Abstract
We recently found that activation of IL17A signaling promotes the development and progression of acute and chronic pulmonary fibrosis, and that the blockade of IL17A activity attenuates pulmonary fibrosis by promoting the resolution of inflammation and the activation of autophagy. Although the induction of autophagy stimulating the collagen degradation in the fibrotic lung tissue has been identified as a mechanism responsible for the antifibrotic role of targeting IL17A, it remains to be clarified how IL17A signaling suppresses autophagy. Here we report that the phosphorylation of B-cell CLL/lymphoma 2 (BCL2), an apoptosis regulatory protein, was inhibited in the presence of IL17A in lung epithelial cells, and this reduction suppressed the ubiquitination degradation of BCL2, which subsequently attenuated autophagy by promoting the interaction of BCL2 and BECN1. We found that IL17A regulated the phosphorylation of BCL2 through activating the phosphoinositide 3-kinase (PI3K)-glycogen synthase kinase 3 β (GSK3B) signaling cascade. In response to IL17A stimulation, PI3K was activated and resulted in phosphorylation of GSK3B at Ser9, which subsequently attenuated the interaction of GSK3B with BCL2. Interrupting the GSK3B and BCL2 interaction precluded the phosphorylation of BCL2 at Ser70, which could trigger the ubiquitination degradation, and restrained the ubiquitination degradation of BCL2. Consequently, a decrease in the BCL2 degradation induced by IL17A resulted in a suppressed autophagy in lung epithelial cells. These findings indicate that the IL17A-PI3K-GSK3B-BCL2 signaling pathway participates in the attenuation of autophagic activity in lung epithelial cells, which is attributed to be primarily responsible for the development and progression of IL17A-induced pulmonary fibrosis.
Introduction
Tissue fibrosis is a core pathogenic change and the underlying reason for a variety of incurable chronic diseases characterized by an excessive accumulation of extracellular matrix (ECM) leading to stiffening and/or scarring of the involved tissue, which destructs the normal architecture, affects function, and causes failure of tissue and organ.Citation1,Citation2 Because no convincing or effective therapeutics are available for the treatment of pulmonary fibrosis, more alarmingly, pulmonary fibrosis has a mortality rate that exceeds that of many cancers.Citation3 Recent studies indicate that tissue fibrosis is mainly driven by chronic inflammation and that the property of immune response is a critical factor for the development and progression of pulmonary fibrosis.Citation4 Indeed, the Th1-type immune response attenuates the development of pulmonary fibrosis by promoting the resolution of chronic inflammationCitation5 while the Th2-type immune response critically contributes to the development of pulmonary fibrosis by suppressing inflammation resolution and promoting tissue repairing.Citation6 Wilson et al. found that interleukin-17A (IL17A) induces pulmonary fibrosis and knockdown of IL17A receptor reduces pulmonary fibrosis.Citation7 Our recent studies indicate that IL17A promotes the development and progression of pulmonary fibrosis through TGFB1-dependent and -independent mechanisms.Citation8 IL17A stimulates not only the expression of collagen by increasing the release of the transforming growth factor β 1 (TGFB1) but also attenuates autophagy to suppress the collagen degradation in the fibrotic lung tissue in a TGFB1-independent manner.Citation8 Thus, regulating the property of immune response may be a promising therapeutic strategy for the prevention and treatment of pulmonary fibrosis.
Autophagy, a self-catabolic process that maintains intracellular homeostasis and determines cell fates under stress, has been recently found to be involved in the regulation of tissue repairing process and fibrosis.Citation6,Citation8 Indeed, autophagy has been recognized as a key mechanism for cellular homeostasis and survival in a variety of lung diseases.Citation9 The autophagy core complex, comprised of B-cell CLL/lymphoma 2 (BCL2), BECN1, phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3), phosphoinositide-3-kinase, regulatory subunit 4 (PIK3R4), ATG14, and AMBRA1, plays a crucial role in the autophagy activation process.Citation10 BCL2 binds to BECN1 by BH3 domain to protect the activation of autophagy under quiescent conditions. When cells are stimulated with a variety of stimuli, BCL2 is isolated from BECN1 and autophagic activity is enhanced in the cells.Citation10 Therefore, BCL2 is a negative switcher of autophagy activation. Our previous studies indicated that neutralizing IL17A reduces pulmonary fibrosis by the activation of autophagy, while IL17A-suppressed autophagy is associated with the overexpression of BCL2 in the fibrotic lung tissue.Citation8 However, the precise regulatory mechanism and signaling pathway of IL17A in the regulation of autophagic process remains unknown.
Glycogen synthase kinase 3 β (GSK3B) is a serine-threonine kinase, and it is involved in energy metabolism, neuronal cell development, and body pattern formation. GSK3B regulates these cellular activities through triggering the degradation of signaling or functional proteins. In the absence of WNT ligand, GSK3B binds to CTNNB1 and triggers the ubiquitination and proteasomal degradation of CTNNB1.Citation11 This situation is analogous to how GSK3B binds to SMAD3 and then triggers the degradation of SMAD3.Citation12 However, IL17A activates phosphoinositide 3-kinase (PI3K) and subsequently stimulates the phosphorylation Ser9 residues on GSK3B to inhibit GSK3B activity.Citation13 It suggests that BCL2 may be a target of GSK3B and that IL17A can inhibit the BCL2 degradation through suppressing the activity of GSK3B. Thus, IL17A may attenuate the activation of autophagy through promoting the interaction of BCL2 and BECN1 to interfere with the BECN1 activation.
In this study, we document that IL17A-activated PI3K-GSK3B signaling cascade is involved in the regulation of the BCL2-BECN1 interaction in lung epithelial cells. In response to IL17A stimulation, PIK3CA, the catalytic subunit of PI3K, is activated to induce the phosphorylation of GSK3B at Ser9, which subsequently attenuates the interaction of GSK3B with BCL2. Interrupting the GSK3B and BCL2 interaction precludes the phosphorylation of BCL2 at Ser70, which triggers the ubiquitination degradation of BCL2. Consequently, the increased expression of BCL2 interferes with the activation of BECN1 and attenuates autophagy in these cells. Our studies provide insight into the molecular mechanism and reveal a novel signal pathway for IL17A-mediated inhibition of autophagy.
Results
IL17A inhibits degradation of BCL2
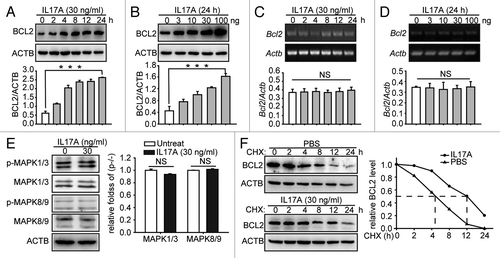
We recently reported that IL17A inhibits the starvation-induced autophagy by increasing the expression of BCL2 and blockade of IL17A activity restores the autophagic flux in the fibrotic lung tissue.Citation8 Here we further determined the exact regulatory effect of IL17A-enhanced BCL2 expression in cultured lung epithelial cells. We found that the treatment of lung epithelial cells with IL17A induced the protein but not mRNA expression of Bcl2 in time- and concentration-dependent manners (). It had been reported that the activation of MAPK1/3Citation14 and MAPK8/9Citation15 induce mRNA expression of Bcl2. We wondered whether the IL17A-induced expression of BCL2 was mediated by the activation of these two kinase enzymes in lung epithelial cells. We found that IL17A did not stimulate phosphorylation of MAPK1/3 and MAPK8/9 in these cells (). We therefore hypothesized that IL17A might increase the BCL2 expression via regulating the stability of BCL2. We found that the treatment of the cells with IL17A delayed the degradation of BCL2 in the presence of protein synthesis inhibitor cycloheximide CHX (). These data suggest that IL17A enhances BCL2 expression through inhibiting the degradation of BCL2 in lung epithelial cells.
Figure 1. IL17A increases BCL2 protein level by supporting the stability of BCL2. (A and B) IL17A promotes the expression of BCL2 in time- and concentration-dependent manners. The MLE-12 cells were treated with 30 ng/ml IL17A for the indicated times (A), or the cells were treated with indicated concentrations of IL17A for 24 h (B). Then the cell lysates were analyzed by immunoblotting. (C and D) The cells were treated and processed as described in (A and B) and total RNA was isolated. The mRNA of Bcl2 was analyzed by semiquantitative RT-PCR as indicated in Materials and Methods. (E) IL17A does not activate MAPK1/3 and MAPK8/9. The MLE-12 cells were treated with 30 ng/ml IL17A for 2 h, then the expression and phosphorylation level of MAPK1/3 and MAPK8/9 were detected by western blotting. (F) IL17A delays the degradation of BCL2. The cells were incubated with CHX (10 μg/ml) for the indicated times after 2 h of IL17A stimulation. Then the cell lysates were isolated for immunoblotting. ACTB was used as the loading control. Data are presented as the mean ± SE of four independent assays. ***p < 0.001; NS, non-significant.
IL17A increases the interaction of BCL2 and BECN1
PIK3C3, PIK3R4, BECN1, and ATG14 are key components of the autophagy core complex.Citation10 We observed previously the regulatory effect of IL17A on these proteins.Citation8 We found that the expression levels of PIK3C3, PIK3R4, BECN1, and ATG14 were decreased when lung epithelial cells were stimulated with IL17A (). These results further suggested that IL17A can inhibit starvation-induced autophagy. Apart from BCL2, BCL2 like 1 (BCL2L1) was also reported to suppress autophagy by interaction with BECN1.Citation16,Citation17 However, we did not find IL17A affecting the expression of BCL2L1 in our current system ().
Figure 2. IL17A enhances the interaction of BCL2 and BECN1. (A) IL17A reduces the expression of autophagy core complex-associated proteins. Cells were starved for 2 h with or without IL17A (30 ng/ml) respectively. Whole cell lysates were collected and the expression of p-BCL2, BCL2, BCL2L1, BECN1, PIK3R4, PIK3C3, and ATG14 detected by western blotting. Data are presented as the mean ± SE of four independent assays. (B) IL17A promotes the association of BCL2 with BECN1. Cells were transfected with BCL2-Myc and BECN1-Flag expression plasmids for 24 h, then were starved for 2 h with or without IL17A (30 ng/ml) for 2 h respectively. Cell lysates were extracted and coimmunoprecipitated with anti-Myc antibody. Precipitates were detected by western blotting with anti-Flag antibody. (C) IL17A enhances the interaction of BCL2 and BECN1 when cells are starved. Cells were starved with or without IL17A (30 ng/ml) for 2 h respectively. The cells were subsequently fixed by 4% paraformaldehyde, stained by fluorochromes, green for BCL2 and red for BECN1, scale bar: 0.5 μm. The interaction of BCL2 and BECN1 was indicated by white arrowheads. Data are representative images of three assays with identical results. **p < 0.01; ***p < 0.001; NS, non-significant.
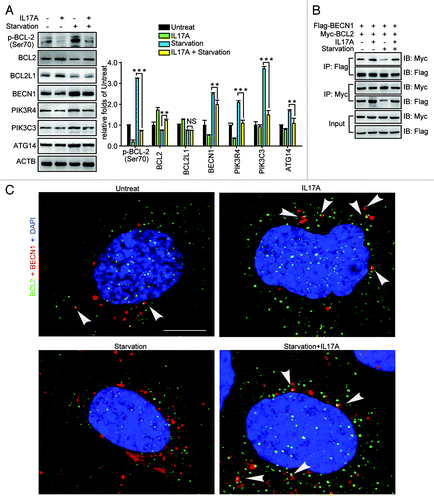
Phosphorylation of Ser70 of BCL2 promotes its dissociation from BECN1 so as to activate autophagy.Citation18 Because IL17A treatment suppressed the phosphorylation of BCL2 induced by starvation (), we speculated that IL17A might regulate the interaction between BCL2 and BECN1. The immunoprecipitation assay revealed that stimulation of lung epithelial cells with IL17A promoted the interaction of BCL2 and BECN1 (). Moreover, IL17A significantly enhanced the colocalization of BCL2 and BECN1 when cells were starved (). Taken together, our data indicate that stimulation of lung epithelial cells with IL17A promoted the interaction of BCL2 and BECN1, which restrains the activation of the autophagy core complex.
GSK3B facilitates the degradation of BCL2
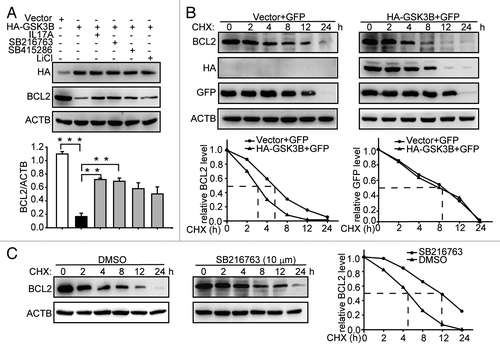
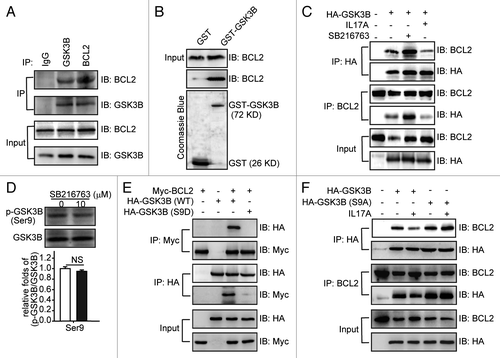
It had been reported that p-GSK3B is downregulated in the IL17A-deficient mouse.Citation19 Additionally, GSK3B promotes the degradation of MCL1, a member of the antiapoptotic BCL2 family.Citation20 We thus supposed that GSK3B might regulate the BCL2 stability induced by IL17A. We found that overexpression of GSK3B decreased the expression of BCL2 (). However, addition of IL17A and GSK3B inhibitors reversed the downregulatory effect of GSK3B on BCL2 (). Then we further determined whether GSK3B participated in the regulation of BCL2 stability. The overexpression of GSK3B promoted the degradation of BCL2 in the presence of CHX (). In contrast, GSK3B inhibitor SB216763 suppressed the degradation of BCL2 (). Taken together, these results indicate that GSK3B promoted the degradation of BCL2.
Figure 3. GSK3B promotes degradation of BCL2. (A) IL17A and GSK3B inhibitors reduce the degradation of BCL2. After transfecting with HA-GSK3B expression plasmids for 24 h, cells were incubated with IL17A (30 ng/ml), SB216763 (10 μM), SB415286 (50 μM), or LiCl (30 mM) for 2 h. Then cell lysates were detected by western blotting with anti-HA antibody and anti-BCL2 antibody. (B) Overexpression of GSK3B promotes degradation of BCL2. After transfecting with HA-GSK3B plasmids for 24 h, cells were incubated with CHX for the indicated times. The cells were also transfected with an identical amount of pEGFP-N1 plasmids to monitor the transfection efficiency. Cell lysates were extracted for immunoblotting. (C) SB216763 delays the BCL2 degradation. Cells were incubated with SB216763 for 2 h before CHX addition. Then cell lysates were extracted for immunoblotting. Data are presented as the mean ± SE (n = 4). **p < 0.01; ***p < 0.001.
IL17A activates PIK3CA to induce the phosphorylation of GSK3B at Ser9
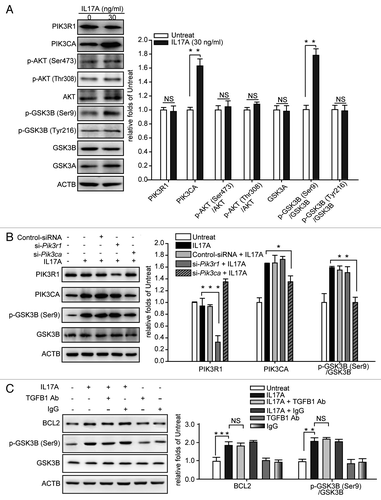
It reports that IL17A activates the PI3K signaling pathway,Citation13 and that PI3K inhibits GSK3B by the phosphorylation of GSK3B at Ser9.Citation21 The PIK3CA subunit is the catalytic subunit of PI3K, Zhao et al. have found that the phosphorylation of several downstream components of PI3K and the lipid kinase activity are attenuated in PIK3CA knockout cells, suggesting that the expression of PIK3CA is positively correlated with the activity of PI3K.Citation22 We found that treatment of lung epithelial cells with IL17A increased the expression of PIK3CA subunit of PI3K and induced the phosphorylation of GSK3B at Ser9, but IL17A did not alter the expression of phosphoinositide-3-kinase, regulatory subunit 1 (α) (PIK3R1), p-AKT (Ser473), p-AKT (Thr308), AKT, p-GSK3B (Tyr216), GSK3B, and GSK3A (). Moreover, IL17A could not induce the phosphorylation of GSK3B at Ser9 in cells, after silencing of Pik3ca but not Pik3r1 by specific siRNAs (). TGFB1 decreases the activity of GSK3B by phosphorylating GSK3B at Ser9.Citation23 However, we found that blocking TGFB1 did not change the IL17A induced expression of BCL2 and phosphorylation of GSK3B (), indicating that IL-17A-inhibted GSK3B-dependent degradation of BCL2 is independent of TGFB1. Together, these data suggest that IL17A inhibits GSK3B via activating the PIK3CA subunit of PI3K in lung epithelial cells.
Figure 4. IL17A inhibits GSK3B by activating PIK3CA to phosphorylate GSK3B at Ser9. (A) IL17A activates PIK3CA but not PIK3R1 and stimulates phosphorylation of GSK3B at Ser9. Cell lysates were collected after 2 h of incubation with or without IL17A (30 ng/ml), then the expression of indicated proteins was detected by immunoblotting. (B) PIK3CA is necessary for GSK3B (Ser9) phosphorylation when stimulated with IL17A. Cells transfected with si-Pik3r1 or si-Pik3ca were incubated with IL17A (30 ng/ml) for 2 h. Cell lysates were prepared, and the expression of indicated proteins was analyzed by western blotting. (C) IL17A inhibits GSK3B independently of TGFB1. After incubation with anti-TGFB1 antibody (2 μg/ml) or isotype- matched Ab (2 μg/ml) for 2 h, cells were treated with IL17A (30 ng/ml) for 2 h and cell lysates were extracted for immunoblotting. Data are presented as the mean ± SE (n = 4). *p < 0.05; **p < 0.01; ***p < 0.001; TGFB1 Ab, anti-TGFB1 antibody; NS, non-significant.
IL17A promotes the dissociation of GSK3B from BCL2
GSK3B functions as a signal molecule through interacting with several proteins and promoting their degradation.Citation12 We had found that GSK3B regulating the degradation of BCL2 in the presence of IL17A and the overexpression of GSK3B facilitated the degradation of BCL2 (), indicating that there might be an interaction between GSK3B and BCL2. We therefore examined if GSK3B interacted with BCL2 in the absence of IL17A. By using coimmunoprecipitation and GST pulldown assays, we found that BCL2 could bind to GSK3B in vivo and in vitro (). Then we examined the regulatory effect of IL17A on the BCL2-GSK3B interaction. As shown in , addition of IL17A reduced BCL2-GSK3B interaction remarkably, while inhibiting of GSK3B activity with SB216763 enhanced the association of BCL2 and GSK3B (). As shown above, both IL17A and SB216763 inhibited degradation of BCL2 induced by GSK3B. Why did they play different roles in the BCL2-GSK3B interaction? SB216763 is an ATP-competitive inhibitor, and we found that it did not impact the expression of GSK3B, Ser9 phosphorylation (). IL17A inhibited GSK3B by stimulating the phosphorylation of GSK3B at Ser9 (). Thus we suspected that phosphorylation of GSK3B at Ser9 might inhibit the association of GSK3B and BCL2. Indeed, we found that GSK3B-S9D mutant, who has the Ser9 residue replaced by aspartate to mimic phosphorylation, could not bind to BCL2 anymore (). On the contrary, the S9A mutant lacking the phosphorylation site, bound to BCL2 as well as the wild-type GSK3B did. Moreover, IL17A treatment could not reduce the binding of S9A mutant to BCL2 (). These data indicate that IL17A promotes the dissociation of GSK3B and BCL2 by stimulating the phosphorylation of GSK3B at Ser9.
Figure 5. IL17A promotes the dissociation of GSK3B and BCL2 by phosphorylating GSK3B at Ser9. (A) BCL2 interacts with GSK3B. Whole cell extracts were immunoprecipitated with anti-BCL2 antibody or equal amount of mouse IgG and blotted with an anti-GSK3B antibody. (B) In vitro binding between BCL2 and GSK3B. Cell lysates were incubated with equal amounts of GST or GST-GSK3B and analyzed by western blotting using the anti-BCL2 antibody. The presence of the GST fusion proteins was confirmed by staining gels with Coomassie Blue. (C) IL17A reduces the association between GSK3B and BCL2, while SB216763 induces it. Cells were transfected with HA-GSK3B plasmids for 24 h and then stimulated with or without IL17A (30 ng/ml) or SB216763 (10 μM) for 2 h. Cell lysates were immunoprecipitated with anti-HA antibody, and then blotted with anti-BCL2 antibody. (D) SB216763 could not phosphorylate GSK3B at Ser9. Cells were incubated with SB216763 (10 μM) for 2 h. Then cell lysates were collected for immunoblotting. Data are represented as the mean ± SE (n = 4). (E) Phosphorylation of GSK3B Ser9 inhibits its association with BCL2. BCL2-Myc was cotransfected with GSK3B-HA or its S9D-HA mutant. After 24 h transfection, whole cell extracts were immunoprecipitated with an anti-HA antibody and blotted with anti-Myc antibody. (F) Phosphorylation of GSK3B Ser9 is necessary for the dissociation of GSK3B and BCL2 regulated by IL17A. Cells were transfected with GSK3B-HA and S9A-HA mutant expression plasmids. After 24 h transfection, cells were stimulated with or without IL17A (30 ng/ml) for 2 h, then cell lysates were prepared and immunoprecipitated with anti-HA antibody. The precipitates were detected by anti-BCL2 antibody. NS, non-significant.
IL17A suppresses the ubiquitination of BCL2
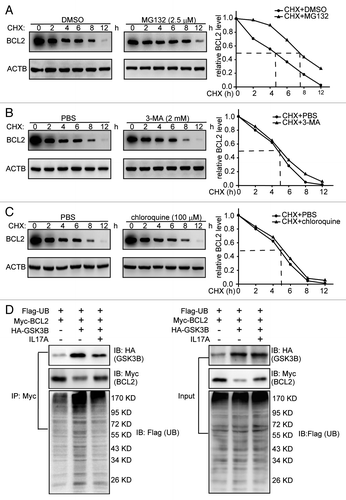
Most cytosolic proteins are degraded by either the ubiquitin-proteasome pathway or the autophagy-lysosomal pathway. We examined which pathway participates in the regulation of the GSK3B induced-BCL2 degradation. In the presence of MG132, a specific proteasome inhibitor, the degradation of BCL2 was delayed (). However, inhibition of autophagy-lysosomal pathway by 3-MA or chloroquine did not change the degradation rate of BCL2 (). We further determined whether the ubiquitination of BCL2 is GSK3B- dependent. Compared with the control cells, more ubiquitinated proteins were immunoprecipitated with an anti-Myc (BCL2-Myc) antibody from GSK3B-transfected cells. Addition of IL17A significantly reduced BCL2 ubiquitination induced by GSK3B (). Taken together, these data suggest that GSK3B promotes degradation of BCL2 via the ubiquitin-proteasome pathway.
Figure 6. IL17A suppresses ubiquitination of BCL2. (A) MG132 inhibition of proteasome blocks degradation of BCL2. Cells were treated with MG132 (2.5 μM) for 2 h, and then added with CHX for the indicated times. BCL2 was immunoblotting with the indicated antibody. (B and C) Inhibition of autophagy-lysosomal pathway by 3-MA or chloroquine does not affect the degradation of BCL2. Cells were treated with 3-MA (2 mM) or chloroquine (100 μM) for 12 h before added with CHX for indicated times. And cell lysates were extracted for western blotting with anti-BCL2 antibody. (D) IL17A eliminates the ubiquitination of BCL2 induced by GSK3B. Cells were transfected with ubiquitin-Flag, BCL2-Myc, and GSK3B-HA expression plasmids for 24 h before added with IL17A (30 ng/ml), for another 2 h. Cell lysates were immunoprecipitated with an anti-Myc antibody. The precipitates were detected by anti-Flag antibody, anti-HA antibody and anti-Myc antibody.
GSK3B triggers the degradation of BCL2 by inducing phosphorylation of BCL2 at Ser70
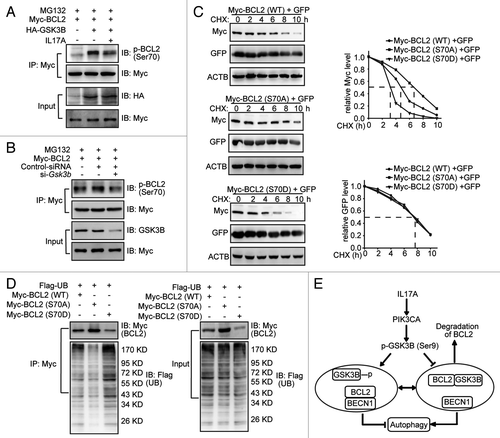
GSK3B has been shown to trigger the ubiquitination degradation of several proteins by direct phosphorylating these proteins.Citation21 GSK3B induces the phosphorylation of CTNNB1 at Ser33, Ser37 and Thr41 and leads to the rapid degradation of CTNNB1.Citation11 Similarly, GSK3B stimulates the phosphorylation of SMAD3 at Thr66 and promotes degradation of SMAD3.Citation12 The phosphorylation of BCL2 at Thr56, Thr74 and Ser87 reduces its degradation.Citation24 But the phosphorylation of BCL2 at Ser70 promotes its degradation.Citation25 Therefore we assumed that GSK3B might phosphorylate BCL2 at Ser70 and then promote BCL2 degradation. We found that overexpression of GSK3B induced the phosphorylation of BCL2 at Ser70 and addition of IL17A attenuated the regulatory effect of GSK3B (). Moreover, siRNA depletion of GSK3B decreased the phosphorylation of BCL2 at Ser70 (). It had been reported that MAPK8 phosphorylates BCL2 at Ser70 to activate autophagy when cells are starved.Citation18 However, we found that IL17A attenuated the phosphorylation of MAPK8 when the cells were starved (Fig. S1). These data suggest that IL17A interferes with the MAPK8-dependent phosphorylation of BCL2 during starvation. In order to define the physiological effect of Ser70 phosphorylation, we compared the protein stability for the wild-type BCL2, the S70A phospho-deficient mutant, and the S70D phospho-mimicking mutant. We found that the half-life of wild-type BCL2 was near 5 h while that of the S70A or S70D mutants was either prolonged to over 6 h or shortened to about 3 h (), respectively. At the same time, the S70D mutant assembled more while the S70A did less ubiquitination than that of wild-type BCL2 (). Together, our data indicate that GSK3B induces the phosphorylation BCL2 at Ser 70 and this modification is critical for BCL2 degradation via the ubiquitin-proteasome system ().
Figure 7. GSK3B phosphorylates BCL2 at Ser70 to trigger the ubiquitination of BCL2. (A) IL17A eliminates the phosphorylation of BCL2 Ser70 induced by GSK3B. Cells transfected with GSK3B-HA were treated with IL17A (30 ng/ml) for 2 h. Then cell lysates were prepared and immunoprecipitated with anti-Myc antibody. The precipitates were detected by anti-p-BCL2 (Ser70) antibody. (B) Depletion of GSK3B decreased the expression of phosphorylation of BCL2 at Ser70. Cells were transfected with si-Gsk3b and BCL2-Myc for 24 h. Then cell lysates were prepared and immunoprecipitated with anti-Myc antibody. The precipitates were detected by anti-p-BCL2 (Ser70) antibody. (C) Mimicking phosphorylation at Ser70 accelerates degradation of BCL2. Cells were transfected with Myc-tagged BCL2 variants (WT, S70A, or S70D) plasmids for 24 h. Then cells were treated with CHX for the indicated times. The cells were also transfected with an identical amount of pEGFP-N1 plasmid to monitor the transfection efficiency. The expression of BCL2 or GFP was examined by western blotting with anti-Myc antibody (for BCL2) or anti-GFP antibody respectively. (D) Mimicking phosphorylation of BCL2 Ser70 promotes ubiquitination. Cells were transfected with Flag-tagged ubiquitin and Myc-tagged BCL2 variants (WT, S70A, or S70D) plasmids. Cell lysates were immunoprecipitated with anti-Myc antibody. The precipitates were blotted with anti-Flag antibody and anti-Myc antibody. (E) Schematic diagram of the mechanism of IL17A-mediated attenuation of autophagy in pulmonary fibrosis. IL17A activates PIK3CA to induce the phosphorylation of GSK3B at Ser9, which causes a decrease in the kinase activity of GSK3B. The suppressed GSK3B attenuates the phosphorylation and degradation of BCL2. Thus, enhanced expression of BCL2 promotes the association of BCL2 and BECN1 to protect against the activation of autophagy.
Blocking IL17A reduces the expression of BCL2 and promotes the interaction of BCL2 and GSK3B in BLM-induced fibrotic lung tissue
Our previous work showed that BLM induces the expression of IL17A in fibrotic lung tissue and that the neutralization of IL17A attenuates BLM-induced pulmonary fibrosis.Citation8 Indeed, blocking IL17A decreased the collagen deposition and the hydroxyproline content in the BLM-induced fibrotic lung tissue (Fig. S2A–S2D). We examined the expression of p-BCL2 (Ser70), BCL2, p-GSK3B (Ser9), and GSK3B in BLM- or anti-IL17A antibody- treated lung tissue. We found that BLM increased, whereas the neutralization of IL17A decreased, the phosphorylation of GSK3B at Ser9 and the expression of BCL2 (Fig. S2E). Moreover, BLM reduced, but blocking IL17A promoted the interaction of BCL2 and GSK3B (Fig. S2F). These data suggest that IL17A inhibits the GSK3B-dependent degradation of BCL2 through phosphorylating GSK3B and attenuating the interaction of BCL2 and GSK3B in BLM-induced pulmonary fibrosis.
Discussion
Th17 cytokine IL17A promotes the development and progression of pulmonary fibrosis through attenuating the autophagy-associated degradation of collagen epithelial-mesenchymal transition accumulated in the fibrotic lung tissue.Citation8 Our current studies indicated that IL17A attenuates the activation of autophagy through regulating the expression or activity of autophagy core complex components. Indeed, we found that treatment of lung epithelial cells with IL17A enhances expression of BCL2 but reduces the expression or activity of BECN1, PIK3C3, PIK3R4, and ATG14. A number of studies demonstrate that enhanced expression of BCL2 suppresses autophagy due to BCL2 binding to and interfering with BECN1, a critical component of the autophagic core complex.Citation10 However, our current observations regarding to the mechanism of IL17A-induced enhanced expression of BCL2 are different from previous reports in where that IL17A activates MAPK1/3 and MAPK8/9,Citation26 and the activation of MAPK3Citation14 and MAPK8Citation15 increase the expression of Bcl2 mRNA. In our current studies, we found that treatment of lung epithelial cells with IL17A neither activates MAPK1/3 and MAPK8/9 nor increases the expression of Bcl2 mRNA. We do not know yet what are the explanations for the different observations in our systems and those of others. A difference between our current studies and others is that the lung epithelial cells have been treated with much lower concentrations of IL17A in our current studies than in the above, quoted studies.
IL17A initiates cellular activities through activating a diversity of the signal transduction pathways including the MAPK14-RELA, MAPK3-AP1, PI3K-CTNNB1 and TRAF3IP2-CEBP pathway.Citation27 Regulation of gene transcription and changing protein stability are major mechanisms for the IL17A-activated signal pathways and cellular biological functions. For example, the activation of RELA results in the rapid degradation of NFKBIA.Citation28 And IL17A stimulates gene transcription of MMP1, but does not have an impact on the degradation of MMP1 to increase the expression of MMP1 through activation of MAPK, AP1 and CEBP.Citation29 However, LuyendykCitation30 and other researchersCitation31,Citation32 have previously shown that pharmacological activation of PI3K reduces degradation of NFKBIA. And Lee and his colleagues report that the stimulation of PI3K inhibits the degradation of muscle protein through regulating the ubiquitin-proteasome systems.Citation33 Indeed, our observation indicated that IL17A suppresses the degradation of BCL2 and enhances the BCL2 expression through the activation of PI3K signal cascade but not MAPK14-RELA, MAPK3-AP1 or TRAF3IP2-CEBP. Our observation is in accordance with the fact that activation of PI3K induces the expression of BCL2 even though the authors did not expose the detailed mechanisms.Citation34 It has demonstrated that the activation of the PI3K-AKT-MTOR signaling pathway attenuates autophagy.Citation35 However, our studies indicated that IL17A does not activate AKT and that IL17A-induced phosphorylation of GSK3B is attenuated in cells silencing PIK3CA, suggesting that phosphorylation of GSK3B is directly induced by PI3K but not by AKT in the IL17A-treated cells. These findings are consistent with our previous observation that IL17A suppresses autophagy through inhibiting the activity of autophagy core complex but not activating AKT-MTOR signalingCitation8 (). Thus, our work dissects a signaling pathway responsible for the attenuation of autophagy by PI3K-GSK3B-BCL2 rather than by a classic PI3K-AKT-MTOR cascade, at least in lung epithelial cells.
GSK3B is an established downstream component of the PI3K signal pathway. GSK3B regulates a variety of cellular activities by facilitating the degradation of signaling proteins such as CTNNB1 and SMAD3. GSK3B binds to CTNNB1 and induces the phosphorylation of CTNNB1, which promotes its degradation.Citation11 Similarly, SMAD3 must be bound and phosphorylated by GSK3B to trigger degradation whereas dephosphorylation of SMAD3 by phosphatases inhibits the SMAD3 degradation.Citation12 Additionally, previous work indicated that GSK3B can enhance the phosphorylation and degradation of MCL1, a member of the antiapoptotic BCL2 family.Citation20 Our current studies indicated that GSK3B also binds to BCL2 and induces the phosphorylation of BCL2 to trigger the degradation of BCL2. It has been reported that the MAPK8 stimulates the BCL2 phosphorylation at Ser70 to promote the dissociation of BCL2 and BECN1, which activates autophagy.Citation18 Similarly, GSK3B induces the BCL2 phosphorylation at Ser70. However, the phosphorylated BCL2 triggers itself degradation and activates autophagy. Our finding is consistent with and supported by the fact that PPP2R1A, a serine/threonine phosphatase, attenuates the degradation of BCL2 by dephosphorylating BCL2 at Ser70.Citation25 However, our findings disagree with the observations of Deng et al. that the phosphorylation of BCL2 at Ser70 suppresses the degradation of BCL2, whereas the dephosphorylation of BCL2 at Ser70 promotes its degradation.Citation36 Thus, it is to be determined if IL17A stimulates phosphatase to reduce the GSK3B-induced phosphorylation of BCL2 in lung epithelial cells.
TGFB1 is a major mediator of pulmonary fibrosis. Our previous work revealed that IL17A increases the synthesis and secretion of collagen and promotes the epithelial-mesenchymal transition by a TGFB1-dependent manner in alveolar epithelial cells.Citation8 Moreover, TGFB1 decreases the activity of GSK3B by phosphorylating GSK3B at Ser9.Citation23 However, our present observation indicated that TGFB1 does not change the IL17A-suppressed GSK3B-dependent degradation of BCL2 in lung epithelial cells. This data is consistent with our previous work that IL17A inhibits autophagy independently of TGFB1.Citation8 Patel et al. have reported that autophagy deficiency is a hallmark of patients with idiopathic pulmonary fibrosis and TGFB1 inhibits autophagy via activation of MTORC1 in fibroblasts.Citation37 These observations indicate the activity of autophagy is suppressed through both TGFB1-dependent and -independent manners in the different types of cells during the development and progression of pulmonary fibrosis.
In summary, our current study showed that IL17A attenuates autophagy through increasing the stability and expression of BCL2, a negative switcher for autophagy activation. Our findings define a novel signaling pathway and the molecular mechanism of IL17A in the regulation of autophagy in lung epithelial cells. Our work indicates that blocking IL17A activity to decrease BCL2 expression and activate autophagy may be a promising therapeutic strategy for the prevention and treatment of fibroproliferative lung diseases such as pulmonary fibrosis.
Materials and Methods
Reagents
The mouse IL17A proteins were purchased from Peprotech (210-17). Cycloheximide (CHX) (01810), 3-methyladenine (3-MA) (M9281) and Chloroquine (C6628) were obtained from Sigma. Alexa Fluor 488 (A-11055) and 647 (A-31573) Abs were obtained from Invitrogen. Mouse IgG (5414) and anti-mouse BCL2 (2870), BECN1 (3495), PIK3C3 (4263), ATG14 (5504), BCL2L1 (2764), GSK3B (9315), p-BCL2 (S70) (2827), p-GSK3B (S9) (9323), GFP (2555), HA (3724), Myc (2278), Flag (2908) and ACTB (4970) antibodies were obtained from Cell Signaling Technology. PIK3R4 (124817) were purchased from Abcam. Pik3r1 siRNA (36218), Pik3ca siRNA (39128), Gsk3b siRNA (35525) and control siRNA (37007) were purchased from Santa Cruz. Other materials were purchased from commercial sources.
Plasmid construction
Mouse Gsk3b was generated by PCR with appropriate primers plasmids and subcloned in frame to the pcDNA6-HA expression plasmid. Mouse Becn1 was generated by PCR and subcloned in frame to the pFLAG-CMV-2 expression plasmid. Myc-tagged Bcl2 plasmid was a gift from Dr W. Stratford May, Jr (University of Florida Shands Cancer Center). HA-tagged Gsk3b (S9A), HA-tagged Gsk3b (S9D), Myc-tagged Bcl2 (S70A) and Bcl2 (S70D) mutants were generated by PCR as described previously.Citation38 Full-length Gsk3b was subcloned in frame to the pGEX4T-1 to make GST fusion proteins. The ubiquitin gene was synthesized as described previouslyCitation39 and cloned into pFLAG-CMVTM-2 expression plasmid.
Cell culture
The mouse type II alveolar epithelial cells (MLE-12) were cultured in DMEM-Ham’s Nutrient Mixture F12 (1:1; Hyclone) supplemented with 2% FBS and L-glutamine (full-nutrient medium). The cells were passaged every 2 d.
Semiquantitative RT-PCR
The expression of Bcl2 mRNA was analyzed as described previously.Citation40 Total RNA was isolated from MLE-12 cells using the TRIzol kit (Invitrogen) according to the manufacturer’s instructions. Next, the RNA was reverse-transcribed and amplified. PCR was performed using a Mycycler thermal cycler, and the amplified products were analyzed by agarose gel. The following specific primer sequences were used: 5′-GAGACAGCCAGGAGAAA TCA-3′ and 5′-CCTGTGGATGACTGAGTACC-3′ (mouse Bcl2); and 5′-TAAC CAACTGGGACGATATG-3′ and 5′-AAACAGGGACAGCACAGCCT-3′ (mouse Actb). All values obtained were normalized to the values obtained for Actb.
Immunoprecipitation and western blotting
Cells were washed three times with phosphate-buffered saline, harvested, and lysed in coimmunoprecipitation buffer that has been described before.Citation39 Total cell lysates (5 mg protein) were subjected to immunoprecipitation with appropriate antibodies, as indicated, overnight at 4°C with gentle agitation, followed by incubation with protein A/G Plus-agarose for 24 h at 4°C. The immunocomplex was washed three times and then mixed with 2 × SDS sample buffer and boiled for 5 min. For western blotting, coprecipitates or whole-cell extracts were resolved by SDS-PAGE and blotted on PVDF membranes (Millipore). The membranes were immunoblotted with the indicated antibodies and developed using an ECL detection system (Amersham Bioscience).
GST pulldown assay
As described previously,Citation41 GST fusion constructs of GSK3B were expressed in Escherichia coli and purified using glutathione-Sepharose 4B beads (Amersham Bioscience). Equal amounts of GST or GST fusion proteins bound to glutathione-Sepharose beads were incubated with lysates from MLE-12 cells. Beads were washed three times and interacting proteins were detected by immunoblotting. Expression of GST fusion proteins was confirmed by Coomassie Blue staining.
Confocal assay
Standard protocols for immunofluorescence microscopy were used as described previously.Citation42 MLE-12 cells were planted on the cover glass-bottom dishes and treated with or without indicated agents respectively. The cells on the dishes were washed two times, fixed by 4% paraformaldehyde for 20 min, and washed three times after fixing. The cells on the dishes (0.5 μm thick) were prepared and stained with indicated primary Abs overnight at 4°C. The sections were washed twice, incubated with fluorochrome-labeled secondary Abs (1:200) for 30 min, and washed three times after staining. Images were obtained with a Leica SP2 confocal microscope (Leica Microsystems, Exton, PA) and analyzed with Leica confocal software.
Statistics
Data are represented as the mean ± SE. Statistical analyses were performed using Student’s t-test was used for two-group comparisons. The p value was set at 0.05. All statistics were analyzed using SPSS 17.0 software.
| Abbreviations: | ||
| BCL2 | = | B-cell CLL/lymphoma 2 |
| BCL2L1 | = | BCL2 like 1 |
| BLM | = | bleomycin |
| CHX | = | cycloheximide |
| ECM | = | extracellular matrix |
| GSK3 | = | glycogen synthase kinase 3 |
| GFP | = | green fluorescent protein |
| IL17A | = | interleukin-17A |
| 3-MA | = | 3-methyladenine |
| MAPK | = | mitogen-activated protein kinases |
| PI3K | = | phosphoinositide-3-kinase |
| PIK3C3 | = | phosphatidylinositol 3-kinase catalytic subunit type 3 |
| PIK3R1 | = | phosphoinositide-3-kinase, regulatory subunit 1 (alpha) |
| PIK3R4 | = | phosphoinositide-3-kinase, regulatory subunit 4 |
| TGFB1 | = | transforming growth factor beta 1 |
| UB | = | ubiquitin |
| WT | = | wild type |
Additional material
Download Zip (319.4 KB)Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81273529, 81030056), Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT, No. IRT1007), and International Corporation Project supported by the Ministry of Science and Technology (2010DFB32900).
We thank Dr W. Stratford May, Jr (University of Florida Shands Cancer Center) for providing mouse BCL2 (WT) and BCL2 (S70A) plasmids.
Disclosure of Potential Conflicts of Interest
The authors declare no competing financial interests.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/24039
References
- Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med 2011; 208:1339 - 50; http://dx.doi.org/10.1084/jem.20110551; PMID: 21727191
- Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest 2012; 122:2756 - 62; http://dx.doi.org/10.1172/JCI60323; PMID: 22850886
- du Bois RM. Strategies for treating idiopathic pulmonary fibrosis. Nat Rev Drug Discov 2010; 9:129 - 40; http://dx.doi.org/10.1038/nrd2958; PMID: 20094055
- Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol 2004; 4:583 - 94; http://dx.doi.org/10.1038/nri1412; PMID: 15286725
- Yang HZ, Cui B, Liu HZ, Chen ZR, Yan HM, Hua F, et al. Targeting TLR2 attenuates pulmonary inflammation and fibrosis by reversion of suppressive immune microenvironment. J Immunol 2009; 182:692 - 702; PMID: 19109203
- Yang HZ, Wang JP, Mi S, Liu HZ, Cui B, Yan HM, et al. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol 2012; 180:275 - 92; http://dx.doi.org/10.1016/j.ajpath.2011.09.019; PMID: 22062220
- Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, et al. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med 2010; 207:535 - 52; http://dx.doi.org/10.1084/jem.20092121; PMID: 20176803
- Mi S, Li Z, Yang HZ, Liu H, Wang JP, Ma YG, et al. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J Immunol 2011; 187:3003 - 14; http://dx.doi.org/10.4049/jimmunol.1004081; PMID: 21841134
- Fernandez IE, Eickelberg O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012; 380:680 - 8; http://dx.doi.org/10.1016/S0140-6736(12)61144-1; PMID: 22901889
- Sinha S, Levine B. The autophagy effector Beclin 1: a novel BH3-only protein. Oncogene 2008; 27:Suppl 1 S137 - 48; http://dx.doi.org/10.1038/onc.2009.51; PMID: 19641499
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science 1996; 272:1023 - 6; http://dx.doi.org/10.1126/science.272.5264.1023; PMID: 8638126
- Guo X, Ramirez A, Waddell DS, Li Z, Liu X, Wang XF. Axin and GSK3- control Smad3 protein stability and modulate TGF- signaling. Genes Dev 2008; 22:106 - 20; http://dx.doi.org/10.1101/gad.1590908; PMID: 18172167
- Huang F, Kao CY, Wachi S, Thai P, Ryu J, Wu R. Requirement for both JAK-mediated PI3K signaling and ACT1/TRAF6/TAK1-dependent NF-kappaB activation by IL-17A in enhancing cytokine expression in human airway epithelial cells. J Immunol 2007; 179:6504 - 13; PMID: 17982039
- Subramanian M, Shaha C. Up-regulation of Bcl-2 through ERK phosphorylation is associated with human macrophage survival in an estrogen microenvironment. J Immunol 2007; 179:2330 - 8; PMID: 17675494
- Yoon SN, Kim KS, Cho JH, Ma W, Choi HJ, Kwon SJ, et al. Phospholipase D1 mediates bFGF-induced Bcl-2 expression leading to neurite outgrowth in H19-7 cells. Biochem J 2012; 441:407 - 16; http://dx.doi.org/10.1042/BJ20110302; PMID: 21916846
- Pattingre S, Levine B. Bcl-2 inhibition of autophagy: a new route to cancer?. Cancer Res 2006; 66:2885 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-05-4412; PMID: 16540632
- Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem 2007; 282:13123 - 32; http://dx.doi.org/10.1074/jbc.M700492200; PMID: 17337444
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678 - 88; http://dx.doi.org/10.1016/j.molcel.2008.06.001; PMID: 18570871
- Hyun YS, Han DS, Lee AR, Eun CS, Youn J, Kim HY. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis 2012; 33:931 - 6; http://dx.doi.org/10.1093/carcin/bgs106; PMID: 22354874
- Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 2006; 21:749 - 60; http://dx.doi.org/10.1016/j.molcel.2006.02.009; PMID: 16543145
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 2004; 29:95 - 102; http://dx.doi.org/10.1016/j.tibs.2003.12.004; PMID: 15102436
- Jean JZ, Hailing C, Shidong J, Li W, Ole VG, Aki M, et al. The p110α isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci USA 2006; 103:44
- Caraci F, Gili E, Calafiore M, Failla M, La Rosa C, Crimi N, et al. TGF-β1 targets the GSK-3β/β-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol Res 2008; 57:274 - 82; http://dx.doi.org/10.1016/j.phrs.2008.02.001; PMID: 18346908
- Dimmeler S, Breitschopf K, Haendeler J, Zeiher AM. Dephosphorylation targets Bcl-2 for ubiquitin-dependent degradation: a link between the apoptosome and the proteasome pathway. J Exp Med 1999; 189:1815 - 22; http://dx.doi.org/10.1084/jem.189.11.1815; PMID: 10359585
- Lin SS, Bassik MC, Suh H, Nishino M, Arroyo JD, Hahn WC, et al. PP2A regulates BCL-2 phosphorylation and proteasome-mediated degradation at the endoplasmic reticulum. J Biol Chem 2006; 281:23003 - 12; http://dx.doi.org/10.1074/jbc.M602648200; PMID: 16717086
- Pickens SR, Volin MV, Mandelin AM 2nd, Kolls JK, Pope RM, Shahrara S. IL-17 contributes to angiogenesis in rheumatoid arthritis. J Immunol 2010; 184:3233 - 41; http://dx.doi.org/10.4049/jimmunol.0903271; PMID: 20173024
- Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol 2009; 9:556 - 67; http://dx.doi.org/10.1038/nri2586; PMID: 19575028
- de Moissac D, Mustapha S, Greenberg AH, Kirshenbaum LA. Bcl-2 activates the transcription factor NFkappaB through the degradation of the cytoplasmic inhibitor IkappaBalpha. J Biol Chem 1998; 273:23946 - 51; http://dx.doi.org/10.1074/jbc.273.37.23946; PMID: 9727009
- Cortez DM, Feldman MD, Mummidi S, Valente AJ, Steffensen B, Vincenti M, et al. IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK- and ERK1/2-dependent C/EBP-beta, NF-kappaB, and AP-1 activation. Am J Physiol Heart Circ Physiol 2007; 293:H3356 - 65; http://dx.doi.org/10.1152/ajpheart.00928.2007; PMID: 17921324
- Luyendyk JP, Schabbauer GA, Tencati M, Holscher T, Pawlinski R, Mackman N. Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J Immunol 2008; 180:4218 - 26; PMID: 18322234
- Díaz-Guerra MJ, Castrillo A, Martín-Sanz P, Boscá L. Negative regulation by phosphatidylinositol 3-kinase of inducible nitric oxide synthase expression in macrophages. J Immunol 1999; 162:6184 - 90; PMID: 10229863
- Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem 2002; 277:32124 - 32; http://dx.doi.org/10.1074/jbc.M203298200; PMID: 12052830
- Lee SW, Dai G, Hu Z, Wang X, Du J, Mitch WE. Regulation of muscle protein degradation: coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. J Am Soc Nephrol 2004; 15:1537 - 45; http://dx.doi.org/10.1097/01.ASN.0000127211.86206.E1; PMID: 15153564
- Barata JT, Silva A, Brandao JG, Nadler LM, Cardoso AA, Boussiotis VA. Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J Exp Med 2004; 200:659 - 69; http://dx.doi.org/10.1084/jem.20040789; PMID: 15353558
- Wu YT, Tan HL, Huang Q, Ong CN, Shen HM. Activation of the PI3K-Akt-mTOR signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy 2009; 5:824 - 34; PMID: 19556857
- Deng X, Gao F, Flagg T, May WS Jr.. Mono- and multisite phosphorylation enhances Bcl2’s antiapoptotic function and inhibition of cell cycle entry functions. Proc Natl Acad Sci U S A 2004; 101:153 - 8; http://dx.doi.org/10.1073/pnas.2533920100; PMID: 14660795
- Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, et al. Autophagy in idiopathic pulmonary fibrosis. PLoS One 2012; 7:e41394; http://dx.doi.org/10.1371/journal.pone.0041394; PMID: 22815997
- Liu X, Sun Y, Constantinescu SN, Karam E, Weinberg RA, Lodish HF. Transforming growth factor β-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc Natl Acad Sci U S A 1997; 94:10669 - 74; http://dx.doi.org/10.1073/pnas.94.20.10669; PMID: 9380693
- Hua F, Mu R, Liu J, Xue J, Wang Z, Lin H, et al. TRB3 interacts with SMAD3 promoting tumor cell migration and invasion. J Cell Sci 2011; 124:3235 - 46; http://dx.doi.org/10.1242/jcs.082875; PMID: 21896644
- Yang HZ, Cui B, Liu HZ, Mi S, Yan J, Yan HM, et al. Blocking TLR2 activity attenuates pulmonary metastases of tumor. PLoS One 2009; 4:e6520; http://dx.doi.org/10.1371/journal.pone.0006520; PMID: 19654875
- Hua F, Zhou J, Liu J, Zhu C, Cui B, Lin H, et al. Glycogen synthase kinase-3beta negatively regulates TGF-beta1 and Angiotensin II-mediated cellular activity through interaction with Smad3. Eur J Pharmacol 2010; 644:17 - 23; http://dx.doi.org/10.1016/j.ejphar.2010.06.042; PMID: 20599907
- Salazar M, Carracedo A, Salanueva ÍJ, Hernández-Tiedra S, Lorente M, Egia A, et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest 2009; 119:1359 - 72; http://dx.doi.org/10.1172/JCI37948; PMID: 19425170