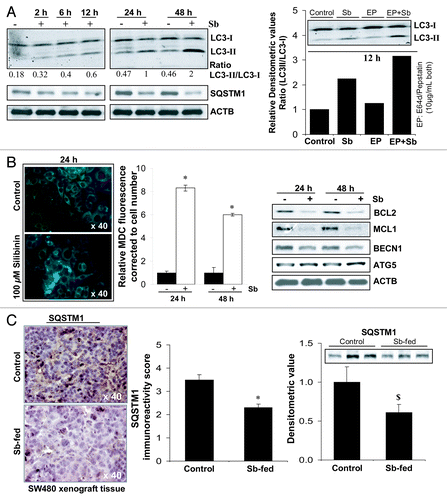
To further examine in vivo involvement of silibinin-induced autophagy in its anticancer efficacy, we analyzed silibinin-treated CRC xenograft tissues for the expression of SQSTM1. Results indicated that the immunoreactivity score of SQSTM1decreased by ~28% (p < 0.001) in SW480 tumor tissue by silibinin feeding (), indicating induction of starvation-caused autophagy in treated tissues. Western blots for SQSTM1 in tumor lysates with densitometric data (adjusted with ACTB as loading control) are shown in , which further confirmed the same.
Silibinin-induced autophagic death is mediated via endoplasmic reticulum stress
To further elucidate the type and origin of vacuoles and other cytological changes induced by silibinin, we examined SW480 cells morphology by transmission electron microscopy (EM, and ), which is the gold standard for assessing autophagic activity within the cells.Citation35 Silibinin treatment either under serum-starved () or serum () conditions resulted in the formation of double- or multi-membrane type autophagic vacuoles with sequestered cellular organelles such as mitochondria, lamellar structures and digested material (; ). The vacuoles were either electron dense or electron lucent. The outer lining of these vacuoles was made up of smooth membrane. Fusion between lysosome-like and autophagosome-like structures could also be identified. Interestingly, a different kind of vacuole was also observed, which was identified as swollen rough ER as recognized by the presence of ribosomes on membrane surface (; ). Furthermore, a relatively intact nucleus with partially condensed chromatin, intact cytoskeleton, slightly swollen as well as fragmented mitochondria were also observed in silibinin-treated cells. However, an increase in autophagic events was seen with an increase in the exposure time to silibinin. Ultimately total collapse in the cellular system indicative of autophagic cellular death was observed at later time points (). In order to delineate whether the silibinin-caused ROS generation played any role in autophagy induction, the cells were cotreated with antioxidant AA and silibinin; this combination was earlier seen to interfere with oxidative stress induction by silibinin. Interestingly, the cells after this cotreatment looked healthier with no swelling of rough ER, there was also no sign of chromatin condensation and fragmented mitochondria, and the autophagic vacuoles formed were significantly decreased, although their formation was not completely inhibited, and they were more electron dense in nature (). Most importantly, intense autophagic events were absent in the cytoplasm and no morphological events indicative of autophagic death could be observed in cells cotreated with AA and silibinin. We then treated the cells with silibinin and autophagy inhibitors 3MA (which blocks early induction of autophagy) or bafilomycin A1 (BAFA1, which blocks fusion of autophagosomes and lysosomes),Citation8 and then determined cell morphology (). Cells cotreated with 3MA and silibinin did not inhibit vacuole formation completely, although vacuoles (including swollen ER) were not as big as with silibinin alone. In addition, the cytoplasmic blebbing with intact nuclear membrane was also observed in cells cotreated with 3MA and silibinin (). In summary, no morphological features indicative of intense apoptosis or autophagy associated cellular death could be observed when cells were cotreated with autophagy inhibitor 3MA and silibinin (). Conversely, when cells were cotreated with BAFA1 and silibinin, completion of autophagy was interrupted as indicated by a significant increase in the number of double-layered autophagic vacuoles, which did not fuse with lysosomes (). There was also no effect on the swelling of rough ER, though no increase in apoptotic events relative to untreated controls could be observed. Interestingly, the buildup of vacuoles was so significant that silibinin irreversibly committed the cells to autophagic death once autophagy was initiated, as indicated by the failure of late autophagy inhibitor BAFA1 to rescue cells from silibinin-induced death events. Furthermore, cell death detection ELISA experiments corroborated with above findings indicating that the cotreatment with autophagy inhibitors BAFA1or 3MA with silibinin, could not initiate significant apoptosis (). Specifically, these results indicated that treatment with late autophagy inhibitor BAFA1, while partially inhibiting autophagy, could not initiate apoptosis in the cells as compared with when the cells were treated with BAFA1 alone (; ). Similarly, cotreatment with the early autophagy inhibitor 3MA initiated a slight apoptotic response in silibinin-treated cells compared with when 3MA was used alone, although, overall it tended to increase cell survival in terms of total percentage of cells being affected (; ). Furthermore, to delineate whether the specific death type induced by silibinin was dose-dependent, dose-escalation studies were performed under serum conditions. The results indicated that, unlike the 100 μM dose, the 200 μM silibinin dose led to a significant apoptotic death in a time-dependent manner ().
Figure 4. Visualization of autophagy induction and cellular morphology under transmission electron microscopy (EM) after treatment of SW480 cells with silibinin under serum-starved conditions. All experimental procedures and statistical analysis were performed as detailed in Materials and Methods. N, nucleus; C, cytoplasm; ER, endoplasmic reticulum with ribosomes as beads on membrane; M, mitochondria; AC, autophagic compartment; AP, autophagosomes.
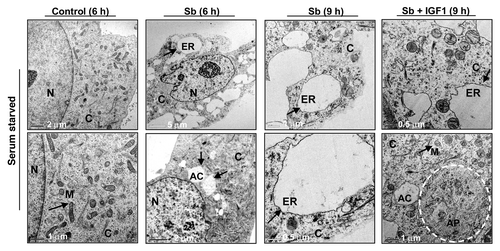
Figure 5. Visualization of autophagy induction and cellular morphology under transmission electron microscopy (EM) after treatment of SW480 cells with silibinin alone or cotreatment with antioxidant AA, or autophagy inhibitors 3MA, and BAFA1 under serum conditions. All experimental procedures and statistical analysis were performed as detailed in Materials and Methods. N, nucleus; C, cytoplasm; ER, endoplasmic reticulum with ribosomes as beads on membrane; M, mitochondria; AC, autophagic compartment; DAC, electron dense autophagic compartment; AP, autophagosomes.
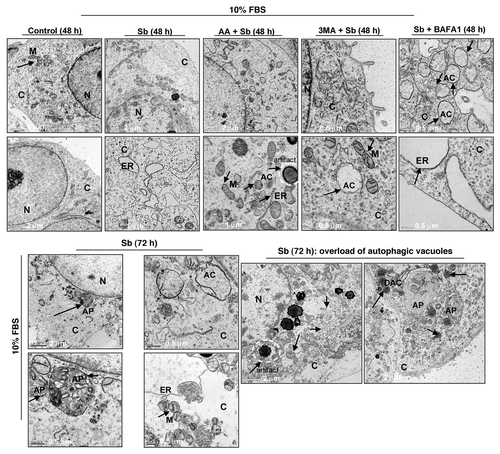
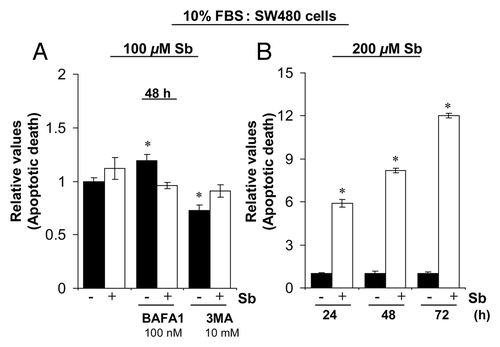
Figure 6. Effect of silibinin on apoptotic induction in SW480 cells. (A) Effect of cotreatment with autophagy inhibitors BAFA1 and 3MA on apoptotic induction by silibinin in SW480 cells. (B) Effect of high dose (200 μM) silibinin on apoptotic induction in SW480 cells. All experimental procedures and statistical analysis were performed as detailed in Materials and Methods. *p < 0.001.
Silibinin inhibits IGF or EGF-induced mitogenic signaling
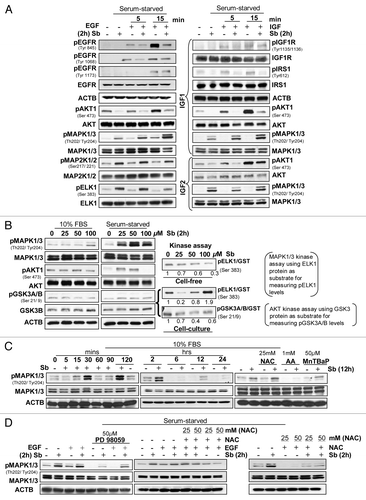
While autophagy pathways depend on nutrient availability, upstream signaling (growth factor signaling, PRKAA1/2, calcium signaling and others) regulates them.Citation6 Growth factor signaling, often deregulated in cancer cells, is mediated via receptor tyrosine kinases (RTKs) regulating two main pathways, the PIK3CA-AKT-MTOR (inhibiting autophagy) and the RAS-RAF1-MAP2K1/2-MAPK1/3 pathway (promoting it).Citation8,Citation36,Citation37 The potential role of these pathways in silibinin-induced autophagy was studied by experimental conditions wherein both pathways where specifically activated in SW480 cells (). Importantly, 100 μM silibinin inhibited both constitutive and EGF- or IGF1/IGF2-induced AKT activation () by inhibiting Tyr phosphorylation of EGFR (significant inhibition) and IGF1R (moderate inhibition). Though IGF1R activation was not dramatically decreased, activation of its downstream mediator IRS1 was significantly reduced by silibinin (, right panel). The above results suggested silibinin would also inhibit MAPK1/3 activation, however, it resulted in increased MAPK1/3 phosphorylation. Similarly, both pMAP2K1/2 and pELK1/2, the up- and down-stream MAPK1/3 kinase and effector, respectively, were also enhanced by silibinin (, left panel). Importantly, silibinin also increased pMAPK1/3 levels under both serum and serum-starved conditions with EGF/IGF (, left and middle panel). Silibinin effect, however, was differential on pAKT1 where lower silibinin doses (25–50 μM) had either no effect or increased pAKT1 levels, 100 μM silibinin strongly decreased pAKT1 with increased levels of activated GSK3A/B (, left and middle panel). Since silibinin increased pMAPK1/3 levels and differentially affected pAKT1 levels, silibinin effect on kinase activity of these two molecules was also analyzed (, right panel). Whereas in a cell-free system silibinin dose-dependently inhibited MAPK1/3 kinase activity to ELK1, under cell culture conditions, silibinin increased it, suggesting that certain specific conditions were generated by silibinin in the cell culture that dramatically enhanced MAPK1/3 kinase activity to ELK1 (, right panel). Importantly, silibinin inhibited AKT kinase activity to GSK3A/B under cell culture conditions (, right panel). The mechanism delineating silibinin-induced MAPK1/3 activation () showed that silibinin induces a cyclic pattern of MAPK1/3 activation starting as early as 15 min and lasting 12 h and then subsiding by 24 h (, left and middle panel). Since ROS induces MAPK1/3 activationCitation38 and silibinin dramatically caused ROS generation, we next examined this association (). Interestingly, the antioxidants, which significantly inhibited silibinin-caused ROS generation (), also displayed similar inhibition of MAPK1/3 phosphorylation by silibinin, suggesting that MAPK1/3 activation by silibinin is mediated by its ROS generation (, right panel). These results were further corroborated by the fact that while PD98059, an inhibitor of MAP2K1/2, was able to completely inhibit the EGF induced activation of MAPK1/3, it was only partially capable of inhibiting the increase in pMAPK1/3 levels induced by a combination of EGF and silibinin (). Similarly, the antioxidant NAC showed only partial inhibition of pMAPK1/3 levels as induced by a combination of EGF and silibinin but was able to significantly reduce MAPK1/3 activation as induced by silibinin alone, thereby reconfirming that ROS generated by silibinin was responsible for MAPK1/3 activation ().
Figure 7. Effect of silibinin on mitogen-induced cell signaling pathways. Effect of silibinin on (A) receptor tyrosine kinase (RTK’s) regulated activation of AKT and MAPK1/3 levels; (B) constitutively activated levels of AKT and MAPK1/3, under serum (left panel) and serum starved (middle panel) conditions, and kinase activity of MAPK1/3 and AKT (right panel). (C) Time-dependent effect of silibinin and antioxidants on MAPK1/3 activation under serum conditions. (D) Effect of MAP2K1/2 inhibitor (PD98059) and antioxidant NAC on silibinin-caused or EGF induced activation of MAPK1/3 levels under serum-starved conditions. All experimental procedures and data analysis were performed as detailed in Materials and Methods.
Silibinin inhibits the AKT-MTOR pathway and interferes with cellular protein synthesis
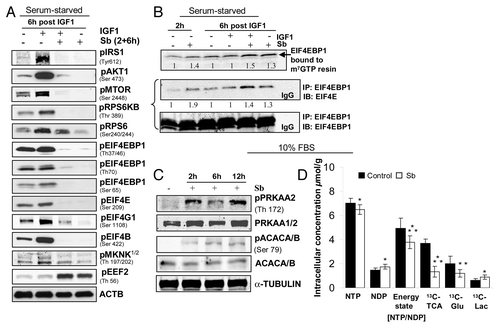
As a sensor for amino acids and ATP, TOR kinase is a gatekeeper for initiation of autophagy.Citation3 Via its connection with AKT pathway, TOR kinase also integrates growth factor-induced signals to autophagic pathway.Citation3 Mechanistic TOR (MTOR)’s downstream effect is mediated by RPS6KB and eukaryotic initiation factor 4E binding protein-1 (EIF4EBP1), which controls protein translation.Citation3 To determine whether silibinin-induced autophagy was due to interference in cellular protein translation via modulation of MTOR pathway, we performed studies in presence of IGF1 to understand the specific effect of silibinin on the activated AKT-MTOR pathway (). Silibinin significantly inhibited IGF1-induced MTOR phosphorylation as well as those of other proteins in MTOR pathway, RPS6KB and its target the ribosomal protein S6 (RPS6), which coincides with induction of autophagy in mammalian cellsCitation3 (). Activated MTOR also phosphorylates EIF4EBP1, which inactivates the protein; conversely on being hypophosphorylated, EIF4EBP1 binds to and inhibits the rate-limiting translation initiation factor EIF4E (eukaryotic translation initiation factor 4E) from assembling with other translation initiation factors to form the active complex EIF4F, which is essential to initiate cap-dependent translation.Citation39-Citation41 Along similar lines, the affinity of EIF4E for cap is increased upon its phosphorylation by upstream kinase MKNK1/2, while its dephosphorylated form can be inhibitory for translation initiation.Citation41 In this regard, silibinin significantly decreased IGF1-induced EIF4EBP1 phosphorylation at various sites as well as those of various translation initiation factors viz., EIF4E, EIF4G1 and EIF4B and also that of the EIF4E kinase, MKNK1/2 (). Consistent with these results, silibinin also affected cap-mediated translation as observed by an increase in EIF4EBP1 level bound to m7GTP resin (). Additional immunoprecipitation studies indicating increased binding of EIF4EBP1 to EIF4E () further confirmed the inhibitory effect of silibinin on translation initiation. Consistent with profound suppression of translation initiation, silibinin also strongly increased the inhibitory phosphorylation of EEF2 (eukaryotic translation elongation factor 2) at Thr56 (), indicating that it interferes with cellular protein synthesis by blocking both the initiation and elongation steps of translation.
Figure 8. Effect of silibinin treatment on cellular energy state in SW480 cells. Effect of silibinin on (A) activated PtdIns3K-AKT-MTOR pathway and (B) cap-dependent translation. (C) Effect of silibinin on activation of PRKAA2 due to altered AMP to ATP ratio. (D) Effect of silibinin on mitochondrial metabolism in SW480 cells by as early as 4 h: as indicated by the effect on high energy phosphates, de novo 13C TCA cycle production (including 13C-glutamate) and activation of glycolysis (de novo 13C-lactate) assessed by 31P- and 13C-NMR. All experimental procedures and data analysis were performed as detailed in Materials and Methods. *p < 0.05; **p < 0.03; ***p < 0.001.
Silibinin causes energy restrictions in CRC cells as assessed by PRKAA2 activation
PRKAA2 functions as a fuel sensor, wherein it gets activated by phosphorylation due to a decrease in cellular ATP levels with a concomitant increase in AMP levels, resulting in inhibition of anabolic pathways via inhibition of MTOR activity and related downstream targets.Citation40,Citation42 Accordingly, we also examined silibinin effect on PRKAA2 activation, and found that it significantly activated PRKAA2 which was also corroborated with an increase in phospho-ACACA/B, a substrate of PRKAA2Citation40 (). This result was quite significant since ACACA/B plays a major role in fatty acid synthesis and is inactivated upon phosphorylation.Citation40 All together, these results suggest potential of silibinin in inducing energy restrictions within SW480 cells, resulting in decreased ATP/AMP ratio.
Silibinin causes metabolic alterations in CRC cells
We also examined the global metabolic profile, including glucose metabolism, energy state and lipid metabolism in CRC SW480 cells after silibinin treatment. The earliest metabolic effect observed in silibinin-treated cells was impairment of mitochondrial oxidation as seen by de novo fate of [1-13C]glucose (). Specifically, the mitochondrial glucose metabolism in SW480 cells was significantly inhibited by silibinin, as observed by decreased [2,3,4-13C]glutamate formation (by 13C-NMR) and accumulation of Krebs cycle substrates like pyruvate and succinate (1H-NMR), indicating that normal functioning of Krebs cycle was impaired (). As calculated from the 13C-NMR spectra of silibinin-treated cell extracts, the concentration of all de novo produced 13C-mitochondrial Krebs cycle-derived metabolites was consequently significantly decreased (). Inhibition of the Krebs cycle by silibinin was accompanied by the decrease in intracellular 13C-glucose with a concomitant increase in de novo produced [13C]lactate indicated that glucose metabolism shifted to the cytosolic glycolysis type upon silibinin treatment (). The time-course of silibinin effects on glucose uptake and lactate release was also studied in the media of silibinin-treated SW480 cells at 4, 24 and 48 h of treatment. While glucose uptake was markedly reduced as a function of time, there was also a significant increase in extracellular lactate export implying an increase in cellular death with increased silibinin exposure-time (; ). Furthermore, the shutdown of mitochondrial metabolism by silibinin accompanied a lower energy state of cells as indicated by 31P-NMR spectra of cell extracts (). The energy state (cell energy balance), calculated as nucleoside triphosphate to nucleoside diphosphate (NTP/NDP) ratios,Citation43 was significantly decreased by silibinin (; ). Silibinin also significantly decreased the levels of phosphocholine (PC), a phosphatidylcholine (Ptd-Cho) precursor:Citation43 the major phospholipid of cell membrane, and altered its turnover as evident by decreased ratio of PC to GPC (Ptd-Cho catabolic product) (). Total choline levels were also reduced by silibinin, indicating its antiproliferative effect (). Furthermore, decreased nucleosides level implied a decreased dividing cell population, which was consistent with silibinin-caused S-phase arrest.
Table 1. Quantitative metabolic profile of untreated (control) and silibinin-treated (100 μM for 4 h) SW480 cells after incubation with [1-13C] glucose by high-resolution multinuclear NMR spectroscopya
Discussion
To survive extreme physiological conditions such as nutrient starvation, and in response to certain developmental and pathological situations, cells employ the catabolic process of autophagy.Citation8,Citation9 Whether the autophagy is initiated in response to prodeath or prosurvival signals is, however, controversial.Citation3,Citation8 Nevertheless, new evidence establishes the potential of macroautophagy (referred to as autophagy in the text) to lead to “type II programmed cell-death,” a degradative process that involves sequestration of cytoplasmic components in double-membrane vesicles (autophagic vesicles/autophagosomes) that fuse with lysosomes to form autolysosomes, wherein the engulfed material is hydrolyzed.Citation3,Citation8,Citation9 In conditions when the total cellular area encompassed by autophagic vacuoles becomes more than or is equal to that of cytosol and organelles outside the autophagic vesicles, autophagic activity results in total collapse of the cellular functions which leads to an irreversible state of ‘absolute cannibalization’ of the cell.Citation3 Thus, when autophagy cannot restore normal functioning of cells, cell death is inevitable.Citation8
Since autophagy can block apoptosis (due to organelle sequestration) and both autophagy and apoptosis are known to cause cell death, their regulation is not only coordinated, but recent studies show that molecules that are core components of the apoptosis or autophagic machinery like AKT and BCL2 family members, regulate both processes directly.Citation3,Citation6,Citation8 Apoptosis and autophagy may either manifest themselves in a mutually exclusive manner or may occur simultaneously in a cellular system.Citation3,Citation6 In such instances, we can use autophagy to our benefit, by two possible strategies: i) by inducing autophagy and enhancing the antitumor effect, ii) by inhibiting autophagy and inducing apoptosis; thus both induction and inhibition of autophagy, in such cases, lead to augmentation of antitumor effects.Citation6,Citation8 The current study identifies silibinin, for the first time, as one such natural agent capable of inducing both types of cell death.
EGF and IGFI are potent mitogens that regulate proliferation and survival of CRC cells via autocrine and paracrine loops involving both MAPK- and AKT-mediated signaling.Citation44 Therefore, the initial aim of our study was to assess silibinin effect on these mitogens-stimulated signaling and associated biological responses in human CRC cells. Over the course of the study, we observed intense vacuolization and cytoplasmic contractile features in SW480 cells by silibinin treatment under serum conditions, eventually leading to cell death, which was neither apoptotic nor necrotic. Even more intriguing was the observation that silibinin induces similar phenomenon more acutely over a shorter time period under serum-starved conditions. Thus, we hypothesized that the mechanism by which silibinin was causing vacuolization leading to cell death was being aggravated by the absence of nutrients/serum in the media which hastened the vacuolization process and eventually led to early cell death compared with when the treatment was carried under nutrient/serum conditions. The pattern of morphological changes observed was indicative of stress-like conditions that the cells had to endure, ultimately resulting in their death. Different possible explanations could be attributed to such a phenomenon. We hypothesized that the cells were under oxidative stress after silibinin treatment, which in turn was causing enlargement of cellular organelles, especially, mitochondria, reflected in terms of vacuolar appearance. The other possibility was that silibinin was interfering with the nutrient supply/uptake in the cells and inducing starvation-like conditions, which in turn was inducing autophagic death; explaining the appearance of vacuoles within the cells, the hallmark of autophagy. We, therefore, investigated both possibilities by carrying out specific experiments that could help identify the mechanism via which silibinin was causing its effect.
As summarized in , we found that silibinin rapidly induces oxidative stress in SW480 cells due to ROS generation with a concomitant dissipation of Δψm, CYCS release and apoptosis. Since mitochondrial changes occurred well before any other morphological changes, it was evident that disruption of Δψm was not a result but preceded cell death. Longer-time silibinin exposure caused ‘S’ phase arrest, intensified cytoplasmic vacuolization followed by sequestration of the organelles, inhibiting further release of CYCS (). Interestingly, decrease in apoptotic response correlated with increased autophagic events as evidenced by tracking the dynamics of LC3-II within the cells. EM-based assessment of cellular morphology further confirmed that together with autophagy induction and maturation to completion, the rough ER was considerably swollen in treated cells indicating silibinin potential to also induce ER stress in conjunction with autophagy (). Further mechanistic studies revealed that silibinin strongly inhibits the activation of ligand-induced PIK3CA-AKT–MTOR pathway but activates the MAP2K1/2- MAPK1/3 pathway. Coincident with this effect, silibinin induced energy restriction wherein an enhanced AMP/ATP ratio caused an increase in PRKAA2 activation, which transduced inhibitory signals to the MTOR pathway for autophagy induction. Downregulation of MTOR activity by silibinin also resulted in suppression of protein translation initiation complex leading to suppression of Cap-dependent translation. Together, these findings made for an intriguing observation, as it has been recognized that signals conveyed from damaged mitochondria to stimulate autophagy may involve MTOR since this protein was recently found associated to the mitochondrial membrane and helped sense stress-induced mitochondrial dysfunction.Citation6,Citation45 Whereas molecular link between autophagy and ER stress remains largely undefined, evidence suggests that stimulation of PRKAA2 may play a major role.Citation6,Citation45,Citation46 Starvation-induced autophagic death by silibinin was further confirmed by the metabolic profile of the cells where NMR results revealed that silibinin does shut down oxidative metabolism in mitochondria as an early effect, followed by significant decrease in cellular glucose uptake, causing an overall decrease in energy state within the cells eventually leading to cell death and accumulation of lactate ().
Figure 9. Model depicting the possible molecular mechanisms of CRC cell death by silibinin.
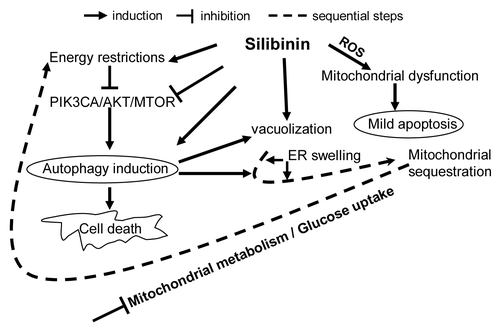
The close connections between apoptotic and autophagic machineries would be expected to lead to a situation where by manipulating the continuum it may be possible to alter the relative amount of type of cell death that occurs as cells die. This is due to the fact that if everything is commonly regulated, alteration of one mechanism should also affect the other or there could be simultaneous activation of these processes.Citation6 Consistent with this, in dose-escalation, a 200 μM silibinin dose led to significant release of CYCS, caused by a rapid disruption of mitochondrial membrane leading to significant apoptotic death, even before significant autophagic events could take over the cellular machinery (). Thus in summary, following silibinin treatment, CRC cells would die by apoptosis when autophagy is not yet initiated or is inhibited; while in conditions when silibinin induces autophagy, apoptosis would be blocked and cells would die by a nonapoptotic mechanism. However, in both cases, cellular damage to tumor cells by silibinin is so severe that it cannot be impaired due to sustained interference in essential cellular processes such as mitochondrial metabolism, glucose uptake and protein synthesis; the ultimate result is that no matter what, the tumor cells cannot survive in the long-term. These CRC cell results are important as we have found that silibinin has no toxic effects on normal colonic tissueCitation19,Citation20 and normal human colon NCM460 cells (data not shown). Most importantly, silibinin was able to specifically induce oxidative stress in the CRC cells as evident by a lack of ROS generation in normal colon NCM460 cells (data not shown). One possibility is that in normal colon cells, silibinin, a known antioxidant, displays a protective antioxidant effect; while in cancerous cells, the inherent antioxidant guard mechanisms to counteract ROS are inadequate and thus these cells display redox vulnerability which on exposure to silibinin causes them to generate more oxidative stress so as to compromise cellular viability. We do realize that more mechanistic studies need to be performed in the future to determine the mechanism associated with the differential response of CRC cells and normal colon cells to silibinin.
Together the findings in the present study are both novel and highly significant in establishing, for the first time, silibinin potential to cause both apoptotic and autophagic types of programmed cell-death in CRC cells. This suggests that silibinin harbors a deadly ‘double-edged sword’ against CRC cells, thereby, further advocating its clinical effectiveness against this malignancy. Considering that silibinin consumption is safe with high bioavailability in colonic tissue of CRC patients (more notable it is in clinical trials in CRC patients at least in Europe), silibinin can be useful in significantly lowering the morbidity and mortality associated with CRC.
Materials and Methods
Reagents
IGF1 (01-208) was from Millipore and EGF (13247-051) was from Invitrogen. N-acetyl-l-cysteine (A-9165), AA (A4544), ACTB antibody (A2228), E 64d (E8640), Pepstatin A (P5318), BAFA1 (B1793), 3MA (M9281) and MDC (30432) were from Sigma-Aldrich. Antibody for LC3B (NB600-1384) was from Novus Biologicals. Antibodies for BECN1 (612112) and CYCS (556433) were from BD PharMingen. Antibodies for HMOX1 (SPA-895D) and GPX (SPA-541E) were from Stressgen. Antibody for VDAC1 (ab16816) was from Abcam. Antibody for CAT (H000008471B1) was from Abnova. FeTCP (FeTCP) was from Frontier Scientific Inc. MnTBAP (475870) and PD98059 (513000) were from Calbiochem. Antibodies for pEGFR (2231, 2234 and 4407), pIGF1R (3024), IGF1R (3027), pMTOR (2971), pRPS6KB (9234), pRPS6 (2215), pEIF4EBP1(9451, 9455 and 2855), pEIF4E (9741), pEIF4G1 (2441), pEIF4B (3591), pMKNK1/2 (2111), pEEF2 (2331), pPRKAA2 (2535), PRKAA1/2(2532), pACACA/B (3661), ACACA/B (3662), ATG5 (2630), pAKT (9271), AKT (9272), pMAPK1/3 (9101), MAPK1/3, (9102), pMAP2K1/2 (9121), MAP2K1/2 (9122), pELK1 (9181), ELK1(9182), BCL2 (2876), pGSK3A/B (9331) and GSK3B (9338) were from Cell Signaling. Antibody for EGFR (06-129) was from Upstate and antibody for IRS1 (44-816G) was from Invitrogen. DHE (D-11347), MitoTracker Red (M7512) and DiOC6(3) (D-273) were from Molecular Probes. Antibodies for α-Tubulin (sc-5546) and MCL1 (sc-12756) were from Santa Cruz Biotechnology.
Cell culture and treatments
SW480, HT29 and LoVo cells were from ATCC and cultured in Leibovitz L-15, DMEM and F12 media (Gibco) respectively, containing 10% FBS and 1% penicillin-streptomycin. All three cell lines were tested and authenticated by polymorphic short tandem repeat profiling. At 60% confluency, cells were serum starved (SS) for 24 h, treated with 100 μM silibinin and after 2 h, stimulated with EGF or IGF1 (50 ng/mL), and harvested as described previously.Citation47 In experiments, using inhibitors like antioxidants, PD98059, and 3MA, cells were pretreated with inhibitors for 2 h followed by silibinin treatment. For autophagic flux determination, lysosomal inhibitors: E64d and pepstatin A were added to the media with silibinin and incubated for respective time periods. BAFA1 was added to the media in the last 4 h of treatment. Trypan blue dye-exclusion and/or MTT assay was used to assess cell viability.Citation47 For in vitro clonal analysis, single cells were plated at a density of 1000 cells/well in a six well plate and cultured for 1 week in media under serum conditions with or without silibinin, which was then replenished after every 72 h. Number of cells/ individual clones was counted at experiment end, and clones with 50 or > 50 cells scored as true colony. In other experiments, following desired treatments, cell pellets were suspended in 0.5 ml of saponin/PtdIns solution followed by FACS analysis for cell cycle distribution.Citation16 DNA fragmentation (apoptotic population), after desired treatments, was determined using Cell death Detection ELISA plus kit (Roche). Cellular kinase assays were performed using specific non-radioactive kits (MAPK1/3, 9800 and AKT, 9840) from Cell Signaling, while the cell-free kinase assay was done as described previously.Citation48 Briefly, for measuring MAPK1/3 kinase activity, ELK1 protein was used as a substrate. Following the kinase activity, the level of ELK1 phosphorylation was detected by western blotting using pELK1 (Ser383) antibody. For measuring AKT kinase activity, GSK-3 fusion protein was used as a substrate, and following the kinase activity assay, the GSK3A/B phosphorylation was detected by western blotting using pGSK3A/B (Ser21/9) antibody. Cytosolic, mitochondrial, and total cellular lysates were prepared, protein concentrations determined, and western blots performed followed by chemiluminescence ECL detection as described previously.Citation16
Identification of ROS generation
Generation of intracellular superoxide radical and dissipation of Δψm in SW480 cells was measured using DHE dye and cationic dye DiOC6(3), respectively. Briefly, after desired treatment, cells were incubated with 5 μM DHE or 40 nM DiOC6(3) for 20 min at 37°C and observed under fluorescent microscope, or harvested and analyzed immediately via FACS analysis.Citation25
Identification of autophagy induction
To correctly identify autophagy induction, step by step verification was done to avoid misinterpretation, as per published guidelines.Citation32,Citation35,Citation49,Citation50 For visualization of intracellular vacuoles, MDC was applied to the cells as described previously.Citation33,Citation34 Briefly, MDC was added to the cells at 50 μM for 30–45 min followed by 10 min incubation with NH4Cl (to reduce lysosomal staining) and washed twice with PBS plus 10% FBS and unfixed cells were directly observed under fluorescent microscope. For measurement of intracellular MDC fluorescence, cells were collected in 10 mM TRIS-HCl/0.1%Triton X-100, pH 8 and quantified by fluorescence photometry (Ex380nm and Em 525nm); values were normalized to number of live cells and expressed as specific activity relative to control.Citation33,Citation34 EM for visualization of cellular vacuoles was performed, as described previously, using FEI Technai G2 BioTwin (FEI Company) at 80 KV, and images were captured with Gatan First Lite digital camera (Gatan, Inc.).Citation35,Citation51-Citation53
Cap-dependent translation
It was done by assessing the association of EIF4EBP1and EIF4E by determining how much EIF4E co-immunoprecipitated with EIF4EBP1 antibody and by measuring how much EIF4EBP1 was recovered when EIF4E was purified with m7GTP-Sepharose resin (GE Amersham, 27-5025).Citation41,Citation54,Citation55 Briefly, cell lysates were incubated with EIF4EBP1 antibody coupled to protein A/G-agarose (Santacruz Biotechnology) and amount of EIF4E coimmunoprecipitated was determined by immunoblot using EIF4E antibody. For affinity purification of EIF4EBP1-EIF4E, m7GTP-Sepharose resin was incubated with cell lysates and EIF4EBP1 bound to EIF4E was detected by immunobloting.Citation41,Citation54,Citation55 m7GTP-Sepharose was chosen because it resembles 5′ mRNA cap structure and is successfully used to isolate EIF4E, the cap-binding protein.Citation41,Citation54,Citation55 The relative amounts of EIF4E and EIF4EBP1 were determined by densitometric analysis of appropriate bands.
NMR spectroscopy experiments
Cells (10–30 × 107) were incubated with 5 mM [1-13C] glucose (Cambridge Isotope Laboratories, CLM-420-0) for 4 h with or without silibinin, and then extracted with perchloric acid; both water soluble and lipid extracts were then subjected to high resolution 1H-, 13C- and 31P-NMR experiments as described previously.Citation43,Citation56 In addition, cell culture media were collected after 4, 24 and 48 h of silibinin treatment (last 4 h in the presence of [1-13C] glucose) for glucose uptake/ lactate export studies. All experiments were performed at the Metabolomics NMR Core at University of Colorado Cancer Center.
Total RNA extraction and gene expression analysis
Total RNA was extracted from SW480 cells treated with 100 μM silibinin or DMSO for 48 h using Trizol extraction method. Reverse transcription was performed using 2–3 μg of RNA and the First strand system for RT-PCR (SAbiosciences). Human Autophagy RTCitation2 Profiler TM PCR Array (SAbiosciences) was used to evaluate the effect of silibinin on expression of genes involved in autophagy. Twenty-five microliters of cocktail mix (cDNA sample and SYBR green probe mix, prepared immediately before the real-time analysis) was added per each well in 96-well plate. A two-step cycling protocol was used on an ABI 7000 cycler (Applied Biosystems, Inc.); 10 min at 95°C followed by 40 cycles of 15 sec at 95°C and 1min at 60°C. The threshold cycle (Ct) values for each well were calculated and genes with Ct values above 35 were considered undetected. Baseline and threshold values were manually set at the same values for comparison of untreated and treated samples. The data was analyzed using the software provided by the manufacturer.
Xenograft tissues
SW480 xenograft tissues from athymic (nu/nu) nude male mice orally gavaged with either silibinin (dose: 200 mg/kg body weight) in 0.2 ml of 0.5% carboxymethyl cellulose (CMC), or with CMC alone, respectively, 5 d/week for 6 weeks, were used in the present study.Citation17 Tumor samples were subjected to immunoblotting and immunohistochemical analysis,Citation17 using specific SQSTM1 primary antibody (Progen Biotechnik, GP62-C). Immunoreactivity (represented by intensity of brown staining) was scored as 0 (no staining), +1 (very weak), +2 (weak), +3 (moderate) and +4 (strong).
Statistical and microscopy analyses
Data shown are mean ± SE of two independent experiments each done in triplicate. The difference between silibinin vs. control or respective ligand group was analyzed by unpaired two-tailed Student’s t-test. Fisher’s Exact test was used to compare incidence of colony formation in control vs. silibinin group. Densitometric analysis of immunoblots (adjusted with ACTB/α-Tubulin as loading control) was done by Scion Image program (NIH) and values are indicated below blots in respective figures. Some blots were multiplexed or stripped and reprobed with different antibodies including those for loading control. All microscopic immunohistochemical analyses were done with a Zeiss Axioscope 2 microscope (Carl Zeiss, Inc.) and photomicrographs were captured by Carl Zeiss AxioCam MrC5 camera. In NMR experiments, all numerical data are presented as means ± SD (n = 4 for each group). All absolute individual concentrations of distinguished biomarkers were analyzed by ANOVA followed by Tukey’s post-hoc test to identify the groups that differed significantly. All statistical analyses were performed with Sigma Plot-version 9.01, Systat Software, and SPSS version 14.0, SPSS Inc., and two sided p values < 0.05 were considered significant.
| Abbreviations: | ||
| CRC | = | colorectal cancer |
| DHE | = | dihydroethidium |
| MDC | = | monodansylcadaverine |
| ER | = | endoplasmic reticulum |
| EM | = | transmission electron microscopy |
| NAC | = | N-acetyl cysteine |
| AA | = | ascorbic acid |
| RTKs | = | receptor tyrosine kinases |
| BAFA1 | = | bafilomycin A1 |
| CMC | = | carboxymethyl cellulose |
| MTT | = | 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide |
| EGF | = | epidermal growth factor |
| EGFR | = | EGF receptor |
| IGF1 | = | insulin-like growth factor-1 |
| IGF1R | = | IGF1 receptor |
Additional material
Download Zip (143.9 KB)Acknowledgments
Supported by RO1 grant CA112304, University of Colorado Cancer Center P30 grant CA046934, and Colorado Clinical and Translational Sciences Institute UL1 award RR025780.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/23960
References
- American Cancer Society. . Cancer Facts and Figures 2012.
- Chau I, Cunningham D. Adjuvant therapy in colon cancer--what, when and how?. Ann Oncol 2006; 17:1347 - 59; http://dx.doi.org/10.1093/annonc/mdl029; PMID: 16524974
- Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004; 23:2891 - 906; http://dx.doi.org/10.1038/sj.onc.1207521; PMID: 15077152
- Li J, Hou N, Faried A, Tsutsumi S, Takeuchi T, Kuwano H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann Surg Oncol 2009; 16:761 - 71; http://dx.doi.org/10.1245/s10434-008-0260-0; PMID: 19116755
- Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004; 6:1221 - 8; http://dx.doi.org/10.1038/ncb1192; PMID: 15558033
- Thorburn A. Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis 2008; 13:1 - 9; http://dx.doi.org/10.1007/s10495-007-0154-9; PMID: 17990121
- Wils J, O’Dwyer P, Labianca R. Adjuvant treatment of colorectal cancer at the turn of the century: European and US perspectives. Ann Oncol 2001; 12:13 - 22; http://dx.doi.org/10.1023/A:1008357725209; PMID: 11249040
- Kondo Y, Kondo S. Autophagy and cancer therapy. Autophagy 2006; 2:85 - 90; PMID: 16874083
- Reggiori F, Klionsky DJ. Autophagosomes: biogenesis from scratch?. Curr Opin Cell Biol 2005; 17:415 - 22; http://dx.doi.org/10.1016/j.ceb.2005.06.007; PMID: 15978794
- Serkova NJ, Niemann CU. Pattern recognition and biomarker validation using quantitative 1H-NMR-based metabolomics. Expert Rev Mol Diagn 2006; 6:717 - 31; http://dx.doi.org/10.1586/14737159.6.5.717; PMID: 17009906
- Serkova NJ, Spratlin JL, Eckhardt SG. NMR-based metabolomics: translational application and treatment of cancer. Curr Opin Mol Ther 2007; 9:572 - 85; PMID: 18041668
- Griffin JL, Kauppinen RA. Tumour metabolomics in animal models of human cancer. J Proteome Res 2007; 6:498 - 505; http://dx.doi.org/10.1021/pr060464h; PMID: 17269706
- Griffin JL, Shockcor JP. Metabolic profiles of cancer cells. Nat Rev Cancer 2004; 4:551 - 61; http://dx.doi.org/10.1038/nrc1390; PMID: 15229480
- Denkert C, Budczies J, Weichert W, Wohlgemuth G, Scholz M, Kind T, et al. Metabolite profiling of human colon carcinoma--deregulation of TCA cycle and amino acid turnover. Mol Cancer 2008; 7:72; http://dx.doi.org/10.1186/1476-4598-7-72; PMID: 18799019
- Wang H, Tso VK, Slupsky CM, Fedorak RN. Metabolomics and detection of colorectal cancer in humans: a systematic review. Future Oncol 2010; 6:1395 - 406; http://dx.doi.org/10.2217/fon.10.107; PMID: 20919825
- Agarwal C, Singh RP, Dhanalakshmi S, Tyagi AK, Tecklenburg M, Sclafani RA, et al. Silibinin upregulates the expression of cyclin-dependent kinase inhibitors and causes cell cycle arrest and apoptosis in human colon carcinoma HT-29 cells. Oncogene 2003; 22:8271 - 82; http://dx.doi.org/10.1038/sj.onc.1207158; PMID: 14614451
- Kaur M, Velmurugan B, Tyagi A, Agarwal C, Singh RP, Agarwal R. Silibinin suppresses growth of human colorectal carcinoma SW480 cells in culture and xenograft through down-regulation of β-catenin-dependent signaling. Neoplasia 2010; 12:415 - 24; PMID: 20454513
- Kaur M, Velmurugan B, Tyagi A, Deep G, Katiyar S, Agarwal C, et al. Silibinin suppresses growth and induces apoptotic death of human colorectal carcinoma LoVo cells in culture and tumor xenograft. Mol Cancer Ther 2009; 8:2366 - 74; http://dx.doi.org/10.1158/1535-7163.MCT-09-0304; PMID: 19638451
- Rajamanickam S, Kaur M, Velmurugan B, Singh RP, Agarwal R. Silibinin suppresses spontaneous tumorigenesis in APC min/+ mouse model by modulating beta-catenin pathway. Pharm Res 2009; 26:2558 - 67; http://dx.doi.org/10.1007/s11095-009-9968-1; PMID: 19779968
- Rajamanickam S, Velmurugan B, Kaur M, Singh RP, Agarwal R. Chemoprevention of intestinal tumorigenesis in APCmin/+ mice by silibinin. Cancer Res 2010; 70:2368 - 78; http://dx.doi.org/10.1158/0008-5472.CAN-09-3249; PMID: 20215518
- Ravichandran K, Velmurugan B, Gu M, Singh RP, Agarwal R. Inhibitory effect of silibinin against azoxymethane-induced colon tumorigenesis in A/J mice. Clin Cancer Res 2010; 16:4595 - 606; http://dx.doi.org/10.1158/1078-0432.CCR-10-1213; PMID: 20823143
- Singh RP, Gu M, Agarwal R. Silibinin inhibits colorectal cancer growth by inhibiting tumor cell proliferation and angiogenesis. Cancer Res 2008; 68:2043 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-07-6247; PMID: 18339887
- Velmurugan B, Gangar SC, Kaur M, Tyagi A, Deep G, Agarwal R. Silibinin exerts sustained growth suppressive effect against human colon carcinoma SW480 xenograft by targeting multiple signaling molecules. Pharm Res 2010; 27:2085 - 97; http://dx.doi.org/10.1007/s11095-010-0207-6; PMID: 20628792
- Hoh C, Boocock D, Marczylo T, Singh R, Berry DP, Dennison AR, et al. Pilot study of oral silibinin, a putative chemopreventive agent, in colorectal cancer patients: silibinin levels in plasma, colorectum, and liver and their pharmacodynamic consequences. Clin Cancer Res 2006; 12:2944 - 50; http://dx.doi.org/10.1158/1078-0432.CCR-05-2724; PMID: 16675592
- Hail N Jr.. Mitochondrial reactive oxygen species affect sensitivity to curcumin-induced apoptosis. Free Radic Biol Med 2008; 44:1382 - 93; http://dx.doi.org/10.1016/j.freeradbiomed.2007.12.034; PMID: 18206126
- Lin X, Li Q, Wang YJ, Ju YW, Chi ZQ, Wang MW, et al. Morphine inhibits doxorubicin-induced reactive oxygen species generation and nuclear factor kappaB transcriptional activation in neuroblastoma SH-SY5Y cells. Biochem J 2007; 406:215 - 21; http://dx.doi.org/10.1042/BJ20070186; PMID: 17542780
- Negre-Salvayre A, Augé N, Duval C, Robbesyn F, Thiers JC, Nazzal D, et al. Detection of intracellular reactive oxygen species in cultured cells using fluorescent probes. Methods Enzymol 2002; 352:62 - 71; http://dx.doi.org/10.1016/S0076-6879(02)52007-3; PMID: 12125377
- Zhang Z, Leonard SS, Huang C, Vallyathan V, Castranova V, Shi X. Role of reactive oxygen species and MAPKs in vanadate-induced G(2)/M phase arrest. Free Radic Biol Med 2003; 34:1333 - 42; http://dx.doi.org/10.1016/S0891-5849(03)00145-X; PMID: 12726921
- Day BJ, Batinic-Haberle I, Crapo JD. Metalloporphyrins are potent inhibitors of lipid peroxidation. Free Radic Biol Med 1999; 26:730 - 6; http://dx.doi.org/10.1016/S0891-5849(98)00261-5; PMID: 10218663
- Javvadi P, Segan AT, Tuttle SW, Koumenis C. The chemopreventive agent curcumin is a potent radiosensitizer of human cervical tumor cells via increased reactive oxygen species production and overactivation of the mitogen-activated protein kinase pathway. Mol Pharmacol 2008; 73:1491 - 501; http://dx.doi.org/10.1124/mol.107.043554; PMID: 18252805
- Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, et al. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1998; 1366:177 - 96; http://dx.doi.org/10.1016/S0005-2728(98)00112-1; PMID: 9714796
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542 - 5; PMID: 17611390
- Høyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jäättelä M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ 2005; 12:1297 - 309; http://dx.doi.org/10.1038/sj.cdd.4401651; PMID: 15905882
- Munafó DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci 2001; 114:3619 - 29; PMID: 11707514
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003
- Pattingre S, Bauvy C, Codogno P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J Biol Chem 2003; 278:16667 - 74; http://dx.doi.org/10.1074/jbc.M210998200; PMID: 12609989
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2:489 - 501; http://dx.doi.org/10.1038/nrc839; PMID: 12094235
- McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal 2006; 8:1775 - 89; http://dx.doi.org/10.1089/ars.2006.8.1775; PMID: 16987031
- Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev 1998; 12:502 - 13; http://dx.doi.org/10.1101/gad.12.4.502; PMID: 9472019
- Jiang W, Zhu Z, Thompson HJ. Dietary energy restriction modulates the activity of AMP-activated protein kinase, Akt, and mammalian target of rapamycin in mammary carcinomas, mammary gland, and liver. Cancer Res 2008; 68:5492 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-07-6721; PMID: 18593953
- Pyronnet S, Dostie J, Sonenberg N. Suppression of cap-dependent translation in mitosis. Genes Dev 2001; 15:2083 - 93; http://dx.doi.org/10.1101/gad.889201; PMID: 11511540
- Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, et al. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem 2006; 281:34870 - 9; http://dx.doi.org/10.1074/jbc.M605488200; PMID: 16990266
- Klawitter J, Anderson N, Klawitter J, Christians U, Leibfritz D, Eckhardt SG, et al. Time-dependent effects of imatinib in human leukaemia cells: a kinetic NMR-profiling study. Br J Cancer 2009; 100:923 - 31; http://dx.doi.org/10.1038/sj.bjc.6604946; PMID: 19259085
- Asghar U, Hawkes E, Cunningham D. Predictive and prognostic biomarkers for targeted therapy in metastatic colorectal cancer. Clin Colorectal Cancer 2010; 9:274 - 81; http://dx.doi.org/10.3816/CCC.2010.n.040; PMID: 21208841
- Desai BN, Myers BR, Schreiber SL. FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc Natl Acad Sci U S A 2002; 99:4319 - 24; http://dx.doi.org/10.1073/pnas.261702698; PMID: 11930000
- Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy. Int J Cell Biol 2010; 2010:930509; http://dx.doi.org/10.1155/2010/930509; PMID: 20145727
- Gu M, Raina K, Agarwal C, Agarwal R. Inositol hexaphosphate downregulates both constitutive and ligand-induced mitogenic and cell survival signaling, and causes caspase-mediated apoptotic death of human prostate carcinoma PC-3 cells. Mol Carcinog 2010; 49:1 - 12; PMID: 19544333
- Tyagi A, Agarwal R, Agarwal C. Grape seed extract inhibits EGF-induced and constitutively active mitogenic signaling but activates JNK in human prostate carcinoma DU145 cells: possible role in antiproliferation and apoptosis. Oncogene 2003; 22:1302 - 16; http://dx.doi.org/10.1038/sj.onc.1206265; PMID: 12618755
- Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy 2005; 1:23 - 36; http://dx.doi.org/10.4161/auto.1.1.1495; PMID: 16874023
- Kimura S, Fujita N, Noda T, Yoshimori T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol 2009; 452:1 - 12; http://dx.doi.org/10.1016/S0076-6879(08)03601-X; PMID: 19200872
- Eskelinen EL. To be or not to be? Examples of incorrect identification of autophagic compartments in conventional transmission electron microscopy of mammalian cells. Autophagy 2008; 4:257 - 60; PMID: 17986849
- Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol 2004; 36:2491 - 502; http://dx.doi.org/10.1016/j.biocel.2004.02.005; PMID: 15325587
- Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol 2009; 452:143 - 64; http://dx.doi.org/10.1016/S0076-6879(08)03610-0; PMID: 19200881
- Graves LM, Bornfeldt KE, Argast GM, Krebs EG, Kong X, Lin TA, et al. cAMP- and rapamycin-sensitive regulation of the association of eukaryotic initiation factor 4E and the translational regulator PHAS-I in aortic smooth muscle cells. Proc Natl Acad Sci U S A 1995; 92:7222 - 6; http://dx.doi.org/10.1073/pnas.92.16.7222; PMID: 7638171
- Haller AA, Sarnow P. In vitro selection of a 7-methyl-guanosine binding RNA that inhibits translation of capped mRNA molecules. Proc Natl Acad Sci U S A 1997; 94:8521 - 6; http://dx.doi.org/10.1073/pnas.94.16.8521; PMID: 9238009
- Raina K, Serkova NJ, Agarwal R. Silibinin feeding alters the metabolic profile in TRAMP prostatic tumors: 1H-NMRS-based metabolomics study. Cancer Res 2009; 69:3731 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-09-0096; PMID: 19366793