Abstract
Failure to engage apoptosis appears to be a leading mechanism of resistance to traditional platinum drugs in patients with ovarian cancer. Therefore, an alternative strategy to induce cell death is needed for the chemotherapy of this apoptosis-resistant cancer. Here we report that autophagic cell death, distinct from cisplatin-induced apoptosis, is triggered by a novel monofunctional platinum (II) complex named Mono-Pt in human ovarian carcinoma cells. Mono-Pt-induced cell death has the following features: cytoplasmic vacuolation, caspase-independent, no nuclear fragmentation or chromatin condensation, and no apoptotic bodies. These characteristics integrally indicated that Mono-Pt, rather than cisplatin, initiated a nonapoptotic cell death in Caov-3 ovarian carcinoma cells. Furthermore, incubation of the cells with Mono-Pt but not with cisplatin produced an increasing punctate distribution of microtubule-associated protein 1 light chain 3 (LC3), and an increasing ratio of LC3-II to LC3-I. Mono-Pt also caused the formation of autophagic vacuoles as revealed by monodansylcadaverine staining and transmission electron microscopy. In addition, Mono-Pt-induced cell death was significantly inhibited by the knockdown of either BECN1 or ATG7 gene expression, or by autophagy inhibitors 3-methyladenine, chloroquine and bafilomycin A1. Moreover, the effect of Mono-Pt involved the AKT1-MTOR-RPS6KB1 pathway and MAPK1 (ERK2)/MAPK3 (ERK1) signaling, since the MTOR inhibitor rapamycin increased, while the MAPK1/3 inhibitor U0126 decreased Mono-Pt-induced autophagic cell death. Taken together, our results suggest that Mono-Pt exerts anticancer effect via autophagic cell death in apoptosis-resistant ovarian cancer. These findings lead to increased options for anticancer platinum drugs to induce cell death in cancer.
Introduction
Ovarian cancer is the third leading cause of cancer death and the fourth most commonly diagnosed in gynecologic malignancies worldwide, accounting for 3.8% (225,500) of the total new cancer cases and 4.1% (140,200) of the total cancer deaths among females in 2008.Citation1 Coupled with the fact that most new cases of ovarian cancer are diagnosed at the advanced stage, there is clearly an urgent need for discovering effective chemotherapeutic agents for advanced-stage patients.Citation2 The discovery of cisplatin more than 30 years ago was one of the most significant therapeutic advances for treating ovarian cancer. However, patients receiving conventional platinum drugs often develop resistance, which diminishes the efficacy of the drugs.Citation3 For example, the TP53/p53-dependent apoptosis was commonly deficient in 50% to 70% of patients with ovarian cancer and the failure to engage apoptotic cell death would thus appear to be an important mechanism of the resistance to this type of cancer.Citation4 Therefore, the development of new platinum compounds with improved anticancer profiles and, more importantly, less likelihood to develop resistance holds much promise for the future.
Although apoptosis (type I programmed cell death) is the primary mechanism of anticancer-drugs-induced cell death, an alternative cell death pathway termed autophagic cell death (type II programmed cell death) has emerged as an important mechanism of cancer cell death induced by chemotherapeutic agents.Citation5-Citation8 Similar to its ‘double-edged sword’ effects on cancer development, autophagy has prodeath or prosurvival roles during cancer therapy.Citation8 On the one hand, autophagy always has cytoprotective roles in response to cancer therapeutics.Citation9,Citation10 In addition, novel therapies such as the use of proteasome inhibitors,Citation11 anti-ERBB2 (HER2) antibodiesCitation12 and kinase inhibitorsCitation13 also induce autophagy which is believed to reduce drug efficacy. On the other hand, excess autophagy can also act as a prodeath mechanism which leads to the destruction of cancer cells since autophagy is a ‘self-cannibalistic’ process. There is evidence that autophagy is required for the death of cancer cells with defects in apoptosis.Citation14,Citation15 It has been reported that autophagy and subsequent cell death can be induced by various anticancer therapies in different cancer cell types.Citation16,Citation17 Although the role of autophagy during cancer therapy is complex, autophagy is becoming an attractive approach for anticancer therapies.
Ever since cisplatin was recognized and employed as an anticancer drug, the cellular mechanisms that lead to apoptosis have been well studied and a series of different platinum anticancer drugs have been clinically applied.Citation18 Classic platinum anticancer drugs including cisplatin cause DNA damages by binding to DNA therefore creating inter- or intra-strand cross links, and, finally, leading to apoptosis.Citation19 In this process, the cis-type of the platinum complexes, but not trans-type or monofunctional platinum complexes, serve as the most contributive type due to the fact that stable cross links with DNA can be formed after the leaving-groups on cis-position leave the complexes. However, to increase the efficacy of non-cis type complexes and to broaden the applicability of platinum complexes, scientists have found that some modifications like introducing aromatic groups into the complexes can optimize the structures and improve the activities of trans-type and monofunctional platinum complexes against cancer cell growth.Citation20-Citation22 Previously, a series of the monofunctional platinum (II) complexes were synthesized by our laboratory, and these platinum analogs were found to exhibit cytotoxicity comparable with, and in some cases, better than cisplatin in human cancer cells.Citation22 The mechanisms of the distinct cytotoxicity profiles of these complexes remained to be found.
In the present study, we compared the sensitivities of different human cancer cells to a monofunctional platinum (II) complex named Mono-Pt and cisplatin, and investigated the mechanism of action of Mono-Pt, revealing the occurring of the autophagic cell death distinct from cisplatin-induced apoptosis. Further examination of the signaling pathways indicated that this Mono-Pt-induced autophagic cell death in ovarian carcinoma cells was subject to either negative regulation of the v-akt murine thymoma viral oncogene homolog 1 (AKT1)-mechanistic target of rapamycin (MTOR) pathway, or positive regulation of the mitogen-activated protein kinase 1/3 (MAPK1/3) pathway. Our findings may provide novel insights into the mechanisms underlying the anticancer effects of the monofunctional platinum (II) complex against human ovarian carcinoma cells.
Results
Mono-Pt is significantly more potent than cisplatin in killing various human cancer cells
First we examined the cytotoxic effects of Mono-Pt and cisplatin (chemical structure shown in ) on human cancer cells. Our results showed that Mono-Pt exhibited a significantly higher cytotoxic effect than cisplatin did on various human cancer cells, including human ovarian carcinoma cell lines Caov-3 and Skov-3, human breast carcinoma cell line MCF-7, human colon carcinoma cell line HCT-116, and human hepatoma cell line BEL-7402 (). Then we further compared the anticancer effects of Mono-Pt and cisplatin in human ovarian carcinoma Caov-3 cells. Mono-Pt and cisplatin both induced cell death in a dose- and time-dependent manner (). Mono-Pt had a significantly more potent cytotoxic effect with an IC50 (half maximal inhibitory concentration) at about 10 μM in Caov-3 cells after 48 h treatment, compared with cisplatin with an IC50 about 50 μM.
Figure 1. Mono-Pt induces cell death in a more efficient way than cisplatin does. (A) Structure of Mono-Pt (N-(tert-butoxycarbonyl)-l-methionine-N′-8-quino-lylamidechloridoplatinum(II), top) and cisplatin (bottom). (B) Different human cancer cell lines were incubated with 10 μM of Mono-Pt or cisplatin for 48 h. Cell death was assessed by MTT assay. Data represent mean ± SEM of three different experiments. *p < 0.05, **p < 0.01. (C) Human ovarian carcinoma Caov-3 cells were incubated with indicated concentrations of Mono-Pt or cisplatin for 12 h, 24 h or 48 h. Cell death was assessed by trypan blue dye exclusion assay. Data represent mean ± SEM of three different experiments. *p < 0.05, **p < 0.01. (D) Cellular uptake in semi-log coordinates. Caov-3 cells were incubated with 10 μM Mono-Pt or 10 μM cisplatin for indicated time. The cellular accumulation of Pt was interpreted as nmol Pt/105 cells. Data represent mean ± SEM of three different experiments. **p < 0.01, as compared with corresponding time point of Mono-Pt-treated group.
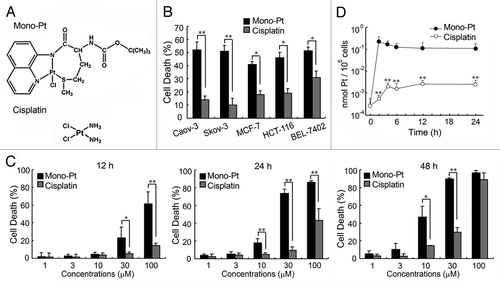
From the time course of the cellular uptake of platinum, it was obvious that Mono-Pt exhibited a faster internalizing pattern than cisplatin did (), indicating that intracellular platinum accumulation was elevated when ligand lipophilicity allowed the complex to rapidly cross the membrane. The accumulation of platinum from cisplatin increased slowly but constantly until reaching the steady-state. With an equal dosage concentration, the accumulation of platinum from Mono-Pt reached the peak quickly in the first 2 h and then steadily dropped until the balance was reached ().
Relative to cisplatin, Mono-Pt barely induces cell cycle arrest or causes DNA damage
As shown in , cisplatin at the dose from 1 μM to 10 μM caused cell cycle arrest at S phase in human ovarian carcinoma Caov-3 cells. In contrast, even with the dose high enough to cause a great extent of cell proliferation inhibition, Mono-Pt did not induce any detectable cell cycle arrest (), indicating that Mono-Pt did not cause DNA damage. Besides, cell cycle arrest induced by cisplatin but not Mono-Pt was also observed in human ovarian carcinoma Skov-3 cells (Fig. S1), which further confirmed the previous results.
Figure 2. Mono-Pt barely induces cell cycle arrest or influences DNA integrity relative to cisplatin. (A and B) Cell cycle analysis. About 5 × 105 synchronized Caov-3 cells were cultured in each well on 6-well plates and treated with various concentrations of cisplatin or Mono-Pt for 24 h. Gates M1, M2 and M3 indicated G0/G1 phase, S phase and G2/M phase, respectively. (C) DNA binding assay. pUC19 DNA (200 ng) was used in each sample and incubated with different concentrations of Mono-Pt or cisplatin at 37°C for 12 h. The conformational changes of the supercoiled DNA caused by the binding of the complexes were displayed as the backward shifting of the band on the 1% agarose gel. All results shown are representative of three different experiments.
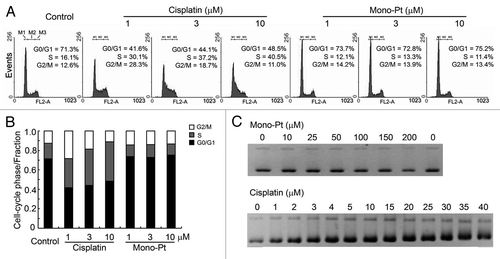
In most cases, DNA damages caused by platinum complexes are attributed to the binding of the complexes to DNA, which leads to DNA conformational changes that block the normal replication and transcription processes.Citation23 So we performed DNA binding assay to verify the DNA conformational changes caused by Mono-Pt or cisplatin. The supercoiled DNA pUC19 was incubated with different concentrations of either Mono-Pt or cisplatin at 37°C for 12 h followed by DNA agarose gel analysis. We found that cisplatin was able to unwind pUC19 DNA at 3 μM or higher concentrations, while there was no observable band shifting for those DNA bound with Mono-Pt even at 200 μM, suggesting that Mono-Pt did not cause conformational change to pUC19 DNA ().
Mono-Pt but not cisplatin induces a nonapoptotic cell death in ovarian carcinoma cells
Then we tried to determine whether a different cell death mechanism as suggested above was induced by Mono-Pt. Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay showed that cisplatin induced significant apoptosis in ovarian carcinoma Caov-3 cells at the indicated concentrations, while Mono-Pt did not (). Cells treated with DNase I were used as the positive control. In addition, Hoechst 33342 staining assay indicated that cells treated with cisplatin rather than Mono-Pt showed typical characteristics of apoptosis, including the condensation of the chromosome, the shrinkage of the whole cell and the appearance of the apoptotic bodies (Fig. S2).
Figure 3. Mono-Pt but not cisplatin induces a nonapoptotic cell death in human ovarian carcinoma Caov-3 cells. (A) Caov-3 cells were treated with 10 μM Mono-Pt or 50 μM cisplatin for indicated time, then stained with TUNEL-FITC and subjected to flow cytometric assay. Data represent mean ± SEM of three different experiments. *p < 0.05, **p < 0.01, as compared with untreated group. (B) Caov-3 cells were treated with 10 μM Mono-Pt or 50 μM cisplatin for 36 h, then stained with DAPI and TUNEL-FITC, and subjected to immunofluorescence imaging. Cells treated with DNase I (1 μg/ml) were used as positive control. The results shown are one of three different experiments. Scale bar: 10 μm. (C) Expression of cleaved CASP3 in Caov-3 cells. Cells were treated with Mono-Pt or cisplatin for 24 h, and then subjected to western blotting. The results shown are representative of three different experiments. (D) CASP3 activity in Caov-3 cells. Cells were treated with 10 μM Mono-Pt or 50 μM cisplatin for indicated time, and then subjected to CASP3 activity analysis. Data represent mean ± SEM of three different experiments. *p < 0.05, **p < 0.01, as compared with untreated group. (E) Mono-Pt-induced cell death was caspase-independent. Caov-3 cells were pretreated with different concentrations of zVAD.fmk for 6 h, and then incubated with 10 μM Mono-Pt or 50 μM cisplatin for 48 h. Cell death was assessed by trypan blue dye exclusion assay. Data represent mean ± SEM of three different experiments. *p < 0.05, **p < 0.01, as compared with the zVAD.fmk-untreated group.
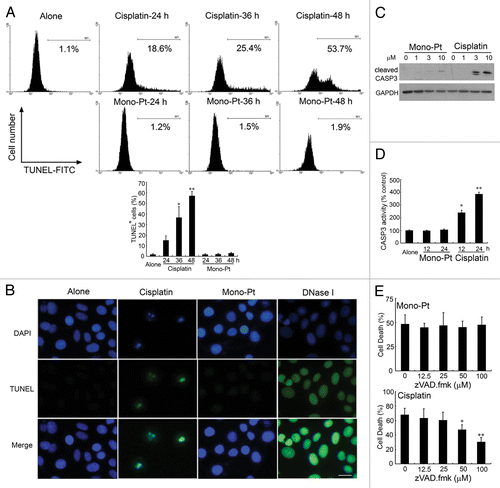
Furthermore, with the treatment of 10 μM cisplatin, there was a great amount of cleaved CASP3/caspase-3 produced. On the contrary, Mono-Pt hardly induced CASP3 cleavage (). The same tendency was also seen in CASP3 enzyme activity analysis (). Interestingly, the growth inhibitory effect of cisplatin but not Mono-Pt was significantly blocked by pan-caspase inhibitor, zVAD.fmk, which provides another strong piece of evidence for a caspase-independent cell death in Mono-Pt-treated cells ().
Next we wondered whether Mono-Pt-induced cell death required de novo protein synthesis during cell death process. We coincubated cells with cisplatin or Mono-Pt in the presence of transcription inhibitor (actinomycin D) or protein biosynthesis inhibitor (cycloheximide). As shown in Figure S3, cells treated with 50 μM cisplatin exhibited a dose-dependent increase of cell death in the presence of either cycloheximide or actinomycin D, suggesting that cisplatin-induced cell death does not involve new protein synthesis. On the contrary, the direct inhibition of protein expression by cycloheximide or mRNA transcription by actinomycin D both decreased the cell death caused by Mono-Pt (Fig. S3), indicating that Mono-Pt-induced cell death process requires mRNA transcription and protein synthesis. These findings demonstrate that Mono-Pt-treated cells exhibit phenotypes of a nonapoptotic cell death.
Mono-Pt, rather than cisplatin, results in a significant induction of autophagy in ovarian carcinoma cells
Since caspase-dependent cell death was not a distinct feature of ovarian carcinoma Caov-3 cells treated with Mono-Pt, we wondered whether the cytotoxic effect of Mono-Pt was due to severe autophagy. We first examined the production of LC3-II, which is a cleaved LC3-phosphatidyl-ethanolamine conjugate and a general autophagosomal marker.Citation24 LC3-I to LC3-II-conversion markedly increased with the rising dose of Mono-Pt over 24 h in Caov-3 cells (). Similar results were also observed in Skov-3 cells (Fig. S4A). However, compared with Mono-Pt-treated group, there was minor but not evident increase in LC3-conversion in cisplatin-treated group as the dose increased (). The LC3-conversion increase also displayed a time-dependent manner in Caov-3 () and Skov-3 cells (data not shown). More importantly, LC3 blots () did suggest that Mono-Pt treatment induced a general increase in LC3 protein expression as well (LC3-I levels increased with longer treatment time/higher dose), which was consistent with the findings that cycloheximide and actinomycin D treatment decreased Mono-Pt-induced cell death (Fig. S3).
Figure 4. Mono-Pt induces evident autophagy in human ovarian carcinoma Caov-3 cells. (A–C) Protein expression of LC3 and SQSTM1 in Caov-3 cells. (A) Caov-3 cells were cultured with indicated concentrations of Mono-Pt or cisplatin for 24 h. (B) Caov-3 cells were cultured with 10 μM Mono-Pt for 6 h, 12 h, 24 h or 1 μM rapamycin for 24 h. (C) Caov-3 cells were cultured with indicated concentrations of Mono-Pt for 24 h. Then cells were subjected to immunoblotting. Rapamycin treatment induced autophagy, and was taken as the positive control. Data represent mean ± SEM of three different experiments. **p < 0.01, as compared with untreated group. (D) Caov-3 cells transfected with GFP-LC3 cDNA were treated with 10 μM Mono-Pt in the presence or absence of 2 mM 3-methyladenine (3-MA) for 24 h. The formation of vacuoles containing GFP-LC3 (dots) was examined by fluorescence microscopy. In another set of experiments, Caov-3 cells were treated with 10 μM Mono-Pt in the presence or absence of 2 mM 3-MA for 24 h, and then incubated with 0.05 mM monodansylcadaverine (MDC) for 10 min. Cells were then analyzed by fluorescence microscopy. Scale bar: 5 μm. Data represent mean ± SEM of three different experiments. **p < 0.01. (E) Immunofluorescence imaging of LC3 in Caov-3 cells. Cells treated with 50 μM cisplatin, 10 μM Mono-Pt or 2 μM rapamycin for 24 h were shown in the figure with DAPI indicating the nuclear area and an Alexa Fluor 488 fluorescent secondary antibody that binds to LC3 primary antibody to indicate LC3 puncta. Scale bar: 10 μm. The results shown are representative of three experiments. (F) The transmission electron microscopy imaging of cells showing numerous double-membraned cytoplasmic vacuolation (arrows) in 10 μM Mono-Pt-treated cells as well as condensed and fragmented nuclei in 50 μM cisplatin-treated cells. The results shown are representative of three different experiments.
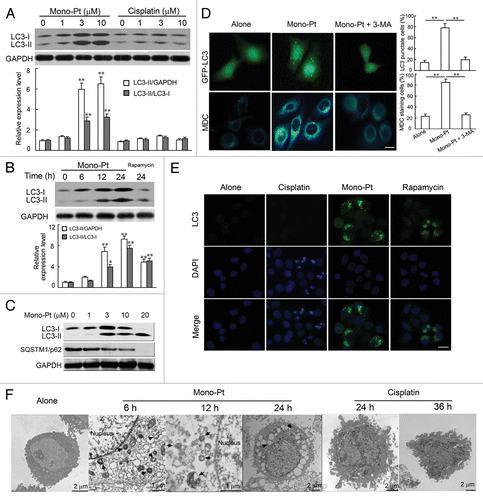
In the context of autophagy, SQSTM1 (sequestosome 1, p62) acts as an adaptor protein that links LC3 with ubiquitin moieties on misfolded proteins. Autophagy therefore mediates the clearance of SQSTM1 together with ubiquitylated proteins.Citation25 In our experiments, we found that expression levels of SQSTM1 were downregulated by Mono-Pt treatment in Caov-3 cells () and Skov-3 cells (Fig. S4A).
Then we used green fluorescent protein (GFP)-fused LC3, a specific marker for autophagosome formation, to detect autophagy. As shown in , the formation of GFP-LC3-labeled vacuoles in Caov-3 cells was markedly increased 24 h after treatment with 10 μM Mono-Pt. The formation of these vacuoles was interfered with by 3-MA, a specific inhibitor of the autophagic process at early stages (). This effect was also confirmed by immunofluorescence assay showing that Mono-Pt treatment remarkably increased the number of vacuoles indicated by endogenous LC3-II (). Consistent with western blot results (), cisplatin did not induce obvious LC3 puncta ().
Monodansylcadaverine (MDC) is another specific marker for autolysosomes that concentrates on the autophagic vacuole membrane structures distributed within the cytoplasm.Citation26 We examined the incorporation of MDC into cells after Mono-Pt treatment, and found that cells treated with Mono-Pt showed an increase of MDC accumulation, indicating the increasing formation of the MDC-labeled vacuoles in comparison with untreated cells (). MDC incorporation was also suppressed by autophagy inhibitor 3-MA (). Similar findings were obtained in Skov-3 cells (Fig. S4B).
Autophagy is a dynamic process of protein degradation characterized by the formation of double-membraned cytoplasmic vesicles.Citation27 Structural analysis via electron microscopy allows the visualization of autophagy with the massive accumulation of autophagic vacuoles (autophagosomes) in the cytoplasm. When we monitored Mono-Pt-induced autophagy using transmission electron microscopy, we observed a time-dependent accumulation of numerous lamellar structures and double-membraned cytosolic autophagic vacuoles in Caov-3 cells starting at 6 h after the treatment with Mono-Pt (). These autophagic vacuoles were not found in cisplatin-treated cells, where only features of apoptosis such as chromatin condensation and fragmentation of the nuclei were observed (). Taken together, these findings suggest that Mono-Pt but not cisplatin treatment significantly induces autophagy in ovarian carcinoma cells.
Increased autophagy is responsible for Mono-Pt-induced cell death in ovarian carcinoma cells
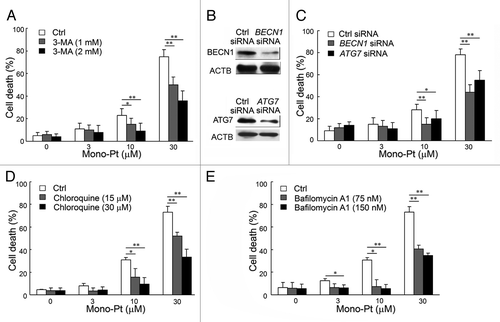
To find out whether autophagy was associated with caspase-independent cell death induced by Mono-Pt in Caov-3 cells, we examined whether inhibiting autophagy by autophagy inhibitors or silencing autophagy-related genes could affect cell death in human ovarian carcinoma cells. First, we found by the trypan blue dye exclusion assay that pretreatment with the autophagy inhibitor 3-MA significantly prevented Mono-Pt-induced cell death in a dose-dependent manner in human ovarian carcinoma Caov-3 cells (). Similar results were observed in another human ovarian carcinoma Skov-3 cells (Fig. S4C). Consistent with the trypan blue exclusion assay data, the MTT assay also showed that Mono-Pt-induced cell death in Caov-3 cells was significantly decreased by 3-MA in a dose-dependent manner (data not shown). Second, we also found that silencing BECN1 or ATG7 by siRNA knockdown () significantly reduced cell death induced by Mono-Pt, as monitored by trypan blue exclusion assay (). These results support autophagy as the major mechanism of the cell death in ovarian carcinoma cells treated with Mono-Pt. To further confirm the role of autophagy in Mono-Pt-induced cell death, we also used autophagolysosome fusion inhibitor chloroquine and lysosomal proteases inhibitor bafilomycin A1 in the present study. Both chloroquine and bafilomycin A1 decreased cell death induced by Mono-Pt (), suggesting autophagy contributes to Mono-Pt-induced cell death.
Figure 5. Mono-Pt induces autophagic cell death in human ovarian carcinoma Caov-3 cells. (A) Caov-3 cells were treated with various concentrations of Mono-Pt and/or 1 or 2 mM 3-MA for 24 h. Cell death was measured by trypan blue dye exclusion assay. *p < 0.05, **p < 0.01. (B) The knockdown of BECN1 or ATG7 was confirmed by western blot. (C) Caov-3 cells were transfected with control siRNA, siRNA targeting BECN1 or siRNA targeting ATG7. After 48 h, cells were treated with 0, 3, 10 or 30 μM Mono-Pt for 24 h, and cell death was measured by trypan blue dye exclusion assay. *p < 0.05, **p < 0.01. (D and E) Caov-3 cells were treated with various concentrations of Mono-Pt and/or chloroquine (D) or bafilomycin A1 (E) for 24 h. Cell death was measured by trypan blue dye exclusion assay. *p < 0.05, **p < 0.01.
Mono-Pt induces autophagic cell death through AKT1-MTOR-RPS6KB1- and MAPK1/3-dependent signaling pathways
There are two well-known pathways involved in the regulation of autophagy: phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit α (PIK3CA/PI3K)-AKT1-MTOR-ribosomal protein S6 kinase, 70 kDa, polypeptide 1 (RPS6KB1) signaling pathway and the RAS-RAF1-MAPK1/3 signaling pathway. Both pathways are often activated in numerous types of cancers.Citation28 Several anticancer agents are known to inhibit the PIK3CA-AKT1-MTOR-RPS6KB1 pathway and simultaneously activate MAPK1/3 signaling, resulting in the induction of autophagy in cancer cells.Citation29-Citation31 So we examined the effect of Mono-Pt on both pathways using western blotting. After the treatment with 10 μM Mono-Pt, there was a time-dependent decrease in the expressions of phosphorylated RPS6KB1, RPS6 (ribosomal protein S6) and EIF4EBP1 (eukaryotic translation initiation factor 4E binding protein 1), compared with the total expressions of the three proteins in Caov-3 cells (). To further investigate the upstream inhibition of MTOR by Mono-Pt, we examined AKT1 phosphorylation at Ser473 and Thr308. As shown in , the treatment with Mono-Pt also suppressed the AKT1 phosphorylation at Ser473 and Thr308 in a time-dependent manner in Caov-3 cells. Similar findings were also obtained in Skov-3 cells (Fig. S5). Subsequently, we examined whether Mono-Pt activated MAPK signaling. As shown in , Mono-Pt specifically increased the phosphorylation of MAPK1 (ERK2)/MAPK3 (ERK1), a key regulator of autophagy, in a time-dependent manner. However, MAPK14 (p38α) and MAPK8 (JNK1) were not activated by the same treatment of Mono-Pt. As expected, MAPK1/3 inhibitor U0126 reversed the increasing LC3-II to LC3-I ratio () and prevented Mono-Pt-induced cell death (). Therefore, we conclude that Mono-Pt induces autophagic cell death through AKT1-MTOR-RPS6KB1- and MAPK1/3-dependent signaling pathways.
Figure 6. Mono-Pt induces autophagic cell death through AKT1-MTOR-RPS6KB1- and MAPK1/3-dependent signaling pathways. (A and B) Caov-3 cells treated with 10 μM Mono-Pt for various time-intervals were analyzed by western blot for phosphorylated and total RPS6KB1, RPS6, EIF4EBP1 (A), and AKT1 expression (B). Densitometry analysis was performed using three independent experiments. *p < 0.05, **p < 0.01, as compared with 0 h point. (C) Caov-3 cells with or without 10 μM Mono-Pt treatment for indicated times were analyzed by western blot for phosphorylated and total MAPK expression. The results shown are representative of three experiments. (D) Caov-3 cells treated with 10 μM Mono-Pt and/or U0126 for 6 h were analyzed by western blot using indicated antibodies. Densitometry analysis was performed using three independent experiments. (E) Caov-3 cells were treated with 0, 3, 10 or 30 μM Mono-Pt and/or 10 or 20 μM U0126 for 24 h. Cell death was measured by trypan blue exclusion assay. (F) Caov-3 cells treated with various concentrations of rapamycin and/or 10 μM Mono-Pt for 24 h were analyzed by western blot. Densitometry analysis was performed using three independent experiments. (G) Caov-3 cells were treated with 0, 3, 10 or 30 μM Mono-Pt and/or 0.5 or 1 μM rapamycin for 24 h. Cell death was measured by trypan blue dye exclusion assay. *p < 0.05, **p < 0.01.
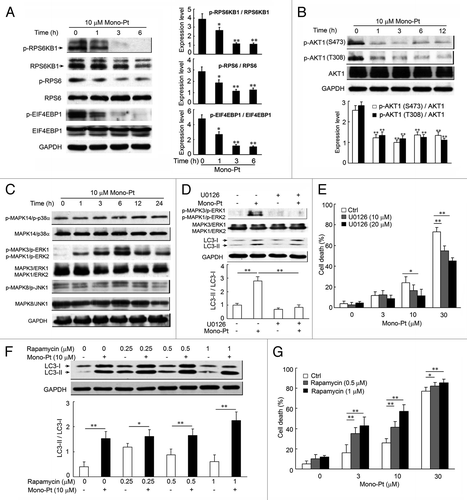
It has been reported that autophagy is indispensable for the death of cancer cells with defects in apoptosis, and lack of autophagy often diminishes the therapeutic effects of anticancer agents. Researchers also found that the MTOR inhibitor rapamycin, which induces autophagy, was able to inhibit malignant glioma cells proliferation.Citation32 This evidence proved the crucial role of autophagy in the mechanism of rapamycin-mediated anticancer effect.Citation33 In the present study, we found that combining Mono-Pt and rapamycin showed a stronger effect on the enhancement of LC3 conversion and cell death compared with the treatment of Mono-Pt or rapamycin alone (). In addition, rapidly growing evidence reinforces the notion that resistance to rapamycin results from a positive feedback loop from MTOR-RPS6KB1 to AKT1, which leads to the enhancement of AKT1 phosphorylation at Ser473.Citation34-Citation36 Importantly, Mono-Pt inhibits AKT1 signaling and thus might counteract the effect of this positive feedback. So it is reasonable that dual suppression of the PIK3CA-AKT1-MTOR pathway by combination of rapamycin with Mono-Pt better facilitates autophagy. Recently, there is evidence showing that using rapamycin and an AKT1 inhibitor simultaneously to inhibit the PIK3CA-AKT1-MTOR pathway facilitated the autophagy and produced synergistic effects on autophagy-mediated apoptosis.Citation37 Taken together, these findings suggest that dual inhibition of PIK3CA-AKT1-MTOR by rapamycin and Mono-Pt may also be a rational choice for cancer treatment.
Discussion
In this study, we provided novel evidence that a monofunctional platinum (II) complex Mono-Pt activates the autophagic cell death, which is fundamentally different from apoptosis induced by cisplatin in human ovarian carcinoma cells. We proposed that autophagic cell death, rather than apoptosis, is a possible mechanism for Mono-Pt-induced cytotoxicity. Our conclusions were based on the following observations: First, we did not observe any apparent signs of apoptosis by transmission electron microscopy and Hoechst staining evaluation in Mono-Pt-treated ovarian carcinoma Caov-3 cells, such as nuclear fragmentation, chromatin condensation, or apoptotic bodies (; Fig. S2). Characteristically, cell death triggered by Mono-Pt was accompanied by deficiency in DNA fragmentation (), independent of caspase activation () and de novo protein synthesis (Fig. S3), indicating that Mono-Pt but not cisplatin induces a nonapoptotic cell death. Furthermore, it was suggested by the punctate distribution of LC3, elevated ratio of LC3-II to LC3-I, increased SQSTM1 degradation and the massive autophagic vacuolization which occurred in Mono-Pt-treated cells () that Mono-Pt but not cisplatin produced a significant induction of autophagy in ovarian carcinoma cells. Second, Mono-Pt-induced cell death was mediated through autophagy: the cell death was significantly suppressed by approaches inhibiting autophagy, such as the pretreatment of various autophagy inhibitors or the transfection of siRNAs targeting the autophagy-related genes ATG7 and BECN1 (). Our data agree with previous studies showing that autophagy may contribute to cancer cell death and that the inhibition of autophagy can reduce cytotoxicity. For instance, the autophagy inhibitor 3-MA or Atg7-siRNA inhibits cell death in caffeine-treated PC12D cells,Citation29 plant alkaloid voacamine-treated human osteosarcoma cells,Citation38 BCL2L1/BCL-XL inhibitor Z36-treated HeLa cells,Citation39 and Bacillus Calmette-Guerin cell wall cytoskeleton plus ionizing radiation-treated colon cancer HCT-116 cells.Citation40 Finally, according to the fact that MTOR inhibitor rapamycin increased while MAPK1/3 inhibitor U0126 decreased Mono-Pt-induced autophagic cell death, it is suggested that AKT1-MTOR-RPS6KB1 pathway and MAPK1/3 signaling were involved. Together, these data strongly suggest that the extent of cell death caused by excessive autophagy seems to be predominant in Mono-Pt-induced cell death. These results, providing a mechanistic characterization of the novel monofunctional platinum complex-induced cancer cell death, shed light on exploring a new tool to overcome apoptosis-resistance in patients with ovarian cancer.
Usually, apoptosis is the major mechanism to eliminate cancer cells. Many chemotherapeutic agents were designed to interfere apoptosis-inducing pathways.Citation41 For example, cisplatin was reported to cause cell cycle arrest followed by apoptosis in various human tumor cells.Citation42 However, because of the resistance which is often developed to apoptosis, researchers have been seeking alternative form of programmed cell death and demonstrated that autophagy plays an important role in cancer therapy.Citation7,Citation8,Citation14,Citation16 So far, several chemotherapeutic agents such as paclitaxel, arsenic trioxide and doxorubicin have been reported to be able to induce caspase-independent cell death.Citation43,Citation44 Thus, targeting the signaling intermediates in autophagy in premalignant and malignant cells may be an effective strategy for cancer therapy, especially for apoptosis-resistant cancer.Citation7,Citation8,Citation14,Citation16 It has also been suggested that autophagy can directly kill cells in the absence of the apoptosis machinery. For example, Atg7 and Becn1 regulated cell death in mouse L929 cells treated with the pan-caspase inhibitor.Citation15 In addition, RNAi against Atg5 and Becn1 also blocked cell death of bax−/− and bak1−/− murine embryonic fibroblasts induced by etoposide.Citation45 Thus, it is important to note that the autophagic cell death induced by monofunctional platinum complex has advantages over cisplatin under conditions of apoptosis deficiency in some cancers.
Most of the platinum complexes cause DNA damages by binding to the DNA, which leads to DNA conformational changes to stop DNA from replication and transcription. Apoptosis often, although not always, follows the DNA damages that could not be repaired.Citation46 For instance, cisplatin was reported to cause DNA damage and apoptosis in various human tumor cells.Citation42 Actually, cisplatin treatment with Caov-3 cells did induce minor but not obvious autophagy as shown in . Indeed there are innumerable examples in the literature over the past decade of DNA damaging agents such as camptothecin, etoposide and, specifically, cisplatin, promoting autophagy which delays apoptosis in cancers.Citation47-Citation50 The point emphasized here is that Mono-Pt induced cell death in tumor cells via autophagy, while autophagy induced by DNA damaging agents such as cisplatin is mainly a cell protective mechanism which delayed cell death. Several lines of evidences support the hypothesis that inhibition of autophagy could enhance cisplatin-induced apoptotic cell death in renal tubular epithelial cells,Citation47 breast cancer MCF-7, 67NR and 4T1 cells,Citation48,Citation49 and cervical cancer HeLa cells.Citation50 So it is suggested that autophagy induced by DNA damaging agents such as cisplatin is a self-protective mechanism by cells for survival. And this is why inhibition of autophagy improves cisplatin chemotherapy. However, in this study, Mono-Pt-induced cell death in human ovary carcinoma Caov-3 cells was significantly inhibited by a knockdown of either BECN1 or ATG7 gene expression, or by various autophagy inhibitors (), suggesting that autophagy here is a kind of cell death mechanism but not a survival response, which is distinct from cisplatin. Interestingly, unlike cisplatin, a novel monofunctional platinum complex Mono-Pt caused neither conformational change of pUC19 DNA nor cell cycle arrest, yet was more potent than cisplatin in killing various human cancer cells in a nonapoptotic pathway. It is of interest to ask what the difference is between the DNA binding with Mono-Pt and the DNA binding with cisplatin. This result was similar to previous report about platinum-phosphato complexes, which did not show any evidence of covalent binding to DNA.Citation51 However, the detailed mechanisms why Mono-Pt barely affects DNA remained to be discovered. Actually, monofunctional and bifunctional platinum complexes lacking aromatic group displayed weaker inhibitory effects on cell proliferation than those with aromatic groups usually did.Citation22 It is worth noticing that Mono-Pt is more lipid-soluble than cisplatin, indicating Mono-Pt possibly enters cells more efficiently than cisplatin does. Here we found in the cellular uptake assay that when the internalization process was stabilized, the amount of Pt accumulated in cells from Mono-Pt was almost 100-fold more than that from cisplatin, which could be one possible contributor to the higher cytotoxic effect of Mono-Pt.
Taken together, our findings suggest that Mono-Pt-induced cell death in human ovarian carcinoma cells is mediated by the caspase-independent autophagic cell death, which is distinct from cisplatin-induced apoptosis. Our study thus provides a useful chemical probe for the investigation of autophagy in cancer cells and offers a rationale for the development of monofunctional platinum complexes as effective chemotherapeutic agents against human ovarian carcinoma in the clinical settings.
Materials and Methods
Cell culture
All tumor cells used in the present study were maintained in HG-Dulbecco’s modified Eagle’s medium (4.5 g/L glucose; Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen) plus 2 mM glutamine, 50 μg/ml streptomycin and 50 U/ml penicillin. Human ovary carcinoma Caov-3 and Skov-3 cell lines, breast adenocarcinoma MCF-7 cell line, colon carcinoma HCT-116 cell line and hepatoma BEL-7402 cell line were purchased from Shanghai Institute of Cell Biology (Shanghai, China), and were cultured at 37°C in a 5% (v/v) CO2 atmosphere.
Chemicals, reagents and antibodies
Novel monofunctional platinum (II) complex Mono-Pt was synthesized by our laboratory as described previouslyCitation22 and the purity is 99.4%. Mono-Pt was dissolved at a concentration of 20 mM in DMSO as a stock solution, stored at -20°C, and diluted with medium before each experiment. Cisplatin (10018) was purchased from Shandong Boyuan Chemical Co., Ltd, dissolved at a concentration of 1 mM in phosphate-buffered saline (PBS) and frozen at -20°C. Rapamycin (R0395), cycloheximide (18079), actinomycin D (01815), Hoechst 33342 (14533), pan-caspase inhibitor zVAD.fmk (C2105), MAPK1/3 inhibitor U0126 (U120), monodansylcadaverine (MDC, 30432), 3-methyladenine (3-MA, M9281), 4’,6-diamidino-2-phenylindole (DAPI, D8417), bafilomycin A1 (B1793) and chloroquine (C6628) were purchased from Sigma Aldrich. The pUC19 DNA (D3219) was purchased from Takara (Dalian, China). GFP-LC3 plasmid was purchased from Yingrun Biotechnologies Inc. (Changsha, China). In Situ Cell Death detection kit (1-684-795) was purchased from Roche Applied Science. The antibodies used were as follows: anti-cleaved CASP3 (9664), anti-LC3B (2775), anti-ATG7 (2631), ATG7 siRNA I (6604), anti-BECN1 (3738), BECN1 siRNA I (6222), anti-RPS6KB1 (ribosomal protein S6 kinase, 2708), anti-phospho-RPS6KB1 (Thr389) (9205), anti-RPS6/S6 ribosomal protein (2217), anti-phospho-RPS6/S6 ribosomal protein (Ser235/236) (2211), anti-EIF4EBP1 (eukaryotic translation initiation factor 4E binding protein 1, 9452), anti-phospho-EIF4EBP1 (Thr37/46) (9459), anti-AKT1 (9272), anti-phospho-AKT1 (Ser473) (4058), anti-phospho-AKT1 (Thr308) (4056), anti-MAPK1 (ERK2)/MAPK3 (ERK1) antibody (9102), anti-phospho-MAPK1/3 antibody (Thy202/Tyr204) (9101), anti-MAPK14 (p38α) (9212), anti-phospho-MAPK14 (Thr180/Tyr182) rabbit mAb (4631), anti-MAPK8 (JNK1) (9252) and anti-phospho-MAPK8 (Thr183/Tyr185) rabbit mAb (4668) were purchased from Cell Signaling Technology. Anti-SQSTM1/p62 (P0067) was purchased from Sigma-Aldrich. Anti-LC3 (clone 4E12) and CASP3 activity kit were purchased from MBL (Woburn, MA). Anti-ACTB (sc-1616) and anti-GAPDH (sc-32233) were purchased from Santa Cruz Biotechnology. Alexa Fluor 488 goat anti-rabbit IgG (A11008) was purchased from Invitrogen. All other chemicals were purchased from Sigma Aldrich.
Assessment of cell death and apoptosis
Cell death was assessed by trypan blue exclusion assay. Both floating and adherent cells were collected and stained with trypan blue dye for 5 min at room temperature. Cells were counted by four independent hemocytometer counts. Cell death was determined as the percentage of dead cells over the total number of cells. Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining was performed using In Situ Cell Death detection kit from Roche Applied Science following the manufacturer’s instructions.
Cellular platinum uptake
Platinum uptake was assessed as described previously.Citation52 Caov-3 cells (2 × 105) were plated into 12-well plates and treated with 10 μM Mono-Pt or 10 μM cisplatin for indicated time. After incubation, cells were harvested and washed twice with PBS. The cell pellets were then heated in nitric acid followed by the addition of hydrogen peroxide and hydrochloric acid, and diluted with Milli-Q water (Millipore Corporation, Billerica, MA). Platinum analysis was performed on an inductively coupled plasma-mass spectrometer (ICP-MS, VG PQ ExCell, Franklin, MA). The experiments were performed in triplicate.
Cell cycle analysis
Cell cycle analysis was determined as described previously.Citation53 The synchronization was performed by incubating cells in DMEM supplemented with 0.5% FBS for 12 h. Then cells were incubated in fresh DMEM supplemented with 10% FBS and treated with Mono-Pt or cisplatin for 24 h. After incubation, cells were harvested, washed with cold PBS and fixed with 70% ethanol at 4°C overnight. The fixed cells were washed with PBS, stained with 50 μg/ml of propidium iodide (PI) containing 100 μg/ml of RNase A and 1% Triton X-100 in the dark for 45 min, and then subjected to flow cytometric analysis.
Immunofluorescence cytochemistry
For TUNEL assay, Caov-3 cells adhered to glass were treated with either 50 μM cisplatin or 10 μM Mono-Pt for 36 h. Then cells were fixed with 4% paraformaldehyde (40 min, room temperature) and stained with TUNEL-FITC (1:100). For the LC3 assay, Caov-3 cells adhered to glass were treated with either 50 μM cisplatin or 10 μM Mono-Pt or 2 μM rapamycin for 24 h. Then cells were fixed with 4% paraformaldehyde (40 min, room temperature), stained with anti-LC3 antibody (1:100), and detected with a secondary antibody (Alexa Fluor 488 goat anti-rabbit IgG, 1:1000, Invitrogen). The coverslips were counterstained with 0.1 μg/ml DAPI and imaged by fluorescence microscopy. In another set of experiments, Caov-3 cells transfected with GFP-LC3 (LC3 tagged with green fluorescent protein) cDNA were treated with 10 μM Mono-Pt in the presence or absence of 2 mM 3-methyladenine (3-MA) for 24 h. The formation of vacuoles containing GFP-LC3 (dots) was examined by fluorescence microscopy (BX51TRF, Olympus, Tokyo, Japan).
Western blot
Proteins were extracted in lysis buffer (30 mM Tris, pH 7.5, 150 mM sodium chloride, 1 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, 1% Nonidet P-40, 10% glycerol, and phosphatase and protease inhibitors), separated by SDS-PAGE and electrophcoretically transferred onto polyvinylidene fluoride membranes (Roche Applied Science). The membranes were probed with antibodies overnight at 4°C, and then incubated with a horse radish peroxidase-coupled secondary antibody. Detection was performed using a LumiGLO chemiluminescent substrate system (KPL, Guildford, UK). Band intensity was quantified by BandScan software (Glyko, Novato, CA).
Transmission electron microscopy assay
Cells were cultured in 100 mm dishes and treated with 10 μM Mono-Pt or 50 μM cisplatin for indicated times. After incubation, approximately 1 × 107 treated cells were harvested in a 1.5 ml microcentrifuge tube for each sample. Cells were washed with cold PBS once and fixed with fixation solution (4% paraformaldehyde, 0.25% glutaraldehyde in 0.1 M phosphate buffer) at 4°C overnight. Then samples were embedded and sectioned, and finally observed under transmission electron microscopy (Hitachi, Tokyo, Japan).
Visualization of MDC-labeled vacuoles
Autophagic vacuoles were labeled with MDC by incubating tumor cells grown on coverslips with 0.05 mM MDC in PBS at 37°C for 10 min. After incubation, cells were washed four times with PBS and immediately analyzed by fluorescence microscopy using an inverted microscope (BX51TRF, Olympus, Japan) equipped with a filter system (excitation, 380 to 420 nm; emission, 450 nm).
Statistical analysis
Data are expressed as mean ± SEM. Student’s t-test and one-way ANOVA test were used for statistical analyses of the data. All statistical analyses were conducted using SPSS 10.0 statistical software (SPSS, Chicago, IL, USA). Cases in which P values of < 0.05 or < 0.01 were considered statistically significant.
| Abbreviations: | ||
| ACTB | = | actin, beta |
| AKT1 | = | v-akt murine thymoma viral oncogene homolog 1 |
| ATG | = | autophagy-related |
| BAK1 | = | BCL2-antagonist/killer 1 |
| BAX | = | BCL2-associated X protein |
| BCL2L1 | = | BCL2-like 1, BCL-XL |
| BECN1 | = | Beclin 1 |
| CASP3 | = | caspase-3 |
| DAPI | = | 4’,6-diamidino-2-phenylindole |
| EIF4EBP1 | = | eukaryotic translation initiation factor 4E binding protein 1 |
| ERBB2 | = | v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, HER2 |
| ERK | = | extracellular signal-regulated kinase |
| GAPDH | = | glyceraldehyde-3-phosphate dehydrogenase |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| 3-MA | = | 3-methyladenine |
| MAPK | = | mitogen-activated protein kinase |
| MDC | = | monodansylcadaverine |
| Mono-Pt | = | a novel monofunctional platinum (II) complex |
| MTOR | = | mechanistic target of rapamycin |
| PIK3CA | = | phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha, PI3K |
| RAF1 | = | v-raf-1 murine leukemia viral oncogene homolog 1 |
| RPS6 | = | ribosomal protein S6 |
| RPS6KB1 | = | ribosomal protein S6 kinase, 70kD, polypeptide 1 |
| SQSTM1 | = | sequestosome 1 |
| TP53 | = | tumor protein p53 |
| TUNEL | = | terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling |
Additional material
Download Zip (279.6 KB)Acknowledgment
This work was supported by Science Fund for Creative Research Groups of NSFC (No. 81121062), National Natural Science Foundation of China (Nos. 90913023, 81173070, 91229109 and 21131003), National Basic Research Program of China (No. 2011CB935800) and National Science & Technology Major Project (No. 2012ZX09304-001).
Disclosure of Potential Conflicts of Interest
The authors state no conflict of interest.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/24407
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69 - 90; http://dx.doi.org/10.3322/caac.20107; PMID: 21296855
- Lutz AM, Willmann JK, Drescher CW, Ray P, Cochran FV, Urban N, et al. Early diagnosis of ovarian carcinoma: is a solution in sight?. Radiology 2011; 259:329 - 45; http://dx.doi.org/10.1148/radiol.11090563; PMID: 21502390
- Piccart MJ, Lamb H, Vermorken JB. Current and future potential roles of the platinum drugs in the treatment of ovarian cancer. Ann Oncol 2001; 12:1195 - 203; http://dx.doi.org/10.1023/A:1012259625746; PMID: 11697824
- Ferreira CG, Tolis C, Giaccone G. p53 and chemosensitivity. Ann Oncol 1999; 10:1011 - 21; http://dx.doi.org/10.1023/A:1008361818480; PMID: 10572598
- White E. Autophagic cell death unraveled: Pharmacological inhibition of apoptosis and autophagy enables necrosis. Autophagy 2008; 4:399 - 401; PMID: 18367872
- Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species?. Autophagy 2011; 7:457 - 65; http://dx.doi.org/10.4161/auto.7.5.14226; PMID: 21150268
- Kondo Y, Kondo S. Autophagy and cancer therapy. Autophagy 2006; 2:85 - 90; PMID: 16874083
- Levy JM, Thorburn A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol Ther 2011; 131:130 - 41; http://dx.doi.org/10.1016/j.pharmthera.2011.03.009; PMID: 21440002
- O’Donovan TR, O’Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011; 7:509 - 24; http://dx.doi.org/10.4161/auto.7.5.15066; PMID: 21325880
- Li J, Hou N, Faried A, Tsutsumi S, Kuwano H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur J Cancer 2010; 46:1900 - 9; http://dx.doi.org/10.1016/j.ejca.2010.02.021; PMID: 20231086
- Zhu K, Dunner K Jr., McConkey DJ. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010; 29:451 - 62; http://dx.doi.org/10.1038/onc.2009.343; PMID: 19881538
- Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS One 2009; 4:e6251; http://dx.doi.org/10.1371/journal.pone.0006251; PMID: 19606230
- Wu Z, Chang PC, Yang JC, Chu CY, Wang LY, Chen NT, et al. Autophagy blockade sensitizes prostate cancer cells towards Src family kinase inhibitors. Genes Cancer 2010; 1:40 - 9; http://dx.doi.org/10.1177/1947601909358324; PMID: 20811583
- Mujumdar N, Saluja AK. Autophagy in pancreatic cancer: an emerging mechanism of cell death. Autophagy 2010; 6:997 - 8; http://dx.doi.org/10.4161/auto.6.7.13334; PMID: 20818166
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004; 304:1500 - 2; http://dx.doi.org/10.1126/science.1096645; PMID: 15131264
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17:654 - 66; http://dx.doi.org/10.1158/1078-0432.CCR-10-2634; PMID: 21325294
- Liu EY, Ryan KM. Autophagy and cancer--issues we need to digest. J Cell Sci 2012; 125:2349 - 58; http://dx.doi.org/10.1242/jcs.093708; PMID: 22641689
- Wong E, Giandomenico CM. Current status of platinum-based antitumor drugs. Chem Rev 1999; 99:2451 - 66; http://dx.doi.org/10.1021/cr980420v; PMID: 11749486
- Hambley TW. Platinum binding to DNA: structural controls and consequences. J Chem Soc, Dalton Trans 2001; 19:2711 - 8; http://dx.doi.org/10.1039/b105406f
- Okada T, El-Mehasseb IM, Kodaka M, Tomohiro T, Okamoto K, Okuno H. Mononuclear platinum(II) complex with 2-phenylpyridine ligands showing high cytotoxicity against mouse sarcoma 180 cells acquiring high cisplatin resistance. J Med Chem 2001; 44:4661 - 7; http://dx.doi.org/10.1021/jm010203d; PMID: 11741483
- Zhang JY, Liu Q, Duan CY, Shao Y, Ding J, Miao ZH, et al. Structural evidence for the facile chelate-ring opening reactions of novel platinum(II)-pyridine carboxamide complexes. J Chem Soc, Dalton Trans 2002; 4:591 - 7; http://dx.doi.org/10.1039/b108378n
- Zhang J, Wang X, Tu C, Lin J, Ding J, Lin L, et al. Monofunctional platinum complexes showing potent cytotoxicity against human liver carcinoma cell line BEL-7402. J Med Chem 2003; 46:3502 - 7; http://dx.doi.org/10.1021/jm020593j; PMID: 12877588
- Ramachandran S, Temple BR, Chaney SG, Dokholyan NV. Structural basis for the sequence-dependent effects of platinum-DNA adducts. Nucleic Acids Res 2009; 37:2434 - 48; http://dx.doi.org/10.1093/nar/gkp029; PMID: 19255091
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy 2007; 3:181 - 206; PMID: 17224625
- Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009; 137:1001 - 4; http://dx.doi.org/10.1016/j.cell.2009.05.023; PMID: 19524504
- Biederbick A, Kern HF, Elsässer HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol 1995; 66:3 - 14; PMID: 7750517
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al, Nomenclature Committee on Cell Death 2009. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16:3 - 11; http://dx.doi.org/10.1038/cdd.2008.150; PMID: 18846107
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Saiki S, Sasazawa Y, Imamichi Y, Kawajiri S, Fujimaki T, Tanida I, et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy 2011; 7:176 - 87; http://dx.doi.org/10.4161/auto.7.2.14074; PMID: 21081844
- Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol 2007; 72:29 - 39; http://dx.doi.org/10.1124/mol.106.033167; PMID: 17395690
- Ellington AA, Berhow MA, Singletary KW. Inhibition of Akt signaling and enhanced ERK1/2 activity are involved in induction of macroautophagy by triterpenoid B-group soyasaponins in colon cancer cells. Carcinogenesis 2006; 27:298 - 306; http://dx.doi.org/10.1093/carcin/bgi214; PMID: 16113053
- Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res 2005; 65:3336 - 46; PMID: 15833867
- Iwamaru A, Kondo Y, Iwado E, Aoki H, Fujiwara K, Yokoyama T, et al. Silencing mammalian target of rapamycin signaling by small interfering RNA enhances rapamycin-induced autophagy in malignant glioma cells. Oncogene 2007; 26:1840 - 51; http://dx.doi.org/10.1038/sj.onc.1209992; PMID: 17001313
- Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther 2005; 4:1533 - 40; http://dx.doi.org/10.1158/1535-7163.MCT-05-0068; PMID: 16227402
- O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006; 66:1500 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-05-2925; PMID: 16452206
- Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007; 26:1932 - 40; http://dx.doi.org/10.1038/sj.onc.1209990; PMID: 17001314
- Cirstea D, Hideshima T, Rodig S, Santo L, Pozzi S, Vallet S, et al. Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol Cancer Ther 2010; 9:963 - 75; http://dx.doi.org/10.1158/1535-7163.MCT-09-0763; PMID: 20371718
- Meschini S, Condello M, Calcabrini A, Marra M, Formisano G, Lista P, et al. The plant alkaloid voacamine induces apoptosis-independent autophagic cell death on both sensitive and multidrug resistant human osteosarcoma cells. Autophagy 2008; 4:1020 - 33; PMID: 18838862
- Lin J, Zheng Z, Li Y, Yu W, Zhong W, Tian S, et al. A novel Bcl-XL inhibitor Z36 that induces autophagic cell death in Hela cells. Autophagy 2009; 5:314 - 20; http://dx.doi.org/10.4161/auto.5.3.7888; PMID: 19242113
- Yuk JM, Shin DM, Song KS, Lim K, Kim KH, Lee SH, et al. Bacillus calmette-guerin cell wall cytoskeleton enhances colon cancer radiosensitivity through autophagy. Autophagy 2010; 6:46 - 60; http://dx.doi.org/10.4161/auto.6.1.10325; PMID: 19901560
- Sun SY, Hail N Jr., Lotan R. Apoptosis as a novel target for cancer chemoprevention. J Natl Cancer Inst 2004; 96:662 - 72; http://dx.doi.org/10.1093/jnci/djh123; PMID: 15126603
- Vakifahmetoglu H, Olsson M, Tamm C, Heidari N, Orrenius S, Zhivotovsky B. DNA damage induces two distinct modes of cell death in ovarian carcinomas. Cell Death Differ 2008; 15:555 - 66; http://dx.doi.org/10.1038/sj.cdd.4402286; PMID: 18064041
- Bröker LE, Kruyt FAE, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res 2005; 11:3155 - 62; http://dx.doi.org/10.1158/1078-0432.CCR-04-2223; PMID: 15867207
- Chen TS, Wang XP, Sun L, Wang LX, Xing D, Mok M. Taxol induces caspase-independent cytoplasmic vacuolization and cell death through endoplasmic reticulum (ER) swelling in ASTC-a-1 cells. Cancer Lett 2008; 270:164 - 72; http://dx.doi.org/10.1016/j.canlet.2008.05.008; PMID: 18547714
- Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004; 6:1221 - 8; http://dx.doi.org/10.1038/ncb1192; PMID: 15558033
- Wang JY. DNA damage and apoptosis. Cell Death Differ 2001; 8:1047 - 8; http://dx.doi.org/10.1038/sj.cdd.4400938; PMID: 11687882
- Kaushal GP, Kaushal V, Herzog C, Yang C. Autophagy delays apoptosis in renal tubular epithelial cells in cisplatin cytotoxicity. Autophagy 2008; 4:710 - 2; PMID: 18497570
- Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 2007; 14:500 - 10; http://dx.doi.org/10.1038/sj.cdd.4402039; PMID: 16990848
- Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012; 8:200 - 12; http://dx.doi.org/10.4161/auto.8.2.18554; PMID: 22252008
- Xu Y, Yu H, Qin H, Kang J, Yu C, Zhong J, et al. Inhibition of autophagy enhances cisplatin cytotoxicity through endoplasmic reticulum stress in human cervical cancer cells. Cancer Lett 2012; 314:232 - 43; http://dx.doi.org/10.1016/j.canlet.2011.09.034; PMID: 22019047
- Bose RN, Maurmann L, Mishur RJ, Yasui L, Gupta S, Grayburn WS, et al. Non-DNA-binding platinum anticancer agents: Cytotoxic activities of platinum-phosphato complexes towards human ovarian cancer cells. Proc Natl Acad Sci U S A 2008; 105:18314 - 9; http://dx.doi.org/10.1073/pnas.0803094105; PMID: 19020081
- Harris AL, Ryan JJ, Farrell N. Biological consequences of trinuclear platinum complexes: comparison of [[trans-PtCl(NH3)2]2mu-(trans-Pt(NH3)2(H2N(CH2)6-NH2)2)]4+ (BBR 3464) with its noncovalent congeners. Mol Pharmacol 2006; 69:666 - 72; http://dx.doi.org/10.1124/mol.105.018762; PMID: 16275707
- Luo Q, Gu Y, Zheng W, Wu X, Gong F, Gu L, et al. Erlotinib inhibits T-cell-mediated immune response via down-regulation of the c-Raf/ERK cascade and Akt signaling pathway. Toxicol Appl Pharmacol 2011; 251:130 - 6; http://dx.doi.org/10.1016/j.taap.2010.12.011; PMID: 21195724