Abstract
Beyond its role in recycling intracellular components nonselectively to sustain survival in response to metabolic stresses, autophagy can also selectively degrade specific cargoes such as damaged or dysfunctional organelles to maintain cellular homeostasis. Mitochondria, known as the power plant of cells, are the critical and dynamic organelles playing a fundamental role in cellular metabolism. Mitophagy, the selective autophagic elimination of mitochondria, has been identified both in yeast and in mammalian cells. Moreover, defects in mitophagy may contribute to a variety of human disorders such as neurodegeneration and myopathies. However, the role of mitophagy in development and cancer remains largely unclear. In this review, we summarize our current knowledge of the regulation and function of mitophagy in development and cancer.
Introduction
As an evolutionarily conserved and genetically programmed lysosomal degradation pathway, autophagy principally self-digests part of the cytoplasmic constituents to maintain homeostasis in response to various metabolic stresses such as nutrient deprivation, growth factor depletion, and hypoxia.Citation1,Citation2 Although it was originally considered as a nonselective bulk degradative process, autophagy was recently found to selectively degrade several specific cargos, either damaged or superfluous organelles or protein aggregates, even under nutrient-rich conditions, to maintain cell viability and organelle quality and quantity control.Citation3,Citation4 To date, several selective types of autophagy have been confirmed, including the cytoplasm-to-vacuole targeting (Cvt) pathway,Citation5 pexophagy,Citation6 ribophagy,Citation7 and xenophagy.Citation8
Mitophagy, another well-characterized type of cargo-specific autophagy, is recently capturing more and more attention. As critical and dynamic organelles in eukaryotic cells, mitochondria are primarily responsible for aerobic energy production through oxidative phosphorylation (OXPHOS).Citation9 However, accumulation of the by-product reactive oxygen species (ROS) during metabolism is detrimental and inseparably leads to mitochondrial dysfunction. Therefore, it is essential to remove dysfunctional mitochondria in a timely manner so as to maintain mitochondrial turnover and cellular homeostasis. The presence of mitochondria within the double-membrane structure that will become an autophagosome in mammalian cells was reported as early as 1957.Citation10 Subsequently, the selective elimination of damaged or excessive mitochondria by mitophagy was found to be an important mitochondrial quality control mechanism.Citation9,Citation11 As mitochondria are the essential organelles indispensable for ATP production and cellular homeostasis, mitochondrial dysfunction has been closely linked to many pathophysiological conditions and diseases. Recent studies have also demonstrated that mitophagy defects are indeed associated with a wide array of human disorders including neurodegenerative diseases,Citation12 myopathies,Citation13 aging,Citation14 cardiac diseases, and autoimmune diseases.Citation15,Citation16 Despite recent substantial research on autophagy, the molecular mechanisms with regard to regulating the fate of mitophagy destined for degradation remain poorly understood. In this review, we summarize recent advances on the regulation and biological significance of mitophagy and highlight its complicated role in development and cancer.
Overview on the Regulation of Mitophagy
Mitophagy serves to degrade damaged or dysfunctional mitochondria to match metabolic demand and orchestrate mitochondrial quality and quantity control in cellular homeostasis. The budding yeast Saccharomyces cerevisiae, a key model organism for understanding the molecular aspects of autophagy, has been an ideal system for exploring the regulation of mitophagy. Mitophagy in yeast can generally be induced under two different conditions: nitrogen starvation and culturing cells to the post-logarithmic phase in a medium with a nonfermentable carbon source.Citation17 Currently, more than 30 autophagy-related (ATG) genes have been identified in yeast, of which the majority have clear mammalian counterparts.Citation18 Being a selective type of autophagy, mitophagy shares core machinery with canonical starvation-induced autophagy. Accordingly, many Atg proteins also play indispensable roles in mitophagy. Herein we briefly review the roles of the main Atg proteins in the regulation of mitophagy.
Yeast Atg proteins in mitophagy
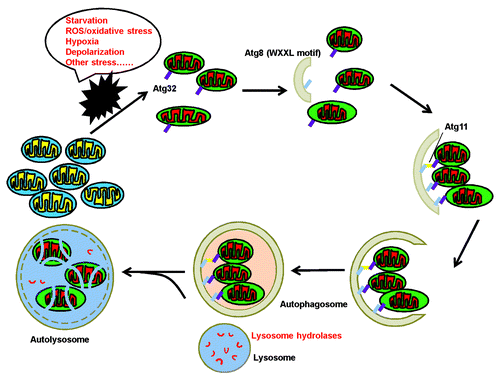
Recently, two independent groups using a genome-wide screen in yeast for mitophagy-related genes almost simultaneously demonstrated that Atg32 is exclusively devoted to mitophagy rather than nonselective macroautophagy or other types of specific autophagy.Citation17,Citation19 Under mitophagy-inducing conditions, the N terminus of the receptor protein Atg32 (residues 51–150) interact with the C terminus of the scaffold protein Atg11 to deliver mitochondria into the vacuolar lumen. In addition to Atg11 binding, Atg32 can also interact with Atg8 through a conserved WXXL motif in its cytosolic domain.Citation19 Therefore, Atg32 mediates mitophagy through direct or indirect interaction with Atg8 (). However, it has been reported that no defects in mitochondrial and vacuolar morphology are detected in ATG32 null cells,Citation19 indicating that there may be other, yet-unidentified mechanisms for Atg32-independent mitophagy.
Figure 1. The general process of selective autophagic degradation of mitochondria. When cells suffer different kinds of stress, damaged or excessive mitochondria are removed by mitophagy to maintain cellular homeostasis. First, damaged mitochondria are recruited into phagophores. Then the phagophore membranes are elongated and sequester targeted mitochondria to form an autophagosome. Finally, an autophagosome fuses with a lysosome to eliminate the engulfed mitochondria through the action of lysosomal hydrolases.
Strikingly, posttranslational modification such as phosphorylation is implicated in mitophagy induction. Atg32 can be phosphorylated upon mitophagy induction by nitrogen starvation and the phosphorylation of Ser114 and Ser119 is critical for Atg11Atg32 interaction.Citation20 It is worth noting that Hog1 and Pbs2 have been validated to regulate Atg32 phosphorylation, and both Atg32 phosphorylation and mitophagy are severely blocked after depletion of either Hog1 or Pbs2.Citation20,Citation21 Unfortunately, it remains unclear which kinase downstream of Hog1-Pbs2 can directly phosphorylate Atg32. Moreover, Hog1 is not always activated even under mitophagy-inducing circumstances. Consequently, it was proposed that the initial mitophagy-inducing triggers coming from mitochondria could activate some yet unrecognized kinase(s) to phosphorylate Atg32 at Ser114 and/or Ser119 and facilitate its interaction with Atg11.Citation20
Different from Hog1 and Pbs2 that are specifically dedicated to mitophagy, another mitogen-activated protein kinase (MAPK), Slt2, is responsible for both mitophagy and pexophagy.Citation21 Intriguingly, both Hog1 and Slt2 remain in the cytoplasm throughout mitophagy, highlighting that both of them might activate certain undiscovered cytoplasmic targets to eventually phosphorylate Atg32.Citation21 Additionally, the timing of their activation is of subtle difference: Slt2 is activated earlier than Hog1, and Slt2 but not Hog1 is essential for recruitment of mitochondria to the mitophagy-specific phagophore assembly site (PAS), indicating that Slt2 might participate in the initiation of mitophagy while Hog1 acts at a later stage.Citation22
Similar to Atg32, Atg33 was identified from a genomic screen for yeast mutants deficient in mitophagy and is another exclusive mitophagy-specific protein.Citation23 Notably, deletion of ATG33 obstructs mitophagy almost completely at the post-log phase but reduces starvation-induced mitophagy by only approximately 50%, suggesting that different mechanisms are dedicated to mitophagy induced under different conditions.Citation23
Mammalian ATG proteins in mitophagy
Mitophagy is highly conserved from yeast to mammals. Consequently, ATGs (in particular, the homologs of the core Atg proteins) are also indispensable for mitophagy in higher eukaryotes. However, the mammalian homologs of Atg32 and Atg33, if they exist, have not yet been identified. Other ATGs required for mitophagy in mammals and their yeast homologs are summarized in . Atg19, Atg22, Atg25, Atg26, Atg28, and Atg30 do not appear to play any role in mitophagy. Moreover, Atg11, Atg20, and Atg24 participate specifically in the Cvt pathway, pexophagy, and mitophagy, but are not essential for nonselective macroautophagy (for example, Atg11 facilitates, but is not absolutely needed for, nonselective macroautophagy),Citation37 highlighting the differences among the Atg proteins in the regulation of macroautophagy and mitophagy.
Table 1. Mitophagy-related Atg proteins in yeast and their mammalian homologs
Other proteins important in the regulation of mitophagy
Uth1
Initially identified in a genetic screen as a protein implicated in life-span determination,Citation38 Uth1 also participates in the regulation of mitophagy.Citation39 The Uth1 protein is a member of the SUN gene family (SIM1, UTH1, and NCA3), mainly anchored on the outer mitochondrial membrane (OMM) and is responsible for mitophagy induced by rapamycin or nitrogen starvation.Citation39 Intriguingly, there are two distinct Uth1-related mitochondrial autophagic pathways: Uth1-dependent and Uth1-independent mitophagy. The first one is a selective process of mitophagy that cannot occur in the absence of Uth1. The second one is a nonselective mitophagy present in both wild-type and UTH1-deleted strains, in which mitochondria are engulfed nonexclusively with a large amount of surrounding cytosol.Citation40
Ptc6/Aup1
Besides Uth1, Ptc6/Aup1, a member of the protein phosphatase 2C superfamily, is required for mitophagy in stationary phase yeast and plays a prosurvival role. It was identified in a protein phosphatase homolog screen aiming to capture proteins that can functionally interact with the autophagy-dedicated protein kinase Atg1.Citation41,Citation42 Notably, there appear to be different roles for Uth1 and Ptc6 in mitophagy, although both of them are required for this process: UTH1 knockout promotes the survival of cells cultured in nitrogen-deprivation medium with lactate but not glucose, whereas knockout of PTC6 leads to decreased cell survival in long-term stationary phase cultures even under sustained gluconeogenic conditions, suggesting that distinct regulatory networks might be involved in the process of mitophagy mediated by these proteins.Citation41 Therefore, further investigation will be needed to shed light upon the possible relationship between these mitophagy-associated proteins and their detailed contributions in the regulation of mitophagy.
PINK1 and PARK2
The best-studied mitophagy-related pathway in mammals is mediated by PINK1 (PTEN induced putative kinase 1) and PARK2/Parkin. Loss-of-function mutations in the PINK1 and PARK2 genes have been substantiated to significantly cause the early onset of autosomal recessive Parkinson disease.Citation43 PINK1 and PARK2 encode a mitochondrially targeted Ser/Thr kinase and a cytosolic RING-domain containing E3 Ub ligase, respectively. Genetic analysis of PINK1 and PARK2 in Drosophila melanogaster indicates that they work together to regulate mitochondrial integrity and suppress mitochondrial damage.Citation44,Citation45
PARK2 also regulates mitochondrial morphology and integrity in mammalian cells and is widely expressed in many tissues including brain, skeletal muscle, heart, and liver.Citation46,Citation47 PARK2 translocates to depolarized mitochondria in cells treated with carbonyl cyanide m-chlorophenyl hydrazone (CCCP), a mitochondrial uncoupler that dissipates the mitochondrial membrane potential to induce mitochondrial damage.Citation46 Other mitochondrial poisons such as paraquat and azide can also induce the translocation of PARK2 to damaged mitochondria,Citation46,Citation47 which is required for depolarization-mediated fragmentation.Citation48 In addition, a novel role of PARK2 as a tumor suppressor has been verified in breast and ovarian cancer, highlighting that PARK2-dependent mitophagy might be associated with cancer biology.Citation49
Translocation of PARK2 to impaired mitochondria and induction of mitophagy are indispensably required for PINK1 activity even in the absence of mitochondrial uncoupling.Citation3,Citation50 PINK1 locates either in the mitochondrial intermembrane space (IMS) or OMMCitation51,Citation52 and may be maintained at a low expression level in healthy mitochondria due to two potential mechanisms; either rapid proteolytic destabilization by proteases such as PMPCA (peptidase [mitochondrial processing] α)Citation53 and the inner membrane PARL (presenilin associated, rhomboid-like),Citation54 or import to the inner mitochondrial membrane through the translocase complexes of the outer and inner mitochondrial membrane (TOMM and TIMM, respectively).Citation55 Once the mitochondrial membrane potential is decreased, proteolysis or import of PINK1 is blocked so that it is stabilized and accumulated on the OMM where it recruits PARK2 to induce mitophagy.Citation52 However, some conflicting results have recently reported that PINK1 deficiency may contribute to mitochondrial fragmentation,Citation56,Citation57 and knockdown of PINK1 with RNA interference induces mitochondrial fragmentation and autophagy in neuronal SH-SY5Y cells, whereas overexpression of wild-type PINK1 suppresses mitophagy,Citation57 consistently supporting the opposite view that inhibition of PINK1 activity may promote mitophagy. These paradoxical findings are likely due to different testing times and/or different organisms used in the studies. Following translocation, PARK2-mediated ubiquitination of a subset of mitochondrial substrates accelerates mitophagy.Citation58,Citation59 PARK2 polyubiquitinates itself and its mitochondrial substrates predominantly at Lys63 residues, producing the docking site for the Ub-binding adaptor SQSTM1/p62 to trigger autophagic degradation by binding to the LC3-interacting region (LIR) motif of LC3/GABARAP (GABA(A) receptor-associated protein).Citation60 Surprisingly, some divergent results propose that SQSTM1 deletion in mouse embryonic fibroblasts facilitates rather than hinders mitochondrial degradation in a PARK2-dependent manner,Citation61 and recruitment of SQSTM1 to mitochondrial-anchored Ub is not sufficient to promote mitophagy,Citation62 implying that SQSTM1 is not indispensable for mitophagy. The most plausible explanation for this discrepancy is the so-called “SQSTM1 functional redundancy”:Citation62 Some other intracellular proteins may substitute for SQSTM1 during mitophagy when it is lost or the corresponding gene is deleted. For instance, HDAC6 was recently identified as another Ub-binding autophagic component for mitochondrial elimination and autophagosome–lysosome fusion.Citation63,Citation64 Specially, some PARK2 substrates can be degraded through a Lys63-linked ubiquitination-independent manner. For instance, ubiquitination of MFN1 (mitofusin 1) and MFN2 is mediated by PARK2 upon membrane depolarization and contributes to degradation in a proteasome-dependent manner via linking with Lys48 polyubiquitin chainsCitation59 or in a VCP/p97-dependent mannerCitation65 and VDAC1 (voltage-dependent anion channel 1) is degraded by the autophagy–lysosomal pathway through recruiting SQSTM1 with Lys27-linked ubiquitin (Ub) chains.Citation66
The molecular mechanism underlying how PINK1 recruits PARK2 for the subsequent induction of mitophagy remains obscure. It is plausible that PINK1 phosphorylates cytosolic PARK2 with its kinase domain that faces the cytoplasm.Citation51 Indeed, PINK1-triggered phosphorylation of two conserved serine/threonine residues, T175 and T217 in human PARK2, and its intact RING1 domain are crucial for its translocation to mitochondria.Citation67 Moreover, S65 in the PARK2 Ub-like domain was recently found to be directly phosphorylated in a PINK1-dependent manner upon the depolarization of mitochondria.Citation68 Intriguingly, several lines of evidence recently indicate that there may exist a crosstalk between PINK1-PARK2-dependent mitophagy and mitophagy receptor-mediated pathways.Citation69 For example, BNIP3L/NIX (BCL2/adenovirus E1B 19 kDa interacting protein 3-like) appears to promote CCCP-induced mitochondrial depolarization and PARK2 translocation to mediate mitophagy in mouse embryonic fibroblasts,Citation70 and the newly identified mitophagy receptor FUNDC1 (FUN14 domain containing 1) in mammalian cells may be associated with FCCP-mediated mitophagy,Citation71 suggesting that identification of more cofactors and coregulators implicated in PINK1-PARK2-mediated mitophagy may be beneficial to further pursue the mechanism of this pathway. Other mitophagy inducers, receptors and their interacting partners and related regulators are listed in .
Table 2. Mitophagy inducers, receptors, and their interacting partners and related regulators
Collectively, an increasing number of regulators have been recognized to play crucial roles in mitophagy, raising some pending questions as to whether different regulators actually regulate mitophagy functionally depending on context-specific conditions and distinct initiating signals, or all regulators ultimately converge at a canonical model of mitochondrial degradation, or even if these differences are only attributed to different experimental procedures.
Mitophagy in Development
Apart from degradation of damaged mitochondria, removal of nondamaged or superfluous mitochondria occurs as a pivotal process for organ and tissue development. Thus, mitophagy during some specialized developmental stages is deemed as a critical quantity control mechanism for maintaining the proper amount of mitochondria.
Mitophagy in early embryogenesis
One intriguing role of mitophagy in development is selective degradation of paternal mitochondria in fertilized oocytes. Mitochondrial DNA (mtDNA) is maternally inherited in almost all eukaryotes despite the fact that, at least in some organisms, sperm-derived paternal mitochondria enter into the oocyte cytoplasm upon fertilization.Citation88 Indeed, several studies in Caenorhabditis elegans embryos have recently demonstrated that fertilization-triggered autophagy is essential for removal of paternal mitochondria and maintenance of maternally inherited mtDNA.Citation89-Citation91 After fertilization, this induction of mitophagy is triggered by penetrating sperm components.Citation89,Citation90 Importantly, such degradation seems to be associated with protein ubiquitination in mammals.Citation92,Citation93 PHB (prohibitin), a highly conserved inner mitochondrial membrane protein, can be ubiquitinated by the Ub-proteasome system to recognize paternal mitochondria and facilitate their degradation.Citation94,Citation95 However, its counterpart in C. elegans cannot be ubiquitinated. Instead, the nematode-specific sperm membranous organelles are ubiquitinated and removed by autophagic machinery together with paternal mitochondria.Citation90 Nevertheless, it remains unknown whether paternal mitochondria are incorporated into autophagosomes for degradation by virtue of their close association with ubiquitinated membranous organelles, or mitophagy is able to degrade nonubiquitinated paternal mitochondria.
Mitophagy in erythrocyte differentiation
In mammals, terminal differentiation of reticulocytes during erythropoiesis requires enucleation (the expulsion of the nucleus) and organelle removal (such as removal of mitochondria by mitophagy).Citation96 BNIP3L is required for programmed mitochondrial clearance since its expression is highly upregulated during reticulocyte maturation and bnip3l−/− mice retain their mitochondria in circulating reticulocytes.Citation77-Citation80 Like wild-type reticulocytes undergoing maturation, autophagy can also be induced in bnip3l−/− reticulocytes. However, the mitochondria are not eliminated. This failure to eliminate mitochondria reveals that they are not properly targeted to autophagosomes in the absence of BNIP3L.
Surprisingly, although BNIP3L was originally thought to be a BH3-only member of the pro-apoptotic BCL2 (B-cell CLL/lymphoma 2) family essential in the regulation of programmed cell death, some proapoptotic proteins including BAX, BAK1, BCL2L1, BCL2L11/BIM, or BBC3/PUMA are not involved in BNIP3L-mediated mitophagy, implying that it may be independent of the proapoptotic pathways during reticulocyte differentiation.Citation30,Citation77,Citation78
Indeed, BNIP3L has been verified as a mitophagy receptor in mammalian cells.Citation80 It is anchored at the OMM and contains a WXXL motif near its N terminus facing the cytosol, which binds to the mammalian homolog of Atg8, LC3, and the LC3 homolog GABARAP.Citation3,Citation80,Citation81 The WXXL (35–38) motif is indispensable for BNIP3L-LC3/GABARAP interaction and BNIP3L-mediated mitophagy, although this interaction can be enhanced under mitochondrial stress conditions such as treatment with rotenone or CCCP.Citation80 Interestingly, depolarizing mitochondria with the uncoupling agent FCCP, or the BH3 mimetic ABT-737, restores mitophagy even in bnip3l−/− reticulocytes,Citation79 demonstrating that mitochondrial depolarization-dependent mitophagy functions through BNIP3L-independent pathways. However, the initiating signals for mitophagy and the relationship between depolarization- and BNIP3L-mediated mitophagy during reticulocyte maturation remain poorly understood.
A mammalian homolog of the yeast mitophagy receptor, Atg32, has not been identified; however, several common characteristics are shared between Atg32 and BNIP3L, suggesting that BNIP3L might be the equivalent of Atg32 in mammals. First, both of them are OMM proteins with their N-terminal domains in the cytosol and C-terminal regions in the IMS. Second, both of them contain a WXXL motif for binding to Atg8 or LC3/GABARAP. Nevertheless, since the mammalian counterpart of Atg11, which interacts with Atg32 as a scaffold protein, remains undiscovered, it is still an open question as to how BNIP3L functions to regulate mitophagy in mammalian cells.
In addition to BNIP3L, ULK1 (Unc51 like autophagy activating kinase 1)Citation97 and Atg7Citation30,Citation96 are also implicated in mitophagy during reticulocyte maturation since Ulk1-deficient mice show delayed mitochondrial clearance, although this defect can be rescued by treatment with CCCP in vitro, emphasizing that there are other Ulk1-independent mechanisms to compensate for mitophagy in Ulk1-deficient reticulocytes. Similarly, there is an accumulation of damaged mitochondria in atg7−/− mice erythrocytes, eventually leading to differentiation failure and cell death, indicating that ATG7 is a critical mitophagy regulator in mammalian hematopoietic cells.Citation96 However, a recent study showed that mitophagy is partially impaired but not completely blocked in atg7−/− reticulocytes, and that ATG7 deficiency has no effect on the expression of BNIP3L and vice versa.Citation30
More regulators of mitophagy in reticulocytes are being discovered. For example, inhibition of ALOX15 (arachidonate 15-lipoxygenase) delays mitophagy in reticulocytes.Citation72 In addition, an evolutionarily conserved KRAB-TRIM28/KAP1-miRNA regulatory cascade controls mitophagy in reticulocytes.Citation98 Once this cascade is disrupted, the expression of mitophagy-associated genes is repressed so that mitochondria are retained in erythroblasts and the host animal displays severe hypoproliferative anemia.
Mitophagy in adipose tissue differentiation
Several studies have underscored that autophagy is closely associated with adipose tissue development and differentiation.Citation99-Citation101 Based on their different colors, morphology, and functions, adipose tissues can be classified into white adipose tissue (WAT) and brown adipose tissue (as well as a more recently characterized beige adipose tissue). Recently it has been reported that alterations in structures and numbers of mitochondria occur in the differentiation of WAT. A dramatic increase in mitochondrial biogenesis is observed in the early stage of adipogenesis, whereas the majority of mitochondria are scattered around a few lipid droplets and disappear in the late stage. Importantly, atg5−/− and atg7−/− mouse embryonic fibroblasts or pre-adipocytic 3T3-L1 cells with either Atg5 or Atg7 knockdown fail to eliminate mitochondria and complete this differentiation process,Citation99-Citation101 implying that mitophagy is indispensable for adipocyte differentiation. Consistently, the white adipocytes in adipose-specific Atg7 conditional knockout mice show abnormal morphological characteristics such as an increased number of mitochondria with small lipid droplets. However, about half of the white adipocytes in atg7−/− mice display normal morphology, indicating that other Atg7-independent autophagic pathways are also involved in mitophagy during WAT differentiation.Citation101 Of note, Atg7 conditional knockout mice display reduced adiposity with highly diet-induced obesity resistance and insulin sensitivity, decreased concentration of blood triglycerides and cholesterol, and higher basal metabolic rate.Citation101 Therefore, novel therapeutics targeting mitophagy may be appropriate for patients with metabolic diseases such as obesity and type II diabetes.
Mitophagy in T lymphocyte differentiation
Mitophagy is also necessary for the clearance of superfluous mitochondria in T lymphocyte differentiation and survival. Both Atg5 and Atg7 are crucial for this process in mice.Citation102,Citation103 Remarkably, mitochondrial content is developmentally regulated only in T rather than B lymphocytes and the decrease in mitochondrial content marks T lymphocyte maturation from precursor thymocytes. Accordingly, autophagy-deficient T lymphocytes are unable to reduce their mitochondrial content and are eventually susceptible to apoptosis largely due to increased production of ROS.Citation103,Citation104
Mitophagy in Human Cancer
Mitochondria are the critical manipulators for regulating cell energy metabolism and cell life/death decisions; thus, alterations in mitochondrial homeostasis have been implicated in the pathogenesis of many human diseases including cancers, myopathies, neurodegenerative diseasesCitation12 such as PD,Citation105,Citation106 Alzheimer disease,Citation107 and Huntington disease.Citation106,Citation108 The relevance of mitophagy to the development of human diseases such as neurodegeneration has been elegantly reviewed. Herein we will focus on the implications of mitophagy in cancer.
Overview of mitophagy in cancer development
Autophagy has always been perceived to play a double-faceted role in tumorigenesis; either supporting survival or promoting death, depending on the different cellular contexts. Generally, basal autophagy remains at a low level to maintain cellular homeostasis and sustain prolonged survival by degradation of polyubiquitinated or aggregated proteins and damaged organelles. However, unrestrained autophagy will eventually trigger cell death when the cellular metabolic balance is disturbed.Citation109 Similarly, mitophagy follows this Janus-faced nature of autophagy. As the main powerhouse in eukaryotic cells, mitochondria are crucial for cell survival. The mtDNA exhibits a high susceptibility to damage attributed to heavy exposure to ROS, restricted repair systems, and lack of histone protection.Citation110 Thus, mitophagy serves to remove dysfunctional mitochondria to alleviate oxidative stress and prevent carcinogenesis. Conversely, mitophagy can protect cells from apoptosis or necrosis and promote tumor cell survival under some adverse conditions such as poor nutrient supply and hypoxic stresses.Citation111,Citation112 Therefore, mitophagy emerges as a key quality control factor and decision maker in cancer cells. In the following sections, we will summarize the current understanding of the biological functions of mitophagy during cancer development, provide an overview of how mitophagy is regulated by cancer-associated signaling pathways and proteins, and discuss the possibilities of targeting mitophagy for cancer intervention.
Regulation and function of mitophagy in the tumor microenvironment
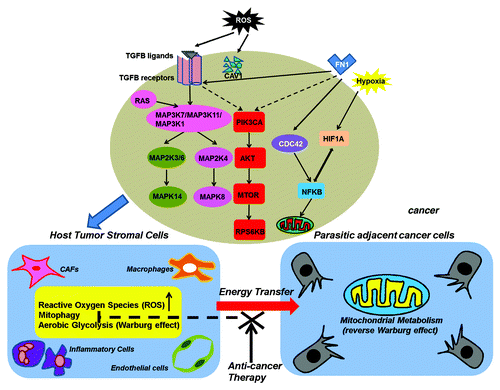
By producing approximately 36 ATPs per molecule of glucose, OXPHOS is primarily used by normal cells for supplying energy. Under anaerobic conditions, however, glycolysis is used as the main source of energy production, but generates only 2 ATPs per molecule of glucose.Citation113 However, cancer cells predominantly utilize glycolysis but not OXPHOS to generate energy even in the presence of oxygen, which is known as the “Warburg effect.”Citation114,Citation115 In the past several decades, the regulation and relevance of the Warburg effect with regard to cancer has been extensively studied. Otto Warburg in his seminal report first proposed the role of aerobic glycolysis in cancer and attributed this phenomenon to dysfunctional mitochondria.Citation114,Citation115 However, later studies found little evidence to support mitochondrial dysfunction in cancer cells. This Warburg paradox has hindered advances in this field for many years. Recently, a new model of tumor metabolism termed “two-compartment tumor metabolism” or “parasitic cancer metabolism” proposed that autophagy/mitophagy and aerobic glycolysis indeed physically converge in the tumor microenvironment.Citation116,Citation117 According to this model, ROS produced by cancer cells are transferred to adjacent cancer-associated fibroblasts or other stromal cells and initiate the onset of a stromal oxidative stress responses including mitophagy. The consequent mitochondrial dysfunction in the stromal cells leads to the production of sufficient high-energy metabolites such as L-lactate, ketones, glutamine, and free fatty acids to support cancer cell survival. This kind of “host-parasite” relationship formed between tumor stromal cells and epithelial cancer cells was coined as the “reverse Warburg effect.”Citation118,Citation119
It has been previously shown that loss of stromal CAV1 is a powerful predictive biomarker of tumor progression and metastasis in human cancers.Citation120,Citation121 ROS released from cancer cells can induce the loss of stromal CAV1, which in turn induces metabolic reprogramming of tumor stromal cells with increased mitophagy and mitochondrial dysfunction.Citation122,Citation123 Notably, a recent miRNA profiling of cav1−/− stromal cells revealed the upregulation of two key cancer-related miRNAs, MIR31 and MIR34C, both of which are sufficient to drive mitophagy due to their close association with the acknowledged mitophagy inducers oxidative stress and activation of the hypoxia response/HIF1A, respectively.Citation124 This finding suggests that miRNA regulation provides new mechanistic insights into the role of mitophagy during tumorigenesis and is attracting increasing attention. Furthermore, since CAV1 can negatively regulate TGFB (transforming growth factor, β 1) signaling, activation of TGFB signaling seems to be important for the induction of mitophagy in tumor stromal cells.Citation125 Indeed, ligand-dependent paracrine or cell–autocrine activation of TGFB signaling in stromal cells rather than in cancer cells induces metabolic reprogramming of the tumor microenvironment.Citation126
Additionally, hydrogen peroxide produced by BRCA1-null ovarian cancer cells can also trigger mitophagy by activating NFKB (nuclear factor of kappa light polypeptide gene enhancer in B-cells) signaling in stromal cells.Citation127 As a well-recognized tumor suppressor gene, BRCA1 normally suppresses tumor growth by maintaining genome integrity. However, BRCA1-null cancer cells exert an influence on the metabolic reprogramming of their adjacent tumor-associated stromal fibroblasts by producing a large amount of hydrogen peroxide that activates NFKB signaling to induce mitophagy and glycolysis in tumor stromal cells.Citation127 In addition to ROS, some cytokines or other bioactive factors enriched in the tumor-permissive microenvironment such as migration stimulating factor, a genetically truncated N-terminal isoform of fibronectin, can also induce mitophagy in tumor stromal cells by activating TGFB and CDC42-NFKB signaling.Citation128
Collectively, mitophagy can be activated by oncogenic signaling pathways, mainly including the TGFB and NFKB pathways, to boost tumor cell growth through reprogramming cancer cell metabolism. Therefore, inhibition of mitophagy in the tumor stroma by neutralizing ROS or inactivating TGFB and NFKB signaling in the tumor microenvironment via metabolically uncoupling tumor cells from their surrounding and supportive stroma may be feasible strategies to inhibit tumor growth, which provides useful hints into targeting mitophagy as a novel and potential anticancer strategy ().
Figure 2. Two-compartment tumor metabolism: Mitophagy in tumor stromal cells supports the metabolism of cancer cells. Cancer cells secrete ROS or cytokines to stimulate mitophagy in tumor stromal cells by the activation of TGFB and NFKB signaling. The consequent mitochondrial dysfunction in the stromal cells leads to the production of sufficient high-energy metabolites such as L-lactate, ketones, glutamine, and free fatty acids to support cancer cell survival. CAFs, cancer-associated fibroblasts.
Regulation and function of mitophagy in cancer cells
RAS-MAPK signaling
Mounting reports have confirmed the dysregulation of mitophagy in cancer cells, in which many oncogenic signalings and proteins are implicated in the regulation of mitophagy. The first oncogene isolated from human tumors, RAS, is frequently and aberrantly activated in many human cancers.Citation129-Citation131 In addition to its well-known transforming potential, oncogenic RAS has been recently found to activate mitophagy as a critical survival strategy through expediting glycolysis to conquer a cellular energy deficit resulting from glucose deficiency.Citation132 Blockage of mitophagy effectively suppresses RAS-induced transformation, further demonstrating the protumorigenic role of mitophagy. Activated RAS can potently induce the expression of glucose transporters such as SLC2A1/GLUT1 and promote the switch of glucose metabolism from OXPHOS into glycolysis by epigenetically reprogramming gene expression.Citation133 Once glycolysis is activated by oncogenic signaling to meet the cellular energy requirement, maintenance of a high amount of mitochondrial mass is no longer essential for ATP production. Therefore, degradation of relatively superfluous mitochondria can be implemented by mitophagy so as to expedite glycolysis and resupply nutrients. Indeed, autophagy facilitates glycolysis during RAS-mediated oncogenic transformation.Citation134 In addition, transformed cells may maintain small numbers of mitochondria during rapid proliferation, minimizing the energy requirement to maintain subcellular organelles.Citation132
Remarkably, activated RAS promotes mitophagy through MAPK/JNK signaling.Citation132 Inhibition of JNK rather than other MAPKs effectively prevents KRAS-induced mitophagy, restores mitochondrial functions, and overcomes energy deficit in RAS-transformed and other cancer cells. Since RAS-JNK signaling can activate mitophagy in the absence of any well-recognized inducers of mitophagy, further studies are required to elucidate the detailed mechanisms of RAS-induced mitophagy. Interestingly, the SQSTM1 protein level is dramatically induced by activated RAS and this is not a result of impaired autophagic turnover because lysosomal inhibitors further enhance the accumulation of SQSTM1.Citation135 It remains largely unexplored whether activated RAS induces SQSTM1 through the activation of JNK signaling. Alternatively, SQSTM1 could be upregulated in response to the initiation of mitophagy induced by RAS-JNK signaling.
AKT2 signaling
The aberrant activation of phosphoinositide 3-kinase-AKT signaling has been well documented as contributing to tumor promotion and progression.Citation136 As a serine/threonine protein kinase, AKT functions to promote cellular proliferation and survival of cancer cells by regulation of downstream signaling molecules such as MTOR and CCND1/cyclin D1. Interestingly, three AKT isoforms, AKT1, 2 and 3, have distinct localizations in human cells, raising the possibility that each isoform may have unique functions and employ different regulatory mechanisms. Among them, AKT2 is the only one localized in mitochondria in a variety of cancer cell lines,Citation137 indicating a potential link between AKT2 and mitochondrial functions during cancer development. Indeed, specific ablation of AKT2 rather than AKT1 or AKT3 has profound effects on mitochondria in MDA-MB231 breast cancer cells.Citation138 Initially, AKT2 ablation increases the volume of mitochondria by activation of PPARGC1A to upregulate mitochondrial biogenesis. However, AKT2 ablation also markedly inhibits the MTOR–RPS6KB pathway to limit protein synthesis. Therefore, the high activity of mitochondrial biogenesis cannot be sustainable for an extended duration, and the persistent inhibition of AKT2 signaling eventually activates mitophagy owing to the collapse of the entire mitochondrial homeostasis system. Alternatively, mitophagy is activated to salvage the mitochondria for enabling cellular survival.Citation138 In addition to mitophagy activation, significant growth inhibition is also induced by AKT2 ablation, indicating that AKT2 might be a promising target for cancer therapy. Actually, selective inhibitors for distinct AKT isoforms have been developed and will soon be studied in clinical trials.
BECN1 complex
The first direct and functional link between autophagy and tumor suppression derived from genetic studies of the mammalian homolog of yeast Vps30/Atg6, BECN1/Beclin 1, which was identified as a haploinsufficient tumor suppressor that is monoallelically deleted in a majority of human cancers including breast, ovarian, and prostate cancers.Citation139 Similar to Vps30 which is an essential autophagy regulator in yeast, BECN1 has the ability to promote autophagy in mammalian cells. Intriguingly, the autophagy-promoting role of BECN1 leads to the inhibition of cell proliferation, clonigenicity, and tumorigenesis both in vitro and in vivo. Furthermore, expression of BECN1 is markedly reduced in human breast carcinoma cell lines.Citation139 Consistently, it has been supported that the heterozygous disruption of Becn1 and/or lacking one copy of Becn1 makes mice susceptible to tumorigenesis and/or they suffer from spontaneous tumors such as B cell lymphoma, hepatocellular carcinoma, and lung adenocarcinoma.Citation140,Citation141 These observations highlight the role of BECN1 as a tumor suppressor. Although the mechanism of tumor suppression by autophagy has not yet been fully determined, it seems to be contradictory to the fact that autophagy sustains cell survival in response to stress conditions. The most plausible explanation is that autophagy-defective cells due to monoallelic loss of BECN1, for instance, may produce a great deal of ROS that accelerates DNA damage or chromosomal instability, leading to the accumulation of oncogenic genetic changes.Citation142-Citation144 Thus, autophagy-mediated tumor suppression is associated with the elimination of damaged mitochondria and peroxisomes, namely through mitophagy and pexophagy.
Notably, BECN1 regulates autophagy by forming multimolecular complexes containing various autophagy-related proteins such as PIK3C3, PIK3R4, UVRAG (UV radiation resistance associated), SH3GLB1 (SH3-domain GRB2-like endophilin B1), ATG14,Citation145 and AMBRA1 (autophagy/Beclin-1 regulator 1).Citation146 BECN1 exerts its function of autophagy induction by interacting with PIK3R4 and PIK3C3, the latter of which corresponds to the lipid kinase component of the class III phosphatidylinositol (PtdIns) 3-kinase (PtdIns3K) complex that is regulated by PIK3R4.Citation147 Similar to BECN1, UVRAG is another candidate tumor suppressor, which is monoallelically deleted in human cancers, and its tumor suppressor activity is dependent on its interaction with BECN1.Citation148 Structurally, UVRAG acts as a bridge to mediate BECN1-SH3GLB1 interaction. SH3GLB1 is a member of the membrane curvature-driving endophilin family, which regulates the post-Golgi trafficking of membrane-integrated ATG9A for autophagy.Citation149,Citation150 In humans, the SH3GLB1 gene is located on chromosome 1p22, a region frequently deleted in neoplasmsCitation151 and its expression is downregulated in a wide variety of cancers.Citation152-Citation156 Furthermore, inhibition of endogenous expression of SH3GLB1 by RNA interference enhances the tumorigenecity of HeLa cells and SH3GLB1-knockout mice are susceptible to the development of spontaneous tumors, indicating that SH3GLB1 is indeed a novel bona fide tumor suppressor.Citation150,Citation157 Notably, the tumor suppressor function of SH3GLB1 seems to be associated with the induction of autophagosome formation.Citation150 SH3GLB1 was recently verified to mediate the clearance of damaged mitochondria by mitophagy to alleviate DNA damage and chromosomal instability in Myc-induced lymphoma cells, highlighting the tumor suppressive role of mitophagy.Citation158 Accordingly, hemizygous deletion of SH3GLB1 results in an increase in mitochondrial mass, accumulation of DNA damage, reduced apoptosis activation, and consequent tumor development in vivo.Citation159
AMBRA1 is a WD40 domain-containing protein crucial for positive regulation of autophagy and nervous system development in mammals.Citation160 As a component of the BECN1 complex, AMBRA1 promotes autophagosome formation by strengthening the interaction of BECN1 with PIK3C3. Both BECN1 and PIK3C3 are two core components of the class III PtdIns3K complex crucial for phagophore nucleation. Moreover, both AMBRA1 and BCL2 bind to BECN1 at the same site. Consequently, it is inevitable that there is a competitive relationship between the binding of BECN1 with AMBRA1 and BCL2. Consistent with this assumption, AMBRA1 can indeed compete with BCL2 both in mitochondria (mito-BCL2) and the endoplasmic reticulum to bind BECN1. Under normal conditions, AMBRA1 can be preferentially docked by the pool of mito-BCL2 to inhibit its autophagic function. Upon autophagy induction, AMBRA1 is released form mito-BCL2 to enhance its binding to BECN1 and reinforce the autophagy-promoting role of BECN1.Citation161 Although the exact role of AMBRA1 in tumorigenesis is still largely unclear, it is conjectured on the one hand that AMBRA1 dysregulation may promote carcinogenesis by virtue of its functional interaction with the BECN1 complex. Indeed, functional deficiency of AMBRA1 in mouse embryos results in excessive cell proliferation and apoptotic cell death.Citation160 On the other hand, it is hypothesized that AMBRA1 might be indirectly implicated in tumorigenesis by balancing cell death/survival owing to its interaction with the anti-apoptotic factor BCL2 to modulate the activity of proapoptotic and proautophagic regulators.Citation161,Citation162 Therefore, AMBRA1 acts as a switch between autophagy and apoptosis during cancer development.
Strikingly, it has been recently reported that AMBRA1 might be a novel PARK2-binding protein implicated in mitophagy.Citation146 Mechanistically, prolonged mitochondrial depolarization markedly enhances the interaction of endogenous PARK2 and AMBRA1, which is recruited to the perinuclear clusters of depolarized mitochondria by PARK2 and activates the class III PtdIns3K complex in the vicinity of those damaged mitochondria and eventually leads to mitophagy. Actually, PARK2 stimulates the perimitochondrial nucleation of new phagophores to boost mitophagy through recruitment of AMBRA1. Consequently, AMBRA1 is indispensable for the final step of PARK2-triggered mitophagy rather than the translocation of PARK2 to depolarized mitochondria.Citation163 These findings unveil a novel mechanism by which AMBRA1 enhances PARK2-mediated mitophagy.
BNIP3
BNIP1, 2, and 3 were initially identified in a yeast two-hybrid screen as BCL2 and adenovirus E1B 19-kDa interacting proteins. They are proapoptotic proteins that can be suppressed by the E1B 19-kDa protein or BCL2. Spatially, BNIP1 and BNIP2 localize to the nuclear envelope and endoplasmic reticulum, whereas BNIP3 resides in the mitochondria. BNIP3 has a putative BH3 domain in addition to its C-terminal transmembrane domain, and BH3-containing proteins can induce both apoptosis and autophagy that converge at mitochondria, indicating that BNIP3 may regulate mitophagy.Citation164 Indeed, BNIP3 interacts with LC3 via its LIR for autophagic degradation of mitochondria.Citation165 Generally, there are three models for the mechanism of BNIP3- or BNIP3L-dependent mitophagy. First, BNIP3 or BNIP3L triggers mitochondrial depolarization and initiates mitophagy. However, whether mitochondrial depolarization is the cause or consequence of mitophagy remains unclear. Second, BNIP3 or BNIP3L functions as a receptor protein to recruit autophagic machinery to mitochondria. Both BNIP3 and BNIP3L can directly interact with LC3 via their LIR. Third, BNIP3 or BNIP3L can compete with BECN1 for the binding to BCL2 or BCL2L1. The increased expression of BNIP3 or BNIP3L will release BECN1 from BCL2 or BCL2L1 to activate mitophagy.Citation166
In response to hypoxia or oxidative stress, cells will undergo rapid mitophagy to enable cell survival, or undergo apoptotic or necrotic cell death. BNIP3 is important to this fate determination. When damage is too severe or persistent, BNIP3 changes its role from enabling survival to promoting cell death.Citation167 However, it remains unconfirmed how BNIP3 can be switched from a mitophagy inducer to an apoptotic mediator. The impact of BNIP3 on mitophagy is regulated by extracellular or intracellular signals. For example, phosphorylation of serine residues 17 and 24 flanking the BNIP3 LIR promote BNIP3-induced mitochondrial sequestration, lysosomal delivery, and, consequently, mitophagy.Citation165 Overexpression of the anti-apoptotic protein BCL2L1 significantly enhances LIR-dependent BNIP3 binding to LC3B and BNIP3-induced mitochondrial sequestration, supporting a prosurvival role of BNIP3-induced mitophagy.Citation165 Additionally, the transcription of BNIP3 and BNIP3L can also be regulated by hypoxia. Actually, BNIP3 was originally identified as an upregulated target in cells exposed to hypoxia. There are two HIF1A binding sites in the BNIP3 promoter. Its expression is suppressed by the VHL (von Hippel-Lindau tumor suppressor, E3 ubiquitin protein ligase) protein in human cancer cells, and deregulated BNIP3 expression in cancer is associated with several aggressive diseases.Citation164
Targeting mitophagy for cancer therapy
Although the transient activation of autophagy including mitophagy may play dual functions to promote tumor regression or enable tumor cell survival, prolonged or robust autophagy activation will eventually lead to the degradation of components essential for cell survival and therefore cause cell death. Accordingly, some agents have been developed to induce mitophagy in cancer cells for anticancer treatment.Citation168 For instance, the lis/lin/GO (glucose oxidase) killer-suicide system has been recognized as a robust mitophagy trigger to induce cell death and growth inhibition of human cancer cells both in vitro and in vivo.Citation169 This oxidase system is composed of linamarase, linamarin, and glucose oxidase. The β-glucosidase enzyme linamarase can hydrolyze its cyanogenic glucoside substrate linamarin into glucose, acetone, and cyanide. Cyanide inhibits cytochrome c oxidase of the mitochondrial respiratory chain, thereby triggering a rapid mitochondrial fission to facilitate mitophagy and eventually autophagic cell death.Citation170-Citation172 The original lis/lin system was sufficient to inhibit the in vitro growth of cancer cells but failed to suppress tumor growth in vivo. Therefore, glucose oxidase was added to increase the therapeutic potential. Glucose oxidase converts glucose into gluconic acid and hydrogen peroxide, thus inducing oxidase stress to activate mitophagy.Citation169 Similar to cyanide, hydrogen peroxide is highly diffusible so as to penetrate freely across membranes and generate a critical bystander effect on tumor regression.Citation173 Either the antioxidant N-acetyl cysteine or inhibitors of the core autophagic machinery such as 3-methyladenine rescue cell death induced by the lis/lin/GO system. Similarly, genetic knockdown of core genes involved in mitophagy such as Atg5, Becn1, and Bnip3 also block cell death.
Another striking example demonstrating the efficacy of mitophagy induction for cancer therapy is ceramide, which acts as a bioactive sphingolipid to induce cell death, growth inhibition, and senescence in various human cancer cellsCitation174,Citation175 Intriguingly, recent studies have proposed that ceramides can promote cell death and tumor regression through inducing mitophagy.Citation176 CERS (ceramide synthase) enzymes CERS1 to CERS6 generate ceramides with distinct biological roles that largely depend on their different localizations. For instance, CERS1 and CERS6 preferentially generate C18- and C16-ceramide, respectively.Citation177 C18- ceramide promotes cell death as observed in preclinical and clinical studies, whereas C16-ceramide is mainly associated with cancer cell proliferation.Citation176,Citation178 Furthermore, exogenous C18-pyridinium ceramide can also trigger mitophagy to promote cell death, and stable knockdown of LC3B expression with shRNA abrogates CERS1- and C18-ceramide-dependent mitophagy, and blocks tumor suppression in vivo.Citation82 After the lipidation of LC3B with phosphotidylethanolamine to form LC3B-II induced by C18-ceramide or C18-pyridinium ceramide, ceramide, and LC3B-II directly interact on mitochondrial membranes via DNM1L/Drp1-dependent mitochondrial fission, thus enabling mithchondria sequestration.Citation82
More recently, it was reported that low-intensity ultrasound therapy in the presence of curcumin enhances the cell death of nasopharyngeal carcinoma CNE2 cells through initiation of mitophagy.Citation179 Therefore, the induction of cell death by combining treatment-triggered mitophagy is another potential and feasible strategy for treating malignancies.
Taken together, an increasing number of approaches are currently being developed to induce tumor regression via mitophagy. As cancer cells have frequently dysregulated oncogenic signaling that stimulates the generation of oxidative stress and vacuole formation, they may have a lower threshold for mitophagy execution and are susceptible to autophagic cell death upon treatment with mitophagy modifiers.
Conclusions and Perspectives
Our understanding of the molecular mechanisms implicated in the regulation of mitophagy has greatly advanced in the past several years. However, there are still many questions to be answered. In different organisms, distinct signals activate their own regulators to induce mitophagy. Different from the inherent conservation of the core autophagic machinery, mitophagy-specific genes differ considerably between yeast and mammals, although the key players in mitochondrial fusion/fission dynamic events are evolutionarily conserved. The relevance of dysregulated mitophagy to neurodegeneration is much clearer, whereas the implication of mitophagy in human carcinogenesis is still very much a mystery, mainly because research in this field is still in its infancy. Meanwhile, more precise and reliable experimental methods to detect and monitor the whole process of mitophagy are urgently needed, which may not only provide further insights into the complex networks involved in mitophagy in detail, but may also help researchers and clinicians explore new therapeutic approaches to disorders that involve dysfunctional mitochondria, such as cancers. The double-faceted role of mitophagy during oncogenesis, either survival-supporting or death-promoting, leads to a novel challenge when targeting it for cancer therapies. Therefore, a better understanding of mitophagy regulation in cancer, and revealing the intrinsic molecular mechanisms of this process, will be crucial for the development of novel anticancer therapeutics.
| Abbreviations: | ||
| AMBRA1 | = | autophagy/Beclin 1 regulator 1 |
| Atg | = | autophagy-related |
| BCL2 | = | B-cell CLL/lymphoma 2 |
| BNIP3L | = | BCL2/adenovirus E1B 19 kDa interacting protein 3-like |
| CCCP | = | carbonyl cyanide m-chlorophenyl hydrazone |
| CERS | = | ceramide synthases |
| Cvt | = | cytoplasm-to-vacuole targeting |
| GABARAP | = | GABA(A) receptor-associated protein |
| IMS | = | intermembrane mitochondrial space |
| LIR | = | LC3-interacting region |
| MAPK | = | mitogen-activated protein kinase |
| mtDNA | = | mitochondrial DNA |
| NFKB | = | nuclear factor of kappa light polypeptide gene enhancer in B-cells |
| OMM | = | outer mitochondrial membrane |
| OXPHOS | = | oxidative phosphorylation |
| PAS | = | phagophore assembly site |
| PINK1 | = | PTEN-induced putative kinase 1 |
| PtdIns3K | = | class III phosphatidylinositol 3-kinase |
| ROS | = | reactive oxygen species |
| SH3GLB1 | = | SH3-domain GRB2-like endophilin B1 |
| TGFB | = | transforming growth factor, beta 1 |
| Ub | = | ubiquitin |
| Ulk1 | = | Unc51 like autophagy activating kinase 1 |
| UVRAG | = | UV radiation-resistance associated |
| WAT | = | white adipose tissue |
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No.81301706), Program for Innovative Research Team in Zhejiang Province (No.2012R10046-15), Ministry of Education of China (No.20120101120044), and Zhejiang Provincial Natural Science Foundation of China (No.Y13H160010 and LR12H16001).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931 - 7; http://dx.doi.org/10.1038/nrm2245; PMID: 17712358
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9:1102 - 9; http://dx.doi.org/10.1038/ncb1007-1102; PMID: 17909521
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12:9 - 14; http://dx.doi.org/10.1038/nrm3028; PMID: 21179058
- Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 2012; 393:547 - 64; http://dx.doi.org/10.1515/hsz-2012-0119; PMID: 22944659
- Monastyrska I, Reggiori F, Klionsky DJ. Harpooning the Cvt complex to the phagophore assembly site. Autophagy 2008; 4:914 - 6; PMID: 18708760
- Oku M, Sakai Y. Pexophagy in Pichia pastoris. Methods Enzymol 2008; 451:217 - 28; http://dx.doi.org/10.1016/S0076-6879(08)03215-1; PMID: 19185723
- MacIntosh GC, Bassham DC. The connection between ribophagy, autophagy and ribosomal RNA decay. Autophagy 2011; 7:662 - 3; http://dx.doi.org/10.4161/auto.7.6.15447; PMID: 21460615
- Kudchodkar SB, Levine B. Viruses and autophagy. Rev Med Virol 2009; 19:359 - 78; http://dx.doi.org/10.1002/rmv.630; PMID: 19750559
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005; 8:3 - 5; http://dx.doi.org/10.1089/rej.2005.8.3; PMID: 15798367
- Clark SL Jr.. Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J Biophys Biochem Cytol 1957; 3:349 - 62; http://dx.doi.org/10.1083/jcb.3.3.349; PMID: 13438920
- Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 2007; 462:245 - 53; http://dx.doi.org/10.1016/j.abb.2007.03.034; PMID: 17475204
- Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther 2012; 342:619 - 30; http://dx.doi.org/10.1124/jpet.112.192138; PMID: 22700435
- Cotán D, Cordero MD, Garrido-Maraver J, Oropesa-Ávila M, Rodríguez-Hernández A, Gómez Izquierdo L, De la Mata M, De Miguel M, Lorite JB, Infante ER, et al. Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J 2011; 25:2669 - 87; http://dx.doi.org/10.1096/fj.10-165340; PMID: 21551238
- Yen WL, Klionsky DJ. How to live long and prosper: autophagy, mitochondria, and aging. Physiology (Bethesda) 2008; 23:248 - 62; http://dx.doi.org/10.1152/physiol.00013.2008; PMID: 18927201
- Zungu M, Schisler J, Willis MS. All the little pieces. -Regulation of mitochondrial fusion and fission by ubiquitin and small ubiquitin-like modifer and their potential relevance in the heart.-. Circ J 2011; 75:2513 - 21; http://dx.doi.org/10.1253/circj.CJ-11-0967; PMID: 22001293
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469:221 - 5; http://dx.doi.org/10.1038/nature09663; PMID: 21124315
- Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 2009; 17:98 - 109; http://dx.doi.org/10.1016/j.devcel.2009.06.014; PMID: 19619495
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 2009; 10:458 - 67; http://dx.doi.org/10.1038/nrm2708; PMID: 19491929
- Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 2009; 17:87 - 97; http://dx.doi.org/10.1016/j.devcel.2009.06.013; PMID: 19619494
- Aoki Y, Kanki T, Hirota Y, Kurihara Y, Saigusa T, Uchiumi T, Kang D. Phosphorylation of Serine 114 on Atg32 mediates mitophagy. Mol Biol Cell 2011; 22:3206 - 17; http://dx.doi.org/10.1091/mbc.E11-02-0145; PMID: 21757540
- Mao K, Wang K, Zhao M, Xu T, Klionsky DJ. Two MAPK-signaling pathways are required for mitophagy in Saccharomyces cerevisiae. J Cell Biol 2011; 193:755 - 67; http://dx.doi.org/10.1083/jcb.201102092; PMID: 21576396
- Mao K, Klionsky DJ. MAPKs regulate mitophagy in Saccharomyces cerevisiae. Autophagy 2011; 7:1564 - 5; http://dx.doi.org/10.4161/auto.7.12.17971; PMID: 22024747
- Kanki T, Wang K, Baba M, Bartholomew CR, Lynch-Day MA, Du Z, Geng J, Mao K, Yang Z, Yen WL, et al. A genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol Biol Cell 2009; 20:4730 - 8; http://dx.doi.org/10.1091/mbc.E09-03-0225; PMID: 19793921
- Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K, Zhang J, Iyengar R, Jung CH, Suen DF, et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell 2011; 43:572 - 85; http://dx.doi.org/10.1016/j.molcel.2011.06.018; PMID: 21855797
- Watanabe Y, Kobayashi T, Yamamoto H, Hoshida H, Akada R, Inagaki F, Ohsumi Y, Noda NN. Structure-based analyses reveal distinct binding sites for Atg2 and phosphoinositides in Atg18. J Biol Chem 2012; 287:31681 - 90; http://dx.doi.org/10.1074/jbc.M112.397570; PMID: 22851171
- Kaiser SE, Mao K, Taherbhoy AM, Yu S, Olszewski JL, Duda DM, Kurinov I, Deng A, Fenn TD, Klionsky DJ, et al. Noncanonical E2 recruitment by the autophagy E1 revealed by Atg7-Atg3 and Atg7-Atg10 structures. Nat Struct Mol Biol 2012; 19:1242 - 9; http://dx.doi.org/10.1038/nsmb.2415; PMID: 23142976
- Yu ZQ, Ni T, Hong B, Wang HY, Jiang FJ, Zou S, Chen Y, Zheng XL, Klionsky DJ, Liang Y, et al. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy 2012; 8:883 - 92; http://dx.doi.org/10.4161/auto.19652; PMID: 22652539
- Walczak M, Martens S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy 2013; 9:424 - 5; http://dx.doi.org/10.4161/auto.22931; PMID: 23321721
- Itakura E, Mizushima N. Atg14 and UVRAG: mutually exclusive subunits of mammalian Beclin 1-PI3K complexes. Autophagy 2009; 5:534 - 6; http://dx.doi.org/10.4161/auto.5.4.8062; PMID: 19223761
- Zhang J, Randall MS, Loyd MR, Dorsey FC, Kundu M, Cleveland JL, Ney PA. Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood 2009; 114:157 - 64; PMID: 19417210
- Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, Simon AK. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci U S A 2010; 107:832 - 7; http://dx.doi.org/10.1073/pnas.0913170107; PMID: 20080761
- Yen WL, Legakis JE, Nair U, Klionsky DJ. Atg27 is required for autophagy-dependent cycling of Atg9. Mol Biol Cell 2007; 18:581 - 93; http://dx.doi.org/10.1091/mbc.E06-07-0612; PMID: 17135291
- Tang F, Watkins JW, Bermudez M, Gray R, Gaban A, Portie K, Grace S, Kleve M, Craciun G. A life-span extending form of autophagy employs the vacuole-vacuole fusion machinery. Autophagy 2008; 4:874 - 86; PMID: 18690010
- Ragusa MJ, Stanley RE, Hurley JH. Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell 2012; 151:1501 - 12; http://dx.doi.org/10.1016/j.cell.2012.11.028; PMID: 23219485
- Mendl N, Occhipinti A, Müller M, Wild P, Dikic I, Reichert AS. Mitophagy in yeast is independent of mitochondrial fission and requires the stress response gene WHI2. J Cell Sci 2011; 124:1339 - 50; http://dx.doi.org/10.1242/jcs.076406; PMID: 21429936
- Legakis JE, Yen WL, Klionsky DJ. A cycling protein complex required for selective autophagy. Autophagy 2007; 3:422 - 32; PMID: 17426440
- Nice DC, Sato TK, Stromhaug PE, Emr SD, Klionsky DJ. Cooperative binding of the cytoplasm to vacuole targeting pathway proteins, Cvt13 and Cvt20, to phosphatidylinositol 3-phosphate at the pre-autophagosomal structure is required for selective autophagy. J Biol Chem 2002; 277:30198 - 207; http://dx.doi.org/10.1074/jbc.M204736200; PMID: 12048214
- Austriaco NR Jr.. Review: to bud until death: the genetics of ageing in the yeast, Saccharomyces. Yeast 1996; 12:623 - 30; http://dx.doi.org/10.1002/(SICI)1097-0061(19960615)12:7<623::AID-YEA968>3.0.CO;2-G; PMID: 8810036
- Kissová I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem 2004; 279:39068 - 74; http://dx.doi.org/10.1074/jbc.M406960200; PMID: 15247238
- Kissová I, Salin B, Schaeffer J, Bhatia S, Manon S, Camougrand N. Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy 2007; 3:329 - 36; PMID: 17377488
- Tal R, Winter G, Ecker N, Klionsky DJ, Abeliovich H. Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J Biol Chem 2007; 282:5617 - 24; http://dx.doi.org/10.1074/jbc.M605940200; PMID: 17166847
- Ruan H, Yan Z, Sun H, Jiang L. The YCR079w gene confers a rapamycin-resistant function and encodes the sixth type 2C protein phosphatase in Saccharomyces cerevisiae. FEMS Yeast Res 2007; 7:209 - 15; http://dx.doi.org/10.1111/j.1567-1364.2006.00160.x; PMID: 17002782
- Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev Mol Med 2009; 11:e22; http://dx.doi.org/10.1017/S1462399409001148; PMID: 19631006
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006; 441:1162 - 6; http://dx.doi.org/10.1038/nature04779; PMID: 16672981
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A 2008; 105:1638 - 43; http://dx.doi.org/10.1073/pnas.0709336105; PMID: 18230723
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795 - 803; http://dx.doi.org/10.1083/jcb.200809125; PMID: 19029340
- Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A 2010; 107:11835 - 40; http://dx.doi.org/10.1073/pnas.0914569107; PMID: 20547844
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A 2008; 105:7070 - 5; http://dx.doi.org/10.1073/pnas.0711845105; PMID: 18443288
- Poulogiannis G, McIntyre RE, Dimitriadi M, Apps JR, Wilson CH, Ichimura K, Luo F, Cantley LC, Wyllie AH, Adams DJ, et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc Natl Acad Sci U S A 2010; 107:15145 - 50; http://dx.doi.org/10.1073/pnas.1009941107; PMID: 20696900
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 2010; 107:378 - 83; http://dx.doi.org/10.1073/pnas.0911187107; PMID: 19966284
- Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci U S A 2008; 105:12022 - 7; http://dx.doi.org/10.1073/pnas.0802814105; PMID: 18687899
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010; 8:e1000298; http://dx.doi.org/10.1371/journal.pbio.1000298; PMID: 20126261
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 2012; 13:378 - 85; http://dx.doi.org/10.1038/embor.2012.14; PMID: 22354088
- Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem 2011; 117:856 - 67; http://dx.doi.org/10.1111/j.1471-4159.2011.07253.x; PMID: 21426348
- Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 2012; 22:320 - 33; http://dx.doi.org/10.1016/j.devcel.2011.12.014; PMID: 22280891
- Lutz AK, Exner N, Fett ME, Schlehe JS, Kloos K, Lämmermann K, Brunner B, Kurz-Drexler A, Vogel F, Reichert AS, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem 2009; 284:22938 - 51; http://dx.doi.org/10.1074/jbc.M109.035774; PMID: 19546216
- Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 2009; 284:13843 - 55; http://dx.doi.org/10.1074/jbc.M808515200; PMID: 19279012
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34:259 - 69; http://dx.doi.org/10.1016/j.molcel.2009.04.026; PMID: 19450525
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 2010; 19:4861 - 70; http://dx.doi.org/10.1093/hmg/ddq419; PMID: 20871098
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 2010; 15:887 - 900; PMID: 20604804
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010; 6:1090 - 106; http://dx.doi.org/10.4161/auto.6.8.13426; PMID: 20890124
- Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol 2010; 189:671 - 9; http://dx.doi.org/10.1083/jcb.201001039; PMID: 20457763
- Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 2010; 29:969 - 80; http://dx.doi.org/10.1038/emboj.2009.405; PMID: 20075865
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 2010; 191:1367 - 80; http://dx.doi.org/10.1083/jcb.201007013; PMID: 21173115
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119 - 31; http://dx.doi.org/10.1038/ncb2012; PMID: 20098416
- Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, Kitada T, Kim JM, Chung J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun 2008; 377:975 - 80; http://dx.doi.org/10.1016/j.bbrc.2008.10.104; PMID: 18957282
- Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep 2012; 2:1002; http://dx.doi.org/10.1038/srep01002; PMID: 23256036
- Feng D, Liu L, Zhu Y, Chen Q. Molecular signaling toward mitophagy and its physiological significance. Exp Cell Res 2013; 319:1697 - 705; http://dx.doi.org/10.1016/j.yexcr.2013.03.034; PMID: 23603281
- Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 2010; 285:27879 - 90; http://dx.doi.org/10.1074/jbc.M110.119537; PMID: 20573959
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 2012; 14:177 - 85; http://dx.doi.org/10.1038/ncb2422; PMID: 22267086
- Grüllich C, Duvoisin RM, Wiedmann M, van Leyen K. Inhibition of 15-lipoxygenase leads to delayed organelle degradation in the reticulocyte. FEBS Lett 2001; 489:51 - 4; http://dx.doi.org/10.1016/S0014-5793(01)02080-4; PMID: 11231012
- Fimia GM, Corazzari M, Antonioli M, Piacentini M. Ambra1 at the crossroad between autophagy and cell death. Oncogene 2013; 32:3311 - 8; http://dx.doi.org/10.1038/onc.2012.455; PMID: 23069654
- Levin DE. Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 2005; 69:262 - 91; http://dx.doi.org/10.1128/MMBR.69.2.262-291.2005; PMID: 15944456
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 2012; 287:19094 - 104; http://dx.doi.org/10.1074/jbc.M111.322933; PMID: 22505714
- Ishihara M, Urushido M, Hamada K, Matsumoto T, Shimamura Y, Ogata K, Inoue K, Taniguchi Y, Horino T, Fujieda M, et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol 2013; 305:F495 - 509; http://dx.doi.org/10.1152/ajprenal.00642.2012; PMID: 23698117
- Zhang J, Ney PA. NIX induces mitochondrial autophagy in reticulocytes. Autophagy 2008; 4:354 - 6; PMID: 18623629
- Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A 2007; 104:19500 - 5; http://dx.doi.org/10.1073/pnas.0708818104; PMID: 18048346
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008; 454:232 - 5; http://dx.doi.org/10.1038/nature07006; PMID: 18454133
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 2010; 11:45 - 51; http://dx.doi.org/10.1038/embor.2009.256; PMID: 20010802
- Kanki T. Nix, a receptor protein for mitophagy in mammals. Autophagy 2010; 6:433 - 5; http://dx.doi.org/10.4161/auto.6.3.11420; PMID: 20200478
- Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, Ramshesh VK, Peterson YK, Lemasters JJ, Szulc ZM, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol 2012; 8:831 - 8; http://dx.doi.org/10.1038/nchembio.1059; PMID: 22922758
- Tang D, Kang R, Livesey KM, Kroemer G, Billiar TR, Van Houten B, Zeh HJ 3rd, Lotze MT. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab 2011; 13:701 - 11; http://dx.doi.org/10.1016/j.cmet.2011.04.008; PMID: 21641551
- Westfall PJ, Ballon DR, Thorner J. When the stress of your environment makes you go HOG wild. Science 2004; 306:1511 - 2; http://dx.doi.org/10.1126/science.1104879; PMID: 15567851
- Itoh H, Komatsuda A, Ohtani H, Wakui H, Imai H, Sawada K, Otaka M, Ogura M, Suzuki A, Hamada F. Mammalian HSP60 is quickly sorted into the mitochondria under conditions of dehydration. Eur J Biochem 2002; 269:5931 - 8; http://dx.doi.org/10.1046/j.1432-1033.2002.03317.x; PMID: 12444982
- Park SJ, Shin JH, Kim ES, Jo YK, Kim JH, Hwang JJ, Kim JC, Cho DH. Mitochondrial fragmentation caused by phenanthroline promotes mitophagy. FEBS Lett 2012; 586:4303 - 10; http://dx.doi.org/10.1016/j.febslet.2012.10.035; PMID: 23123158
- Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet 2005; 14:3477 - 92; http://dx.doi.org/10.1093/hmg/ddi377; PMID: 16207731
- Ankel-Simons F, Cummins JM. Misconceptions about mitochondria and mammalian fertilization: implications for theories on human evolution. Proc Natl Acad Sci U S A 1996; 93:13859 - 63; http://dx.doi.org/10.1073/pnas.93.24.13859; PMID: 8943026
- Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 2011; 334:1141 - 4; http://dx.doi.org/10.1126/science.1210333; PMID: 21998252
- Al Rawi S, Louvet-Vallée S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011; 334:1144 - 7; http://dx.doi.org/10.1126/science.1211878; PMID: 22033522
- Zhou Q, Li H, Xue D. Elimination of paternal mitochondria through the lysosomal degradation pathway in C. elegans. Cell Res 2011; 21:1662 - 9; http://dx.doi.org/10.1038/cr.2011.182; PMID: 22105480
- Sutovsky P. Ubiquitin-dependent proteolysis in mammalian spermatogenesis, fertilization, and sperm quality control: killing three birds with one stone. Microsc Res Tech 2003; 61:88 - 102; http://dx.doi.org/10.1002/jemt.10319; PMID: 12672125
- Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitin tag for sperm mitochondria. Nature 1999; 402:371 - 2; http://dx.doi.org/10.1038/46466; PMID: 10586873
- Thompson WE, Ramalho-Santos J, Sutovsky P. Ubiquitination of prohibitin in mammalian sperm mitochondria: possible roles in the regulation of mitochondrial inheritance and sperm quality control. Biol Reprod 2003; 69:254 - 60; http://dx.doi.org/10.1095/biolreprod.102.010975; PMID: 12646488
- Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitinated sperm mitochondria, selective proteolysis, and the regulation of mitochondrial inheritance in mammalian embryos. Biol Reprod 2000; 63:582 - 90; http://dx.doi.org/10.1095/biolreprod63.2.582; PMID: 10906068
- Mortensen M, Ferguson DJP, Edelmann M, Kessler B, Morten KJ, Komatsu M, Simon AK. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci U S A 2010; 107:832 - 7; http://dx.doi.org/10.1073/pnas.0913170107; PMID: 20080761
- Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008; 112:1493 - 502; http://dx.doi.org/10.1182/blood-2008-02-137398; PMID: 18539900
- Barde I, Rauwel B, Marin-Florez RM, Corsinotti A, Laurenti E, Verp S, Offner S, Marquis J, Kapopoulou A, Vanicek J, et al. A KRAB/KAP1-miRNA cascade regulates erythropoiesis through stage-specific control of mitophagy. Science 2013; 340:350 - 3; http://dx.doi.org/10.1126/science.1232398; PMID: 23493425
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest 2009; 119:3329 - 39; PMID: 19855132
- Baerga R, Zhang Y, Chen PH, Goldman S, Jin S. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy 2009; 5:1118 - 30; http://dx.doi.org/10.4161/auto.5.8.9991; PMID: 19844159
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A 2009; 106:19860 - 5; PMID: 19910529
- Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med 2007; 204:25 - 31; http://dx.doi.org/10.1084/jem.20061303; PMID: 17190837
- Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol 2009; 182:4046 - 55; http://dx.doi.org/10.4049/jimmunol.0801143; PMID: 19299702
- Pua HH, He YW. Mitophagy in the little lymphocytes: an essential role for autophagy in mitochondrial clearance in T lymphocytes. Autophagy 2009; 5:745 - 6; http://dx.doi.org/10.4161/auto.5.5.8702; PMID: 19398889
- Xilouri M, Stefanis L. Autophagic pathways in Parkinson disease and related disorders. Expert Rev Mol Med 2011; 13:e8; http://dx.doi.org/10.1017/S1462399411001803; PMID: 21418705
- Chaturvedi RK, Beal MF. Mitochondria targeted therapeutic approaches in Parkinson’s and Huntington’s diseases. Mol Cell Neurosci 2013; 55:101 - 14; http://dx.doi.org/10.1016/j.mcn.2012.11.011; PMID: 23220289
- Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, Nunomura A, Szweda LI, Aliev G, Smith MA, et al. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy 2007; 3:614 - 5; PMID: 17786024
- Costa V, Scorrano L. Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. EMBO J 2012; 31:1853 - 64; http://dx.doi.org/10.1038/emboj.2012.65; PMID: 22446390
- Mathew R, White E. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev 2011; 21:113 - 9; http://dx.doi.org/10.1016/j.gde.2010.12.008; PMID: 21255998
- Gredilla R, Garm C, Stevnsner T. Nuclear and mitochondrial DNA repair in selected eukaryotic aging model systems. Oxid Med Cell Longev 2012; 2012:282438.
- Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal 2009; 11:777 - 90; http://dx.doi.org/10.1089/ars.2008.2270; PMID: 18828708
- Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, Witkiewicz A, Lin Z, Balliet R, Howell A, et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor micro-environment. Cancer Biol Ther 2010; 10:537 - 42; http://dx.doi.org/10.4161/cbt.10.6.13370; PMID: 20861671
- Wang X, Jin H. The epigenetic basis of the Warburg effect. Epigenetics 2010; 5:566 - 8; http://dx.doi.org/10.4161/epi.5.7.12662; PMID: 20622527
- Warburg O. On the origin of cancer cells. Science 1956; 123:309 - 14; http://dx.doi.org/10.1126/science.123.3191.309; PMID: 13298683
- Warburg O. On respiratory impairment in cancer cells. Science 1956; 124:269 - 70; PMID: 13351639
- Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab 2012; 15:4 - 5; http://dx.doi.org/10.1016/j.cmet.2011.12.011; PMID: 22225869
- Martinez-Outschoorn UE, Pestell RG, Howell A, Tykocinski ML, Nagajyothi F, Machado FS, Tanowitz HB, Sotgia F, Lisanti MP. Energy transfer in “parasitic” cancer metabolism: mitochondria are the powerhouse and Achilles’ heel of tumor cells. Cell Cycle 2011; 10:4208 - 16; http://dx.doi.org/10.4161/cc.10.24.18487; PMID: 22033146
- Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, Martinez-Outschoorn UE, Whitaker-Menezes D, Howell A, Sotgia F, et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal 2012; 16:1264 - 84; http://dx.doi.org/10.1089/ars.2011.4243; PMID: 21883043
- Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009; 8:3984 - 4001; http://dx.doi.org/10.4161/cc.8.23.10238; PMID: 19923890
- Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, Kleer CG, Brody JR, Lisanti MP. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol 2009; 174:2023 - 34; http://dx.doi.org/10.2353/ajpath.2009.080873; PMID: 19411448
- Sloan EK, Ciocca DR, Pouliot N, Natoli A, Restall C, Henderson MA, Fanelli MA, Cuello-Carrión FD, Gago FE, Anderson RL. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am J Pathol 2009; 174:2035 - 43; http://dx.doi.org/10.2353/ajpath.2009.080924; PMID: 19411449
- Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol 2012; 7:423 - 67; http://dx.doi.org/10.1146/annurev-pathol-011811-120856; PMID: 22077552
- Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res 2011; 13:213; http://dx.doi.org/10.1186/bcr2892; PMID: 21867571
- Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, Frank PG, Casimiro MC, Wang C, Pestell RG, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010; 9:3485 - 505; http://dx.doi.org/10.4161/cc.9.17.12721; PMID: 20861672
- Razani B, Zhang XL, Bitzer M, von Gersdorff G, Böttinger EP, Lisanti MP. Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGF-beta type I receptor. J Biol Chem 2001; 276:6727 - 38; http://dx.doi.org/10.1074/jbc.M008340200; PMID: 11102446
- Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, Howell A, Aquila S, Andò S, Martinez-Outschoorn U, et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 2012; 11:3019 - 35; http://dx.doi.org/10.4161/cc.21384; PMID: 22874531
- Martinez-Outschoorn UE, Balliet RM, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Hereditary ovarian cancer and two-compartment tumor metabolism: epithelial loss of BRCA1 induces hydrogen peroxide production, driving oxidative stress and NFκB activation in the tumor stroma. Cell Cycle 2012; 11:4152 - 66; http://dx.doi.org/10.4161/cc.22226; PMID: 23047606
- Carito V, Bonuccelli G, Martinez-Outschoorn UE, Whitaker-Menezes D, Caroleo MC, Cione E, Howell A, Pestell RG, Lisanti MP, Sotgia F. Metabolic remodeling of the tumor microenvironment: migration stimulating factor (MSF) reprograms myofibroblasts toward lactate production, fueling anabolic tumor growth. Cell Cycle 2012; 11:3403 - 14; http://dx.doi.org/10.4161/cc.21701; PMID: 22918248
- Jin H, Sperka T, Herrlich P, Morrison H. Tumorigenic transformation by CPI-17 through inhibition of a merlin phosphatase. Nature 2006; 442:576 - 9; http://dx.doi.org/10.1038/nature04856; PMID: 16885985
- Jin H, Wang X, Ying J, Wong AH, Cui Y, Srivastava G, Shen ZY, Li EM, Zhang Q, Jin J, et al. Epigenetic silencing of a Ca(2+)-regulated Ras GTPase-activating protein RASAL defines a new mechanism of Ras activation in human cancers. Proc Natl Acad Sci U S A 2007; 104:12353 - 8; http://dx.doi.org/10.1073/pnas.0700153104; PMID: 17640920
- Lam EK, Wang X, Shin VY, Zhang S, Morrison H, Sun J, et al. A microRNA contribution to aberrant Ras activation in gastric cancer. American journal of translational research 2011; 3:209-18.
- Kim JH, Kim HY, Lee YK, Yoon YS, Xu WG, Yoon JK, Choi SE, Ko YG, Kim MJ, Lee SJ, et al. Involvement of mitophagy in oncogenic K-Ras-induced transformation: overcoming a cellular energy deficit from glucose deficiency. Autophagy 2011; 7:1187 - 98; http://dx.doi.org/10.4161/auto.7.10.16643; PMID: 21738012
- Liu X, Wang X, Zhang J, Lam EK, Shin VY, Cheng AS, Yu J, Chan FK, Sung JJ, Jin HC. Warburg effect revisited: an epigenetic link between glycolysis and gastric carcinogenesis. Oncogene 2010; 29:442 - 50; http://dx.doi.org/10.1038/onc.2009.332; PMID: 19881551
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 2011; 22:165 - 78; http://dx.doi.org/10.1091/mbc.E10-06-0500; PMID: 21119005
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008; 13:343 - 54; http://dx.doi.org/10.1016/j.ccr.2008.02.001; PMID: 18394557
- Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal 2002; 14:381 - 95; http://dx.doi.org/10.1016/S0898-6568(01)00271-6; PMID: 11882383
- Santi SA, Lee H. The Akt isoforms are present at distinct subcellular locations. Am J Physiol Cell Physiol 2010; 298:C580 - 91; http://dx.doi.org/10.1152/ajpcell.00375.2009; PMID: 20018949
- Santi SA, Lee H. Ablation of Akt2 induces autophagy through cell cycle arrest, the downregulation of p70S6K, and the deregulation of mitochondria in MDA-MB231 cells. PLoS One 2011; 6:e14614; http://dx.doi.org/10.1371/journal.pone.0014614; PMID: 21297943
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672 - 6; http://dx.doi.org/10.1038/45257; PMID: 10604474
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 2003; 100:15077 - 82; http://dx.doi.org/10.1073/pnas.2436255100; PMID: 14657337
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809 - 20; PMID: 14638851
- Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 2007; 14:500 - 10; http://dx.doi.org/10.1038/sj.cdd.4402039; PMID: 16990848
- Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ 2007; 14:548 - 58; http://dx.doi.org/10.1038/sj.cdd.4402030; PMID: 16946731
- Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007; 21:1621 - 35; http://dx.doi.org/10.1101/gad.1565707; PMID: 17606641
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11:385 - 96; http://dx.doi.org/10.1038/ncb1846; PMID: 19270696
- Van Humbeeck C, Cornelissen T, Vandenberghe W. Ambra1: a Parkin-binding protein involved in mitophagy. Autophagy 2011; 7:1555 - 6; http://dx.doi.org/10.4161/auto.7.12.17893; PMID: 21921694
- Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol 2009; 186:773 - 82; http://dx.doi.org/10.1083/jcb.200907014; PMID: 19797076
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 2006; 8:688 - 99; http://dx.doi.org/10.1038/ncb1426; PMID: 16799551
- Takahashi Y, Young MM, Serfass JM, Hori T, Wang HG. Sh3glb1/Bif-1 and mitophagy: Acquisition of apoptosis resistance during Myc-driven lymphomagenesis. Autophagy 2013; 1107 - 9; http://dx.doi.org/10.4161/auto.24817; PMID: 23680845
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mulé JJ, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 2007; 9:1142 - 51; http://dx.doi.org/10.1038/ncb1634; PMID: 17891140
- Knuutila S, Aalto Y, Autio K, Björkqvist AM, El-Rifai W, Hemmer S, Huhta T, Kettunen E, Kiuru-Kuhlefelt S, Larramendy ML, et al. DNA copy number losses in human neoplasms. Am J Pathol 1999; 155:683 - 94; http://dx.doi.org/10.1016/S0002-9440(10)65166-8; PMID: 10487825
- Lee JW, Jeong EG, Soung YH, Nam SW, Lee JY, Yoo NJ, Lee SH. Decreased expression of tumour suppressor Bax-interacting factor-1 (Bif-1), a Bax activator, in gastric carcinomas. Pathology 2006; 38:312 - 5; http://dx.doi.org/10.1080/00313020600820880; PMID: 16916719
- Coppola D, Oliveri C, Sayegh Z, Boulware D, Takahashi Y, Pow-Sang J, Djeu JY, Wang HG. Bax-interacting factor-1 expression in prostate cancer. Clin Genitourin Cancer 2008; 6:117 - 21; http://dx.doi.org/10.3816/CGC.2008.n.018; PMID: 18824435
- Coppola D, Khalil F, Eschrich SA, Boulware D, Yeatman T, Wang HG. Down-regulation of Bax-interacting factor-1 in colorectal adenocarcinoma. Cancer 2008; 113:2665 - 70; http://dx.doi.org/10.1002/cncr.23892; PMID: 18833585
- Kim SY, Oh YL, Kim KM, Jeong EG, Kim MS, Yoo NJ, Lee SH. Decreased expression of Bax-interacting factor-1 (Bif-1) in invasive urinary bladder and gallbladder cancers. Pathology 2008; 40:553 - 7; http://dx.doi.org/10.1080/00313020802320440; PMID: 18752120
- Coppola D, Helm J, Ghayouri M, Malafa MP, Wang HG. Down-regulation of Bax-interacting factor 1 in human pancreatic ductal adenocarcinoma. Pancreas 2011; 40:433 - 7; http://dx.doi.org/10.1097/MPA.0b013e318205eb03; PMID: 21283040
- Takahashi Y, Karbowski M, Yamaguchi H, Kazi A, Wu J, Sebti SM, Youle RJ, Wang HG. Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol Cell Biol 2005; 25:9369 - 82; http://dx.doi.org/10.1128/MCB.25.21.9369-9382.2005; PMID: 16227588
- Takahashi Y, Hori T, Cooper TK, Liao J, Desai N, Serfass JM, Young MM, Park S, Izu Y, Wang HG. Bif-1 haploinsufficiency promotes chromosomal instability and accelerates Myc-driven lymphomagenesis via suppression of mitophagy. Blood 2013; 121:1622 - 32; http://dx.doi.org/10.1182/blood-2012-10-459826; PMID: 23287860
- Cuddeback SM, Yamaguchi H, Komatsu K, Miyashita T, Yamada M, Wu C, Singh S, Wang HG. Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. J Biol Chem 2001; 276:20559 - 65; http://dx.doi.org/10.1074/jbc.M101527200; PMID: 11259440
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007; 447:1121 - 5; PMID: 17589504
- Strappazzon F, Vietri-Rudan M, Campello S, Nazio F, Florenzano F, Fimia GM, Piacentini M, Levine B, Cecconi F. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J 2011; 30:1195 - 208; http://dx.doi.org/10.1038/emboj.2011.49; PMID: 21358617
- Fimia GM, Corazzari M, Antonioli M, Piacentini M. Ambra1 at the crossroad between autophagy and cell death. Oncogene 2013; 32:3311 - 8; http://dx.doi.org/10.1038/onc.2012.455; PMID: 23069654
- Van Humbeeck C, Cornelissen T, Hofkens H, Mandemakers W, Gevaert K, De Strooper B, et al. Parkin interacts with Ambra1 to induce mitophagy. The Journal of neuroscience: the official journal of the Society for Neuroscience 2011; 31:10249-61.
- Burton TR, Gibson SB. The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ 2009; 16:515 - 23; http://dx.doi.org/10.1038/cdd.2008.185; PMID: 19136941
- Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, Novak I, Dikic I, Hamacher-Brady A, Brady NR. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem 2013; 288:1099 - 113; http://dx.doi.org/10.1074/jbc.M112.399345; PMID: 23209295
- Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 2009; 16:939 - 46; http://dx.doi.org/10.1038/cdd.2009.16; PMID: 19229244
- Thomas RL, Kubli DA, Gustafsson AB. Bnip3-mediated defects in oxidative phosphorylation promote mitophagy. Autophagy 2011; 7:775 - 7; http://dx.doi.org/10.4161/auto.7.7.15536; PMID: 21460627
- Kondo Y, Kondo S. Autophagy and cancer therapy. Autophagy 2006; 2:85 - 90; PMID: 16874083
- Gargini R, García-Escudero V, Izquierdo M. Therapy mediated by mitophagy abrogates tumor progression. Autophagy 2011; 7:466 - 76; http://dx.doi.org/10.4161/auto.7.5.14731; PMID: 21270513
- García-Escudero V, Gargini R, Izquierdo M. Glioma regression in vitro and in vivo by a suicide combined treatment. Mol Cancer Res 2008; 6:407 - 17; http://dx.doi.org/10.1158/1541-7786.MCR-07-0243; PMID: 18337448
- García-Escudero V, Gargini R. Autophagy induction as an efficient strategy to eradicate tumors. Autophagy 2008; 4:923 - 5; PMID: 18716458
- Girald W, Collin A, Izquierdo M. Toxicity and delivery methods for the linamarase/linamarin/glucose oxidase system, when used against human glioma tumors implanted in the brain of nude rats. Cancer Lett 2011; 313:99 - 107; http://dx.doi.org/10.1016/j.canlet.2011.08.029; PMID: 21955616
- Cortés ML, García-Escudero V, Hughes M, Izquierdo M. Cyanide bystander effect of the linamarase/linamarin killer-suicide gene therapy system. J Gene Med 2002; 4:407 - 14; http://dx.doi.org/10.1002/jgm.280; PMID: 12124983
- Adan-Gokbulut A, Kartal-Yandim M, Iskender G, Baran Y. Novel agents targeting bioactive sphingolipids for the treatment of cancer. Curr Med Chem 2013; 20:108 - 22; PMID: 23244584
- Rego A, Costa M, Chaves SR, Matmati N, Pereira H, Sousa MJ, Moradas-Ferreira P, Hannun YA, Costa V, Côrte-Real M. Modulation of mitochondrial outer membrane permeabilization and apoptosis by ceramide metabolism. PLoS One 2012; 7:e48571; http://dx.doi.org/10.1371/journal.pone.0048571; PMID: 23226203
- Saddoughi SA, Ogretmen B. Diverse functions of ceramide in cancer cell death and proliferation. Adv Cancer Res 2013; 117:37 - 58; PMID: 23290776
- Venkataraman K, Riebeling C, Bodennec J, Riezman H, Allegood JC, Sullards MC, Merrill AH Jr., Futerman AH. Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast longevity assurance gene 1 (LAG1), regulates N-stearoyl-sphinganine (C18-(dihydro)ceramide) synthesis in a fumonisin B1-independent manner in mammalian cells. J Biol Chem 2002; 277:35642 - 9; http://dx.doi.org/10.1074/jbc.M205211200; PMID: 12105227
- Saddoughi SA, Garrett-Mayer E, Chaudhary U, O’Brien PE, Afrin LB, Day TA, Gillespie MB, Sharma AK, Wilhoit CS, Bostick R, et al. Results of a phase II trial of gemcitabine plus doxorubicin in patients with recurrent head and neck cancers: serum C₁₈-ceramide as a novel biomarker for monitoring response. Clin Cancer Res 2011; 17:6097 - 105; http://dx.doi.org/10.1158/1078-0432.CCR-11-0930; PMID: 21791630
- Wang X, Leung AW, Luo J, Xu C. TEM observation of ultrasound-induced mitophagy in nasopharyngeal carcinoma cells in the presence of curcumin. Exp Ther Med 2012; 3:146 - 8; PMID: 22969860
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature 2003; 425:686 - 91; http://dx.doi.org/10.1038/nature02026; PMID: 14562095
- Bell C, English L, Boulais J, Chemali M, Caron-Lizotte O, Desjardins M, Thibault P. Quantitative Proteomics Reveals the Induction of Mitophagy in Tumor Necrosis Factor-α-activated (TNFα) Macrophages. Mol Cell Proteomics 2013; 12:2394 - 407; http://dx.doi.org/10.1074/mcp.M112.025775; PMID: 23674617
- Jangamreddy JR, Ghavami S, Grabarek J, Kratz G, Wiechec E, Fredriksson BA, et al. Salinomycin induces activation of autophagy, mitophagy and affects mitochondrial polarity: Differences between primary and cancer cells. Biochim Biophys Acta 2013; 1832:2057 - 69
- Wurm CA, Neumann D, Lauterbach MA, Harke B, Egner A, Hell SW, Jakobs S. Nanoscale distribution of mitochondrial import receptor Tom20 is adjusted to cellular conditions and exhibits an inner-cellular gradient. Proc Natl Acad Sci U S A 2011; 108:13546 - 51; http://dx.doi.org/10.1073/pnas.1107553108; PMID: 21799113
- Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol 2013; 14:480 - 8; http://dx.doi.org/10.1038/ni.2563; PMID: 23525089