Abstract
Atg17, in complex with Atg29 and Atg31, constitutes a key module of the Atg1 kinase signaling complex and functions as an important organizer of the phagophore assembly site in the yeast Saccharomyces cerevisiae. We have determined the three-dimensional reconstruction of the full S. cerevisiae Atg17-Atg31-Atg29 complex by single-particle electron microscopy. Our structure shows that Atg17-Atg31-Atg29 is dimeric and adopts a relatively rigid and extended “S-shape” architecture with an end-to-end distance of approximately 345 Å. Subunit mapping analysis indicated that Atg17 mediates dimerization and generates a central rod-like scaffold, while Atg31 and Atg29 form two globular domains that are tethered to the concave sides of the scaffold at the terminal regions. Finally, our observation that Atg17 adopts multiple conformations in the absence of Atg31 and Atg29 suggests that the two smaller components play key roles in defining and maintaining the distinct curvature of the ternary complex.
Introduction
Macroautophagy (hereafter autophagy) is a catabolic pathway involved in the bulk degradation of cellular proteins and organelles,Citation1 and is essential to cell growth, development, and homeostasis. Upregulated during nutrient starvation and cellular stress, autophagy in yeast proceeds through a sequence of steps that begin with the formation of the phagophore at the phagophore assembly site (PAS), expansion of the phagophore, engulfment of cytoplasmic materials, and, finally, generation of a double-membrane vesicle called an autophagosome, which has a size that ranges from ~300 to 900 nm in diameter.Citation1,Citation2 The autophagosome is subsequently transported to the vacuole (in plants and yeast, or the lysosome in mammalian cells) where it fuses, releasing the inner vesicle. When present in the vacuole lumen, this single-membrane vesicle is termed an autophagic body, and it is degraded along with its cargo inside this lytic compartment. The breakdown products are released through permeases for reuse in the cytosol, or for catabolic processes to generate energy.
Genetic screening in the yeast S. cerevisiae and other fungal species led to the identification of 36 ATG (autophagy-related) genes, with more than half of these genes also present in higher eukaryotes. Of the corresponding 36 Atg proteins, 18 of them are essential to initiating and mediating autophagosome formation,Citation3 a process that is tightly regulated. In yeast, these 18, mostly conserved, Atg proteins localize at least transiently to the PAS, and are thought to constitute the core autophagy machinery.Citation4 The 18 core Atg proteins can be categorized into five functional groups: (1) components of the Atg1 protein kinase complex (Atg1, Atg13, Atg17, Atg29, Atg31), (2) components of the autophagy-specific phosphatidylinositol 3-kinase complex (Vps30/Atg6, Atg14), (3) the integral membrane protein Atg9 and the Atg2-Atg18 complex, (4) the Atg8 conjugation system (Atg3, Atg4, Atg7, and Atg8), and (5) the Atg12 conjugation system (Atg5, Atg7, Atg10, Atg12, and Atg16).Citation5 Systematic gene disruption studies established the hierarchical relationships between and within these groups of Atg proteins. Notably, it was shown that Atg17 functions upstream of all other core Atg proteins in this hierarchy.Citation6
Characterization of yeast deletion mutants showed that the atg17Δ strain is almost completely defective in autophagy, and that upon starvation this strain produces few small autophagosomes that are less than half the normal size.Citation7,Citation8 The absence of Atg17 also severely compromises the localization of other core Atg proteins to the PAS.Citation6 Collectively, these results suggest the number and size of the autophagosome are major factors in determining the magnitude of the autophagic response, and that Atg17 plays a key role in regulating these parameters.Citation7 These results further implicate the importance of Atg17’s involvement in recruiting different Atg proteins to the PAS, and in organizing these proteins in a configuration necessary for autophagosome formation.
How Atg17 exerts its biological functions in the autophagy pathway is not fully understood. Atg17 constitutively binds two smaller-sized proteins, Atg29 and Atg31. Although often designated as the Atg17-Atg31-Atg29 complex, this assembly represents one of the core modules of the Atg1 kinase complex.Citation9,Citation10 Recent studies suggest that the five-component Atg1 kinase complex, which also includes the Atg1 kinase and Atg13, is preformed in nonautophagy-inducing conditions and remains intact during autophagy.Citation11 Upon induction of autophagy, Atg1 protein kinase activity is upregulated through an unknown mechanism that requires dephosphorylation of Atg13Citation12 and possibly phosphorylation of the Atg31 and Atg29 components.Citation10 Understanding how changes in post-translational modifications of these proteins lead to activation of Atg1 necessitates a comprehensive understanding of the structural properties of the Atg1 kinase complex.
Recently, Ragusa et al. reported the crystal structure of the core Atg17-Atg31-Atg29 complex from the thermophilic yeast Lachancea thermotolerans.Citation13 This structure revealed that Atg17 is composed of α-helical bundles that form an extended scaffold while Atg31 and a proteolytic fragment of Atg29 assemble a globular β-sandwich domain that binds to the curved scaffold via interaction between Atg17 and Atg31. In vitro reconstituted recombinant Atg17-Atg31-Atg29 is dimeric in solution,Citation10 but several types of Atg17-Atg31-Atg29 dimers are observed from the crystal lattice. Through small angle X-ray scattering and biochemical analysis of the crystallized complex, Ragusa et al. concluded that dimerization likely occurred through a coiled-coil interaction at the C terminus of Atg17, and they generated a structural model that features a double-crescent shaped overall architecture. Further studies indicated that while the unique curvature of the modeled Atg17-Atg31-Atg29 dimer implicates a possible role in binding curved membranes, the actual membrane sensor of the Atg1 kinase complex appears to be the C-terminal domain of Atg1.Citation13 This pioneering work provided a structural framework to delineate the early events of autophagosome formation. However, the physiological relevance of the modeled dimer, the biological function of Atg31 and Atg29, and the relationship between L. thermotolerans Atg17-Atg31-Atg29 and the better characterized S. cerevisiae complex remain to be determined.
To address these important questions, we have purified the full S. cerevisiae Atg17-Atg31-Atg29 complex, in both recombinant and native forms, and analyzed the structural properties of this complex by single-particle electron microscopy (EM). Our data confirm that recombinant Atg17-Atg31-Atg29 adopts a dimeric, S-shaped architecture that spans a length of approximately 345 Å. In agreement with the crystallographic model, EM-based subunit mapping studies indicated that Atg17 mediates dimerization and generates a central scaffold where the terminal globular domains formed by Atg31 and Atg29 bind. Finally and most importantly, our EM studies of purified Atg17 uncovered a possible role of Atg31 and Atg29 in establishing and maintaining a rigid overall architecture.
Results
Purification of native and recombinant S. cerevisiae Atg17-Atg31-Atg29
To investigate the structural properties of the fully assembled S. cerevisiae Atg17-Atg31-Atg29 complex, we first attempted to isolate the native complex from yeast cells using tandem affinity purification (TAP). The TAP method is designed for the purification of native complexes and uses gentle buffers and mild elution conditions to preserve the structural integrity of the protein complex.Citation14 We prepared lysates from the Atg29-TAP yeast strain, which contains the TAP cassette fused to the C terminus of Atg29, and performed the conventional two-step TAP procedure.Citation15 However, due to the low abundance of Atg29, Atg31, and Atg17,Citation16 we could not detect these proteins at the end of the purification. We tested a number of lysis buffers, and found that the yield could be improved by switching the detergent to CHAPS. We also found that the second calmodulin-sepharose chromatography step resulted in massive loss of material. We therefore modified the TAP procedure by replacing the calmodulin-sepharose step with gel filtration chromatography.Citation17 These changes enabled us to obtain a purified sample that contains Atg17, Atg31, and Atg29, as confirmed by silver-stain SDS-PAGE and mass spectrometry analyses (; Table S1).
Figure 1. Purification and negative-stain EM analysis of the S. cerevisiae Atg17-Atg31-Atg29 complex. (A) Silver-stained SDS-PAGE analysis of purified native Atg17-Atg31-Atg29 complex (left). A representative raw image of negatively-stained native Atg17-Atg31-Atg31 (right) with particles in dotted circles (scale bar: 50 nm). (B) Silver-stained SDS-PAGE analysis of purified recombinant Atg17-Atg31-Atg29 complex (left). A representative image of negatively-stained recombinant Atg17-Atg31-Atg29 (right) with particles in dotted circles (scale bar: 50 nm). (C) class average for the top class from classification of 10,251 negatively-stained particles showing an overall S-shape with two circular-shaped domains attached to the terminal regions of the complex (inset). Side length of this panel is 52 nm. (D) The 3D reconstruction of Atg17-Atg31-Atg29 viewed in different orientations (scale bar: 5 nm). Two characteristic features of the reconstruction are labeled.
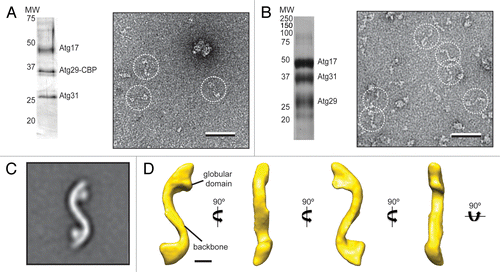
We next prepared negative-stain specimens by adsorbing the purified complexes to glow discharged carbon-coated EM grids and staining these grids with uranyl formate,Citation18 and then examined these specimens with the transmission electron microscope. Although the concentration of the sample was very low (estimated to be < 0.001 mg/ml), we consistently and reproducibly observed elongated particles with an overall shape resembling a stretched out letter “S” ().
Although promising, the native purification could not generate sufficient yield for further single-particle EM-based structural analysis. We therefore reconstituted and isolated recombinant Atg17-Atg31-Atg29 using a purification strategy modified from a previously published procedure.Citation10 In particular, we overexpressed N-terminal His-MBP (hexahistidine-maltose binding protein)-tagged Atg17, N-terminal hexahistidine-tagged Atg31, and untagged Atg29 in the same E. coli expression host, and purified the ternary complex using two affinity chromatography steps (nickel sepharose and amylose sepharose), followed by removal of the His-MBP tag by Tobacco etch virus (TEV) protease cleavage and gel filtration chromatography. The peak fraction from the gel filtration column showed stoichiometric quantities of Atg17, Atg31, and Atg29 (). We examined the purified recombinant complex by negative-stain EM. The raw images showed particles with morphologies that resemble those observed for the native complex (). These initial EM studies established recombinant Atg17-Atg31-Atg29 as a physiologically relevant model to characterize the structural properties of the native complex.
3D reconstruction of the S. cerevisiae Atg17-Atg31-Atg29 complex
To better resolve the structural features of Atg17-Atg31-Atg29, we next determined the three-dimensional (3D) reconstruction of the recombinant ternary complex. Because the ternary complex is too small to be considered for cryo-electron microscopy analysis (calculated mass of approximately 96 kDa), we applied the random conical tilt approach on negatively stained specimens.Citation19 This method is suitable because the negative-staining procedure induces the Atg17-Atg31-Atg29 complex to adopt a preferred orientation on the carbon support layer. We collected pairs of images (55° tilted/untilted) from our negative-stained specimens, selected 10,251 pairs of particles from 150 image pairs using WEB,Citation20 and classified the particles from the images of the untilted specimen into 20 classes using SPIDER.Citation20 The averaged image calculated for the most dominant class showed that the S-shaped Atg17-Atg31-Atg29 complex consists of an elongated central scaffold and has an end-to-end length of approximately 345 Å (). The complex adopts an apparent 2-fold symmetry and contains two circular-shaped domains tethered to the concave side near the terminal regions (). Using the corresponding particles from the images of the tilted specimen, we calculated a 3D reconstruction using the random conical tilt approach implemented in SPIDER.Citation20 The resulting reconstruction, with an estimated resolution of 37 Å according to the Fourier shell correlation = 0.5 cutoff, shows a tube-like density of approximately 35 Å in diameter (). The tubular density forms, along the same plane, two arcs of equal lengths but opposite curvatures (with radii of approximately 90 Å) to generate an overall S-shaped architecture (). The EM structure of S. cerevisiae Atg17-Atg31-Atg29 is reminiscent of the dimeric structural model constructed based on the crystal structure of L. thermotolerans Atg17-Atg31-Atg29.Citation13
Atg17 mediates dimerization and forms the central scaffold of the complex
Because the resolution of our EM reconstruction is insufficient to resolve intermolecular boundaries, we took an unbiased labeling approach to elucidate the subunit organization of the Atg17-Atg31-Atg29 complex. This approach involves fusing a “bulky” tag to the terminus of each subunit, purifying the various tagged complexes, and sequentially localizing the labels in the different versions of the tagged complexes by two-dimensional (2D) negative-stain EM methods.Citation21 We first focused on Atg17, the largest subunit of the complex. Since the His-MBP tag is sufficiently large and globular to be detected by EM, we purified His-MBP-tagged Atg17-Atg31-Atg29 by eliminating the TEV cleavage step in our purification procedure. We selected 8,209 particles from the negatively-stained specimens and classified these particle images into 100 classes. The averaged images calculated for the majority of classes showed the characteristic S-shaped particles with two additional globular densities protruding from the extreme ends (). We confirmed this finding by examining the difference image obtained by subtracting a representative averaged image of untagged Atg17-Atg31-Atg29 from that of the His-MBP-tagged complex ().
Figure 2. Subunit organization of the Atg17-Atg31-Atg29 complex. (A) Localization of Atg17. The left panels are representative class averages of negatively-stained His-MBP-tagged Atg17-Atg31-Atg29 (side length: 59 nm). The middle panel is a difference image generated from subtracting the untagged Atg17-Atg31-Atg29 average from the first His-MBP-tagged Atg17-Atg31-Atg29 average. The right panel depicts difference densities with respect to untagged Atg17-Atg31-Atg29. (B) Overall architecture of Atg17. The left panels show averages of negatively-stained His-MBP-tagged Atg17 that have similar overall shapes as Atg17-Atg31-Atg29. Side length of each panel is 64 nm. Difference image (middle panel), and positions of the difference densities (right panel) show that the terminal circular domains are composed of Atg29 and Atg31. (C) Representative class averages of negatively-stained Atg17-Atg31-Atg29 containing C-terminal GFP-tagged Atg29 (left 3 panels). Localizing the GFP density by difference mapping (middle and right panels). (D) Representative class averages of negatively-stained Atg17-Atg31-Atg29 containing C-terminal GFP-tagged Atg31 (left 3 panels). Localizing the GFP density by difference mapping (middle and right panels). (E) Subunit organization map of Atg17-Atg31-Atg29 with labels depicting the positions of the three subunits.
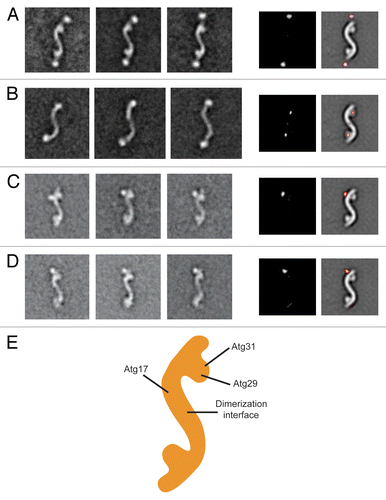
We were unable to assemble a stable Atg17-Atg31-Atg29 complex containing C-terminally green fluorescent protein (GFP)-tagged Atg17. As an alternative approach, we purified His-MBP-tagged Atg17 and examined this purified sample by negative-stain 2D EM. Although the size of Atg17 is near the detection limit of negative-stain EM, we were able to visualize the low contrast Atg17 particles by preserving the His-MBP tag. We performed 2D EM analysis on negatively-stained His-MBP-Atg17, and the obtained class averages not only revealed the broad range of conformations adopted by this protein (see next section) but also clearly showed that Atg17 forms the central S-shaped scaffold (). This result suggests that Atg17 mediates dimerization, while Atg31 and Atg29 constitute the terminal globular domains. To confirm this hypothesis, we calculated a difference image by subtracting a representative averaged image of His-MBP-Atg17 from that of the full Atg17-Atg31-Atg29 complex. Indeed, the positive densities in a difference image overlap perfectly with the globular densities along the terminal regions of the complex (). We also conducted further GFP-labeling studies in which we fused a GFP label to the C terminus of Atg29 or Atg31, purified this GFP-labeled complex, and subjected the purified complex to negative-stain 2D EM analysis. The gallery of class averages showed that the GFP labels, for the C termini of Atg29 and Atg31, are located near the convex side opposite of the globular densities (). We noted that only a single well-defined GFP label was detected in class averages for both Atg29 and Atg31. This observation is likely due to the GFP moiety being more susceptible to proteolytic cleavage, and/or conformational flexibility of the linker between GFP and the tagged protein. Results from the localization studies led to a subunit organization model in which Atg17 forms the central scaffold while Atg31 and Atg29 form the globular domains decorated along this scaffold (). This model is in agreement with the subunit arrangement observed from the crystal structure of L. thermotolerans Atg17-Atg31-Atg29.Citation13
Conformational flexibility of Atg17
Our 2D EM analysis of His-MBP-Atg17 revealed that this dimer can adopt a broad range of conformations; from a canonical S-shape to a fully “stretched out” structure (). To further investigate this flexibility, we compared the radius of curvature and length of His-MBP-labeled Atg17 particles to that of His-MBP-Atg17-Atg31-Atg29 particles. For this analysis, we selected all class averages that contained two discernible His-MBP densities because these averages showed well-defined boundaries for measurements. The length of a particle was measured as a junction-to-junction length at the boundary of the His-MBP linker and Atg17. We found that while the junction-to-junction lengths of His-MBP-labeled Atg17-Atg31-Atg29 particles fell within a narrow range between 275 Å to 310 Å, the distribution of lengths was much broader for His-MBP-Atg17 and ranged from 158 Å to 345 Å (). Despite this variation in the range of lengths observed, the median values of length were 293 Å and 285 Å for the ternary complex and Atg17, respectively. These results indicate that the observed flexibility is an inherent property of Atg17 and is not significantly influenced by the presence of the His-MBP tags.
Figure 3. Analysis of the conformational flexibility of Atg17. (A) Structural segregation of class averages from classification of 5,922 negatively-stained His-MBP-tagged Atg17 particles exhibiting a range of conformations Atg17 can adopt: S-shaped, asymmetric, and extended. Side length of each panel is 64 nm. (B) Distribution of junction-to-junction lengths measured for 17 class averages (1,029 total particles) of His-MBP-labeled Atg17-Atg31-Atg29 (black) and 18 class averages (1,099 total particles) of His-MBP-Atg17 (white). Median length was 293 Å and 285 Å for His-MBP-Atg17-Atg31-Atg29 and His-MBP-Atg17, respectively. (C) Distribution of the ratio of curvature measured for the two arcs within each ternary complex and Atg17 dimer. Seventeen class averages (1,029 total particles) of His-MBP-labeled Atg17-Atg31-Atg29 (black) and 9 class averages (539 total particles) of His-MBP-Atg17 particles (white) were analyzed. Median radius was 75 Å and 213 Å for His-MBP-Atg17-Atg31-Atg29 and His-MBP-Atg17, respectively. (D) Schematic depicting the role of Atg29 and Atg31 in defining and stabilizing the distinct curvature of Atg17.
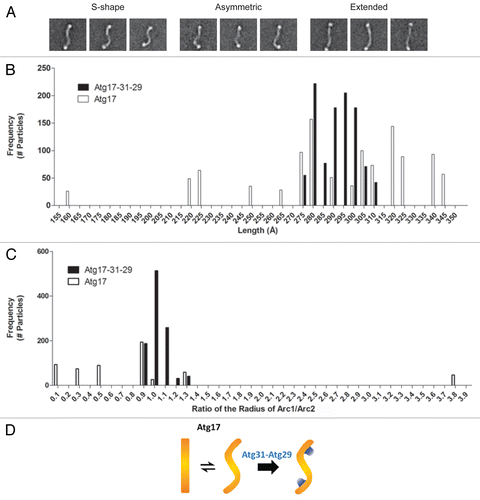
When we compared the measured curvatures, the distribution of values for the His-MBP-labeled Atg17-Atg31-Atg29 was again very narrow, with a median radius of 75 Å, whereas His-MBP-Atg17 exhibited a broader distribution with a median radius of 213Å. In fact, a large number of His-MBP-Atg17 particles exhibited little to no measurable curvature. Furthermore, when the radius of each arc was compared in a single dimer, we observed much lower correlation between the curvatures of each arc, indicating inherent flexibility of the two halves within the Atg17 dimer (). This was not observed in the context of the full complex as the radii of the two arcs correlated well indicating a high degree of symmetry ().
We note that the average junction-to-junction length for the His-MBP-tagged ternary complex is less than the measured end-to-end length of the unlabeled complex, and that the curvature also appears to be slightly underestimated. We attribute these deviations to our inability to accurately locate the true “termini” of the complex because they are partially masked by the His-MBP labels. In spite of this, the general trends we observed do hold, as there are significant differences observed between the measured parameters of His-MBP-labeled Atg17-Atg31-Atg29 particles and that of His-MBP-Atg17 particles. Collectively, results from this analysis confirm that Atg17 is capable of adopting multiple conformations, and implicate a critical role of Atg31 and Atg29 in restricting this flexibility and maintaining the ternary complex in a rigid S-shape with well-defined curvatures ().
Discussion
The structural model of dimeric Atg17-Atg31-Atg29, constructed from small angle X-ray scattering and crystallographic analyses of the core L. thermotolerans, provided a first structural view of this early acting component of the autophagy pathway.Citation13 The EM structure of the full S. cerevisiae Atg17-Atg31-Atg29 complex we reported here provides direct evidence supporting the physiological relevance of the high-resolution model and shows the structural similarities between the orthologous complexes from S. cerevisiae and L. thermotolerans. Notably, we were able to unambiguously fit the high-resolution L. thermotolerans model into our EM density map (Fig. S1). Our docking analysis also showed that the subunit organization, determined in an unbiased manner by single-particle EM, agrees with that observed in the crystal structure. The exception is the location of the C terminus of Atg31, in which our EM data localizes it to the convex side of the arc while in the high-resolution model it localizes to the concave side of the arc. Although the crystallized L. thermotolerans core complex lacks more than 100 C-terminal residues of Atg29,Citation13 there is no unaccounted density observed in our docking analysis. This observation indicates that the C-terminal region of Atg29 is either completely unstructured (as suggested by our recent dataCitation22) or adopts a multitude of conformations that our method cannot resolve.
Atg17-Atg31-Atg29, as a key module of the Atg1 kinase complex, acts in the early steps of the autophagy pathway.Citation5 In addition to being a key positive regulator of the gate-keeping Atg1 kinase, Atg17-Atg31-Atg29 mediates the recruitment of other Atg proteins and protein complexes to the PAS, where the autophagosome is constructed.Citation6 The EM structure of S. cerevisiae Atg17-Atg31-Atg29 and the high-resolution structural model of L. thermotolerans Atg17-Atg31-Atg29 both support a role of this complex as an extended and highly accessible molecular scaffold. These features might allow different Atg proteins to dock and interact with their binding partners and to accommodate the dynamic trafficking of proteins in and out of the PAS during autophagosome formation. Our studies also represent the first single-particle EM analysis of an Atg complex and generate a framework for future investigation of the overall structure and organization of the full Atg1 kinase complex and the Atg1 kinase activation mechanisms.
Our single-particle EM studies confirmed that Atg17 is inherently dimeric and is the primary component involved in dimerization of the complex. Atg17 dimerization doubles the length of the central scaffold and generates two functional modules for protein recruitment. This oligomeric state is critical to Atg17-Atg31-Atg29 function, as an engineered monomeric ternary complex fails to localize to the PAS, and such mutation also results in a complete loss in autophagy activity.Citation13 The mammalian ortholog of Atg17 is RB1CC1/FIP200, and future experiments will focus on examining whether or not this protein also forms a molecular dimer.Citation23
Although dimeric Atg17 forms the bulk of the Atg17-Atg31-Atg29 complex, Atg29, and Atg31 are essential for the proper function of the complex and efficient autophagy.Citation24,Citation25 While Atg29 and Atg31 are not involved in dimerization, results from our length and curvature analysis indicated that their extensive interaction with Atg17 restricts the inherent flexibility of Atg17 and establishes its characteristic curvatures (). Our finding indicates that the overall shape and curvature of Atg17-Atg31-Atg29 is critical to its function. Previous molecular genetics studies on Atg17 have identified two N-terminal regions of Atg17 that are critical for interaction with Atg1 and Atg13, and for initiating autophagosome formation.Citation7,Citation8 According to the high-resolution model, these N-terminal regions localize distal to the point of contact of the Atg31-Atg29 dimer, where curvature is most prominent.Citation13 It is therefore possible that the distinct curvature is required for Atg17-Atg31-Atg29 to interact with Atg1 and Atg13 in the context of the Atg1 kinase complex.
Based on their crystallographic studies, Ragusa et al. initially speculated that the distinct curvatures of Atg17-Atg31-Atg29 are involved in sensing highly curved phagophore membranes much like BAR domain-containing proteins.Citation26 Paradoxically, they found that Atg17-Atg31-Atg29 cannot bind lipid vesicles of any sizes.Citation13 After conducting liposome-binding studies, these authors proposed an alternate model in which the presumably unstructured Atg1 early autophagy targeting/tethering (EAT) domain within the Atg1 kinase complex is involved in tethering two highly curved vesicles in close proximity. According to this model, the observed “S-shaped” conformation of the Atg17-Atg31-Atg29 complex represents an auto-inhibited state, and tethering by the Atg1 EAT domain requires displacement of Atg31 and Atg29 from the Atg17 central scaffold. Although our EM-based analysis is capable of capturing different conformations of a protein complex, we have not observed significant deviations in the positions of the globular densities constituted by Atg31 and Atg29 when we examined the unlabeled and His-MBP-labeled ternary complexes. That being said, Atg31 and Atg29 are phosphoproteins,Citation10,Citation22 and these post-translational modifications may switch the ternary complex to an active state, or relieve the proposed auto-inhibition. Future experiments should focus on mapping these phosphorylation sites, determining whether or not the phosphorylation events are associated with autophagy initiation, and assessing how these post-translational modifications alter the architecture and function of the Atg17-Atg31-Atg29 complex.
Materials and Methods
Purification of native Atg17-Atg31-Atg29
Yeast cells (Atg29-TAP) were grown to an OD600 of ~2.5 and the cell pellets were frozen until purification. Approximately 25 g of frozen cells were resuspended in lysis buffer (100 mM HEPES pH 8, 250 mM KCl, 2 mM EDTA, 1 mM EGTA, 5% glycerol, 1% CHAPS, 2.5 mM DTT, 1 mM PMSF, 0.1 mM sodium vanadate, 2 mM benzamidine, 50 mM NaF) containing EDTA-free protease inhibitor (Roche, 11873580001) and lysed by bead beating. The lysate was clarified by centrifugation at ~30,000 × g for 30 min, and the supernatant fraction was incubated with IgG sepharose (GE Healthcare, 17-0969-01) for 1 h. The resin was washed with buffer (50 mM Tris-HCl pH 8.0, 100 mM NaCl, 5% glycerol, 0.1% CHAPS, 0.5 mM DTT) and bound proteins were eluted by TEV protease (Life Technologies, 12575-015) cleavage for 1.5 h at 16 °C. The eluate was concentrated using an Amicon Ultra 10K centrifugation unit (Millipore, UFC901008) and loaded onto a Superose 6 column equilibrated with buffer (50 mM Tris-HCl pH 8.0, 100 mM NaCl, 5% glycerol, 0.1% CHAPS) and connected to an ÄKTAprime plus (GE Healthcare, 11-0013-13). Purified complexes were TCA-precipitated for SDS-PAGE analysis or examined by EM.
Purification of recombinant untagged and GFP-tagged Atg17-Atg31-Atg29
Atg17 was cloned into the pET28bHMT vector,Citation27 whereas Atg31 and Atg29 were cloned into the two multiple cloning sites of pETDuet-1 vector (Novagen, 71146-3). To purify recombinant untagged Atg17-Atg31-Atg29, N-terminal His-MBP-tagged Atg17, was co-expressed with N-terminal hexahistidine-tagged Atg31, and untagged Atg29 in E. coli BL21 StarTM (DE3) (Life Technologies, C6010-03). The recombinant complex was purified using HisPur nickel-NTA resin (Pierce, 88221), and amylose resin (New England Biolabs, E8021S), followed by TEV protease cleavage to remove the His-MBP tag. The cleaved complex was further purified by gel filtration chromatography using a Superose 6 column connected to an ÄKTAprime plus. We used the same procedure for purifying His-MBP-tagged Atg17-Atg31-Atg29 and His-MBP-tagged Atg17 alone, except that the TEV cleavage step was eliminated to preserve the affinity tag.
For purification of complexes containing GFP-tagged Atg29 or Atg31, we cloned Atg17 into the pGEX6P-1 vector (GE Healthcare, 28-9546-48), and Atg31 and Atg29 into the two multiple cloning sites of pCOLADuet-1 vector (Novagen, 71406-3). The GFP-tag was subsequently engineered into the C terminus of Atg31 or Atg29. To purify the GFP-tagged complex, N-terminal glutathione-S-transferase-tagged Atg17 was co-expressed with C-terminal GFP-tagged Atg31 (or Atg29) together with untagged Atg29 (or Atg31) in E. coli BL21Star, and purified using glutathione agarose (Pierce, 16100), followed by PreScission protease (GE Healthcare, 27-0843-01) cleavage to remove the glutathione-S-transferase tag, and gel filtration chromatography (using a Superose 6 column).
Electron microscopy
Negatively-stained specimens were prepared as previously described.Citation18 Raw images were recorded at a nominal magnification of 49,000× on a 4 K × 4 K Eagle charge-coupled device camera (FEI) with a Tecnai Spirit transmission electron microscope (FEI) operated at an accelerating voltage of 120 kV. Images for 2D analysis were collected using the same instrument with the same operating parameters and at a defocus value of −1.2 μm. For tilt-pair data collection, the same specimen areas were recorded at tilt angles of 55° and untilted under low-dose conditions at a nominal magnification of 49,000 and a defocus value of −1.2 μm on the 4 K × 4 K Eagle charge-coupled device camera. For image processing, 2 × 2 pixels were averaged to yield a pixel size of approximately 4.6 Å at the specimen level.
Image processing
For 2D analysis of untagged Atg17-Atg31-Atg29, particles were interactively selected using Boxer.Citation28 The 10,251 selected particles were windowed into 112 × 112-pixel images, rotationally and translationally aligned, and subjected to ten cycles of multi-reference alignment using SPIDER.Citation20 Each round of alignment was followed by K-means classification specifying 100 classes. We used the same procedure for analyzing His-MBP-tagged Atg17-Atg31-Atg29 (8,209 particles), His-MBP-tagged Atg17 (5,922 particles), the ternary complex containing C-terminal GFP-tagged Atg29 (2,290 particles) and C-terminal GFP-tagged Atg31 (1,991 particles), except the window size was enlarged to 128 pixels, 140 pixels, 128 pixels and 128 pixels, respectively, for these samples.
For 3D reconstruction, a total of 10,251 particle pairs were interactively selected from both the untilted and 55° tilted images (150 pairs) using WEB.Citation20 The selected particles were windowed into 112 × 112-pixel images, and the particles from the untilted specimens were classified into 20 classes. 3D reconstruction was calculated for the largest class from the tilted specimens using the backprojection and angular refinement procedures in SPIDER.
Length and curvature analysis
Measurements of particles were performed on CorelDRAW5. Only class averages containing two visible MBP-labels were selected for analysis. Seventeen class averages for His-MBP-Atg17-Atg31-Atg29 and 18 class averages for His-MBP-Atg17 met these criteria. Measurements of the width and height for each arc of a monomer of Atg17 were measured separately. The width of the arc was determined to begin at the junction of the MBP-linker-Atg17 and end at the center of the Atg17 dimer, while the height was measured at edge of the arc apex to the arc chord.
| Abbreviations: | ||
| 2D | = | two-dimensional |
| 3D | = | three-dimensional |
| Atg | = | autophagy-related |
| EAT | = | early autophagy targeting/tethering |
| EM | = | electron microscopy |
| GFP | = | green fluorescent protein |
| His | = | hexahistidine |
| MBP | = | maltose binding protein |
| PAS | = | phagophore assembly site |
| TEV | = | Tobacco etch virus |
Additional material
Download Zip (892.5 KB)Acknowledgments
We thank Dr Leonard Foster for assistance with mass spectrometry analysis. This work was supported by NIH grant GM053396 to DJK, and by a Canadian Institutes of Health Research Operating Grant, a Natural Sciences and Engineering Research Council of Canada Discovery Grant, a Michael Smith Foundation for Health Research Career Investigator Award, a Canadian Institutes of Health Research New Investigator award, and startup funds from the University of British Columbia to CKY, and by a Natural Sciences and Engineering Research Council of Canada Postgraduate Scholarship award to LHC.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/25687
Related Research Data
References
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 2009; 10:458 - 67; http://dx.doi.org/10.1038/nrm2708; PMID: 19491929
- Kraft C, Martens S. Mechanisms and regulation of autophagosome formation. Curr Opin Cell Biol 2012; 24:496 - 501; http://dx.doi.org/10.1016/j.ceb.2012.05.001; PMID: 22664348
- Noda NN, Ohsumi Y, Inagaki F. ATG systems from the protein structural point of view. Chem Rev 2009; 109:1587 - 98; http://dx.doi.org/10.1021/cr800459r; PMID: 19236009
- Suzuki K, Ohsumi Y. Current knowledge of the pre-autophagosomal structure (PAS). FEBS Lett 2010; 584:1280 - 6; http://dx.doi.org/10.1016/j.febslet.2010.02.001; PMID: 20138172
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107 - 32; http://dx.doi.org/10.1146/annurev-cellbio-092910-154005; PMID: 21801009
- Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 2007; 12:209 - 18; http://dx.doi.org/10.1111/j.1365-2443.2007.01050.x; PMID: 17295840
- Cheong H, Yorimitsu T, Reggiori F, Legakis JE, Wang C-W, Klionsky DJ. Atg17 regulates the magnitude of the autophagic response. Mol Biol Cell 2005; 16:3438 - 53; http://dx.doi.org/10.1091/mbc.E04-10-0894; PMID: 15901835
- Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell 2005; 16:2544 - 53; http://dx.doi.org/10.1091/mbc.E04-08-0669; PMID: 15743910
- Kawamata T, Kamada Y, Kabeya Y, Sekito T, Ohsumi Y. Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol Biol Cell 2008; 19:2039 - 50; http://dx.doi.org/10.1091/mbc.E07-10-1048; PMID: 18287526
- Kabeya Y, Noda NN, Fujioka Y, Suzuki K, Inagaki F, Ohsumi Y. Characterization of the Atg17-Atg29-Atg31 complex specifically required for starvation-induced autophagy in Saccharomyces cerevisiae.. Biochem Biophys Res Commun 2009; 389:612 - 5; http://dx.doi.org/10.1016/j.bbrc.2009.09.034; PMID: 19755117
- Kraft C, Kijanska M, Kalie E, Siergiejuk E, Lee SS, Semplicio G, Stoffel I, Brezovich A, Verma M, Hansmann I, et al. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. EMBO J 2012; 31:3691 - 703; http://dx.doi.org/10.1038/emboj.2012.225; PMID: 22885598
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 2000; 150:1507 - 13; http://dx.doi.org/10.1083/jcb.150.6.1507; PMID: 10995454
- Ragusa MJ, Stanley RE, Hurley JH. Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell 2012; 151:1501 - 12; http://dx.doi.org/10.1016/j.cell.2012.11.028; PMID: 23219485
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 1999; 17:1030 - 2; http://dx.doi.org/10.1038/13732; PMID: 10504710
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Séraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 2001; 24:218 - 29; http://dx.doi.org/10.1006/meth.2001.1183; PMID: 11403571
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature 2003; 425:737 - 41; http://dx.doi.org/10.1038/nature02046; PMID: 14562106
- Yip CK, Walz T. Molecular structure and flexibility of the yeast coatomer as revealed by electron microscopy. J Mol Biol 2011; 408:825 - 31; http://dx.doi.org/10.1016/j.jmb.2011.03.029; PMID: 21435344
- Ohi M, Li Y, Cheng Y, Walz T. Negative Staining and Image Classification - Powerful Tools in Modern Electron Microscopy. Biol Proced Online 2004; 6:23 - 34; http://dx.doi.org/10.1251/bpo70; PMID: 15103397
- Radermacher M, Wagenknecht T, Verschoor A, Frank J. Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli.. J Microsc 1987; 146:113 - 36; http://dx.doi.org/10.1111/j.1365-2818.1987.tb01333.x; PMID: 3302267
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol 1996; 116:190 - 9; http://dx.doi.org/10.1006/jsbi.1996.0030; PMID: 8742743
- Lees JA, Yip CK, Walz T, Hughson FM. Molecular organization of the COG vesicle tethering complex. Nat Struct Mol Biol 2010; 17:1292 - 7; http://dx.doi.org/10.1038/nsmb.1917; PMID: 20972446
- Mao K, Chew LH, Inoue-Aono Y, Cheong H, Nair U, Popelka H, Yip CK, Klionsky DJ. Atg29 phosphorylation regulates coordination of the Atg17-Atg31-Atg29 complex with the Atg11 scaffold during autophagy initiation. Proc Natl Acad Sci U S A 2013; 110:E2875 - 84; http://dx.doi.org/10.1073/pnas.1300064110; PMID: 23858448
- Hara T, Mizushima N. Role of ULK-FIP200 complex in mammalian autophagy: FIP200, a counterpart of yeast Atg17?. Autophagy 2009; 5:85 - 7; http://dx.doi.org/10.4161/auto.5.1.7180; PMID: 18981720
- Kawamata T, Kamada Y, Suzuki K, Kuboshima N, Akimatsu H, Ota S, Ohsumi M, Ohsumi Y. Characterization of a novel autophagy-specific gene, ATG29. Biochem Biophys Res Commun 2005; 338:1884 - 9; http://dx.doi.org/10.1016/j.bbrc.2005.10.163; PMID: 16289106
- Kabeya Y, Kawamata T, Suzuki K, Ohsumi Y. Cis1/Atg31 is required for autophagosome formation in Saccharomyces cerevisiae.. Biochem Biophys Res Commun 2007; 356:405 - 10; http://dx.doi.org/10.1016/j.bbrc.2007.02.150; PMID: 17362880
- Qualmann B, Koch D, Kessels MM. Let’s go bananas: revisiting the endocytic BAR code. EMBO J 2011; 30:3501 - 15; http://dx.doi.org/10.1038/emboj.2011.266; PMID: 21878992
- Van Petegem F, Clark KA, Chatelain FC, Minor DL Jr.. Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature 2004; 429:671 - 5; http://dx.doi.org/10.1038/nature02588; PMID: 15141227
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 1999; 128:82 - 97; http://dx.doi.org/10.1006/jsbi.1999.4174; PMID: 10600563