
Abstract
The combination of temsirolimus (TEM), an MTOR inhibitor, and hydroxychloroquine (HCQ), an autophagy inhibitor, augments cell death in preclinical models. This phase 1 dose-escalation study evaluated the maximum tolerated dose (MTD), safety, preliminary activity, pharmacokinetics, and pharmacodynamics of HCQ in combination with TEM in cancer patients. In the dose escalation portion, 27 patients with advanced solid malignancies were enrolled, followed by a cohort expansion at the top dose level in 12 patients with metastatic melanoma. The combination of HCQ and TEM was well tolerated, and grade 3 or 4 toxicity was limited to anorexia (7%), fatigue (7%), and nausea (7%). An MTD was not reached for HCQ, and the recommended phase II dose was HCQ 600 mg twice daily in combination with TEM 25 mg weekly. Other common grade 1 or 2 toxicities included fatigue, anorexia, nausea, stomatitis, rash, and weight loss. No responses were observed; however, 14/21 (67%) patients in the dose escalation and 14/19 (74%) patients with melanoma achieved stable disease. The median progression-free survival in 13 melanoma patients treated with HCQ 1200mg/d in combination with TEM was 3.5 mo. Novel 18-fluorodeoxyglucose positron emission tomography (FDG-PET) measurements predicted clinical outcome and provided further evidence that the addition of HCQ to TEM produced metabolic stress on tumors in patients that experienced clinical benefit. Pharmacodynamic evidence of autophagy inhibition was evident in serial PBMC and tumor biopsies only in patients treated with 1200 mg daily HCQ. This study indicates that TEM and HCQ is safe and tolerable, modulates autophagy in patients, and has significant antitumor activity. Further studies combining MTOR and autophagy inhibitors in cancer patients are warranted.
Introduction
Autophagy is a mechanism by which intracellular organelles and macromolecules undergo bulk degradation and subsequent recycling, therefore providing the cell with the basic building blocks on which it survives during periods of stress.Citation1 In advanced cancer, autophagy is a survival mechanism that is induced by a wide variety of intra- and extracellular stresses.Citation2 Autophagy inhibition with chloroquine (CQ) derivatives can augment the cytotoxicity of a number of chemotherapies and targeted therapies.Citation3 Growth factor signaling can directly regulate autophagy through the MTORC1 (mechanistic target of rapamycin complex 1)-ULK1 (unc-51 like autophagy activating kinase 1) complex interactions.Citation4 The first protein complex that is considered part of the core autophagy machinery is the ULK1-ATG13-RB1CC1/FIP200-ATG101/C12orf44 complex. When growth factor signaling pathways are activated and extracellular nutrients are abundant, the serine threonine kinase MTORC1 directly phosphorylates ULK1 repressing the activity of this complex and blocking the catabolic autophagy program from becoming activated. When MTORC1 activity is suppressed either through growth factor signaling inhibitors, rapamycin analogs, or nutrient withdrawal, the ULK1 complex becomes derepressed and autophagy is induced. Rapamycin-induced autophagy has been shown to be a cytoprotective mechanism,Citation5 possibly explaining why rapamycin analogs are mainly cytostatic in animal models and have produced low response rates in clinical trials.Citation6 Combining a distal autophagy inhibitor HCQ with the rapamycin analog such as CCI-779 (temsirolimus) was found to produce synergistic cytotoxicity and antitumor activity in preclinical animal models of melanoma and renal cell carcinoma.Citation7,Citation8 Here we report the first clinical trial of a rapamycin analog and HCQ in patients with advanced solid tumors.
Temsirolimus, an intravenous rapamycin analog, is approved by the United States Food and Drug Administration for the treatment of advanced renal cell cancer.Citation9 The pivotal phase III trial that led to its approval, demonstrated that TEM improved survival in patients with renal cell carcinoma. Given the low response rate, this survival benefit is likely due to prolonged stable disease in renal cell carcinoma.Citation10 Given that MTOR inhibition is a potent inducer of autophagy, autophagy may serve as a mechanism by which the cell escapes therapy-induced cell death. Inhibition of autophagy with HCQ may relieve this resistance, induce cell death, and improve the clinical effect of MTOR inhibition.
This phase I study was conducted to determine the recommended phase II dose of daily, oral HCQ administered in combination with fixed dose, weekly TEM in patients with advanced solid malignancies. Initially the trial was conducted in patients with advanced solid tumors, but once a high rate of stable disease was observed in melanoma patients, the protocol was amended to include an expansion cohort of melanoma patients. High-dose HCQ was safely combined with standard dose TEM and produced a high rate of stable disease in patients with advanced melanoma and other advanced malignancies in which rapamycin analogs have previously been found to be inactive.
Results
Patients
Between December 2008 and May 2012, 40 patients were enrolled on this trial, including 27 patients with advanced solid tumors on the phase I dose escalation, and 13 patients with advanced melanoma on the melanoma expansion. Of the 27 patients on the phase I dose escalation portion, 5 were not evaluable for response but were evaluable for toxicity, since they received at least one dose of study medication, but did not receive at least 4 wk of combined treatment. One patient was a screen failure and therefore was not evaluable for toxicity or response. In the melanoma expansion 13 patients were enrolled and 1 patient was not evaluable for response, but was evaluable for toxicity. The most common reasons for not being evaluable was rapid disease progression limiting the ability to take the prescribed medication (5), patient withdrawal from the study for personal reasons (1), or screen failure (1). Patients with advanced solid tumors enrolled and evaluable for toxicity (n = 26) on the phase I dose escalation study had a median age of 62 and 62% of patients had an Eastern Cooperative Oncology Group performance status 1 (). The most common malignancies were melanoma (35%), colorectal carcinoma (15%), head and neck cancer (12%), and breast cancer (8%). The median number of prior therapies was 3 (range 0 to 9). Including the melanoma patients enrolled on the dose escalation portion of the trial, 19 patients with Stage IV melanoma were evaluable on this trial. The median age was 60 with 74% of patients male. The majority of patients had poor prognosis as determined by M1c stage (69%), lactate dehydrogenase > upper limit of normal (53%) and 26% of patients had brain metastases. The majority of patients (84%) were BRAF wild type, and few patients had prior BRAF inhibitor (11%) or ipilumumab (11%) therapy.
Table 1. Dose escalation patient characteristics
Dose-limiting toxicities and adverse events
One patient with advanced prostate cancer extensively involving his bones in the HCQ 200 mg/d cohort (Table S1) experienced grade 4 thrombocytopenia, hematuria, and thrombotic cerebrovascular accident. This was considered a dose-limiting toxicity (DLT) and dose expansion to 6 patients was undertaken. No additional DLT was observed in the 200 or 400 mg (200 mg twice daily oral) cohort. One patient in the 800 mg (400 mg twice daily oral) cohort had grade 5 pneumonia, and sepsis after 3 mo of combined therapy. Death was attributed to a significant delay in the patient reporting symptoms and seeking medical care for pneumonia. Although this was not considered a DLT, dose expansion was undertaken in the 800 mg cohort. No additional DLTs were observed on this trial. Other significant adverse events of note included one patient (HCQ 800 mg cohort) had a grade 3 perirectal abscess in cycle 3 treated successfully with incision drainage and antibiotics. One patient with head and neck cancer and a history of aspiration developed a grade 3 pulmonary tumor abscess treated successfully with antibiotics in cycle 3. One patient with a large gall bladder metastases from melanoma that completely regressed on treatment developed grade 4 cholangitis and sepsis during tumor regression but was successfully retreated for months afterwards. Finally only one patient with uveal melanoma who was known to be borderline diabetic starting study, and treated with temsirolimus and HCQ 600 mg po twice daily, developed symptomatic hyperglycemia, hypertriglyceridemia, known toxicities associated with temsirolimus. The addition of metformin 500 mg 3 times daily alleviated these symptoms without any adverse events, and without the need for dose reduction.
In the total study population evaluable for toxicity (n = 39) grade 3–4 adverse events were rare with anemia (10%), anorexia (8%), fatigue (8%), and nausea (15%) being most common (). The most common grade 1 or 2 adverse events were anorexia (41%), constipation (13%), diarrhea (18%), fatigue (49%), lymphopenia (31%), nausea (33%), rash (38%), stomatitis (41%), and weight loss (18%). These adverse events were more common in the 1200 mg cohort compared with lower cohorts, indicating a dose-dependent increase in grade 2 toxicities. Dose reduction from 1200 mg/d (600 mg twice daily oral) to 1000 mg/d (600 mg in the morning and 400 mg in the evening) was common after 3 mos of treatment at HCQ 1200 mg daily.
Table 2. Adverse events
Antitumor activity, autophagy modulation, and tumor metabolic compromise
No partial responses by response evaluation criteria in solid tumors (RECIST) 1.0Citation11 were observed in this study. In the dose escalation cohort, stable disease was the best response in (14/21) 67% of patients (). Stable disease was achieved in 14/19 (74%) of the melanoma patients treated on this study and 9/13 (69%) of patients treated with HCQ 1200 mg/d (12 expansion patients and one melanoma patient treated with 1200 mg/d in the dose escalation study). All melanoma patients treated on this study had evidence of progressive disease at the time they entered the study. Representative lesions from the melanoma patients treated with HCQ 1200 mg/d are provided in Table S4 to further underscore the clinical significance of the stable disease observed in this study. Waterfall plots demonstrated some degree of tumor shrinkage was observed in 7/19 (37%) patients in the dose escalation (Fig. S1A), and 6/12 (50%) of the patients in the melanoma expansion (Fig. S1B). An example of tumor shrinkage observed on computed tomography (CT) scans with this regimen in a patient with rapidly progressive melanoma is provided in . Serial [18F]-fluorodeoxyglucose-positron emission tomography (FDG-PET)/CT scans from a melanoma patient with massive tumor burden demonstrates the potential clinical impact of tumor stability on maintaining performance status (). The median progression-free survival of evaluable patients on the melanoma expansion was 3.5 mo (95%CI 2–6 mo) (Fig. S2).
Table 3. RECIST response in evaluable patients
Figure 1. Antitumor activity of temsirolimus and hydroxychloroquine. (A) Serial contrast CT (CT) scans of the chest and abdomen in a patient with rapidly progressive melanoma treated with temsirolimus and HCQ. Orange outlines: tumor. (B) Serial [18]-fluordeoxy glucose positron emission tomography (FDG-PET) scans of a melanoma patient with massive tumor burden at baseline, who was able to maintain performance status by achieving stable disease on temsirolimus and hydroxychloroquine. Black signal indicates FDG-avid tumor.
![Figure 1. Antitumor activity of temsirolimus and hydroxychloroquine. (A) Serial contrast CT (CT) scans of the chest and abdomen in a patient with rapidly progressive melanoma treated with temsirolimus and HCQ. Orange outlines: tumor. (B) Serial [18]-fluordeoxy glucose positron emission tomography (FDG-PET) scans of a melanoma patient with massive tumor burden at baseline, who was able to maintain performance status by achieving stable disease on temsirolimus and hydroxychloroquine. Black signal indicates FDG-avid tumor.](/cms/asset/652de648-003b-48a9-b7d4-79cd955de138/kaup_a_10929119_f0001.gif)
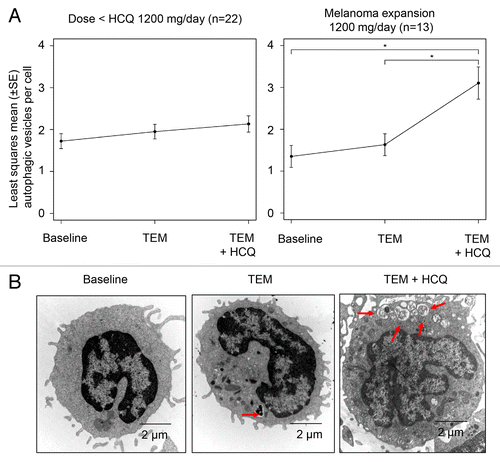
The mean number of AV/cell in serially collected peripheral blood mononuclear cells (PBMC) was measured by electron microscopy (EM; see Patients and Methods) across cohorts to determine if there was evidence of autophagy modulation with TEM or TEM + HCQ. Samples from at least 2 of 3 scheduled timepoints were evaluable for 89/114 (78%) of samples obtained from 32 patients. No samples were obtained for 8 patients due to patient preference, improper handling, or patient never receiving study drug. The most common missing sample in patients with 2 of 3 samples was the third timepoint due to the patient coming off the study (n = 4), patient preference, or improper handling (n = 8). Serial PBMC were collected from the HCQ 200 mg/d and HCQ 400 mg/d cohorts at the following timepoints: 1) pretreatment 2) pre-TEM infusion 1 wk after the first TEM infusion 3) pre-TEM infusion after 4 wk of TEM + HCQ. With subsequent cohorts, the timepoints were changed to post-TEM infusion rather than pre (trough) TEM infusion to determine if this would yield more striking evidence of autophagy modulation. For the 800 mg cohort, the timepoints were changed to 1) pretreatment 2) 24 h after first TEM infusion 3) 24 h after TEM infusion 5 after 4 wk of combined HCQ and TEM treatment. For the 1200 mg cohort the timepoints were changed to 1) pretreatment 2) 4 h after first TEM infusion 3) 4 h after TEM infusion 5 after 4 wk of combined HCQ and TEM treatment. Despite changing the timing of the TEM infusions to coincide with the known pharmacokinetic peak of TEM exposure in patients no significant increase in AV was observed with TEM treatment, so all of the data for TEM was pooled. Evaluating the mean ± SEM number of AV/cell across cohorts () demonstrated that there was a trend for AV accumulation with TEM treatment alone reflecting autophagy induction, and a further accumulation of AV with TEM + HCQ treatment reflecting simultaneous autophagy induction and distal blockade (). There was a significant AV accumulation with TEM + HCQ compared with baseline only in the 1200 mg cohort.
Figure 2. Pharmacodynamic effects of temsirolimus and hydroxychloroquine on autophagic vacuole accumulation in peripheral blood mononuclear cells (PBMC). (A) Mixed-effects model of mean ± SD autophagic vacuoles (AVs)/cell. *P < 0.05. (B) Representative electron micrographs from a patient treated with TEM and TEM + HCQ 600 mg/po bid. Arrows, AVs; scale bar: 2 µm.
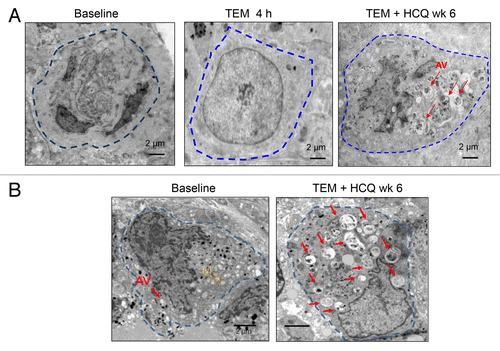
To determine if there was any autophagy modulation with these regimens in tumor tissue, serial tumor biopsies of cutaneous melanoma metastases were obtained from 2 patients treated with TEM + HCQ 1200 mg/d. In one patient that was able to provide 3 timepoints for biopsy () there was no accumulation of AV in tumor tissue after 4 h of treatment with TEM compared with baseline. However after 6 wk of combined TEM + HCQ there was clear accumulation of AV’s with undigested contents. Therapy-induced AV accumulation was also observed in a second patient’s serial tumor biopsy (). This accumulation could be attributed to effective autophagy blockade by HCQ, or induction of autophagy finally achieved over longer term exposure to TEM, or a combination of both TEM-associated induction of autophagy and an HCQ-associated block in the clearance of AV.
Figure 3. Therapy-associated autophagic vacuole accumulation in serial tumor biopsies from melanoma patients. Representative electron micrographs of a melanoma cell from 2 different patients (A and B) at the indicated timepoints. Dotted blue line: border of cytoplasmic membrane of 1 tumor cell. Red arrows, autophagic vacuoles. Yellow arrow, mitochondria.
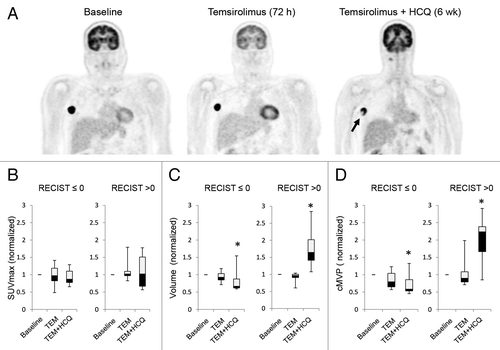
To determine if autophagy modulation with TEM + HCQ was impacting tumor glycolytic metabolism, serial FDG-PET/CT scans were obtained on 11 out of 12 patients on the melanoma expansion treated with TEM + HCQ 1200 mg/d. Patients underwent FDG-PET/CT imaging pretreatment, 72 h after TEM infusion 1, and 72 h after TEM infusion 5 following 4 wk of combined TEM and HCQ. In 2 patients, central photopenia developed on FDG-PET/CT images in tumors only after the addition of HCQ (), possibly reflecting the preclinical finding that the center of tumors are more susceptible to autophagy inhibition than the well-perfused rims.Citation12 We analyzed FDG-PET outcomes in patients that either had clinical benefit as defined as a change from baseline in RECIST measurements ≤ 0% or that did not (RECIST > 0%). No significant differences in maximum standardized uptake value (SUVmax) were identified with either TEM treatment or TEM + HCQ treatment in patients that did or did not have clinical benefit (). However, when tumor metabolically active volume (MAV) was measured, a significant decline compared with baseline was observed in the patients with clinical benefit and a significant increase in total tumor MAV was observed in patients with no clinical benefit only after the addition of HCQ. Similarly, a significant decline in the metabolic volumetric product (MVP) and a significant increase in MVP was observed in patients with clinical benefit and without clinical benefit, respectively, but only after the addition of HCQ (see Patients and Methods for more detailed description of FDG-PET measurement techniques used). Taken together these data suggest that high-dose HCQ blocks autophagy in patients and in melanoma patients, tumor metabolism and growth are impaired when HCQ is added to TEM. Randomized studies are needed to rule out the possibility that the decline in MAV and MVP at 6 wk could have been due to TEM alone. In addition, the clinical significance of metabolic volume (MAV and MVP) measurements and patient outcomes is still being established.
Figure 4. Changes in FDG-PET uptake in patients treated with temsirolimus and HCQ. (A) Serial FDG-PET images in a patient with metastatic melanoma. Arrow: central necrosis. (B–D) Comparison of FDG-PET parameters in patients with no clinical benefit (RECIST measurements > 0) or clinical benefit (RECIST measurement ≤ 0). (B) SUVmax normalized to baseline. (C) Tumor volume normalized to baseline. (D) Partial volume corrected metabolic volumetric product (cMVP); *P < 0.05.
HCQ pharmacokinetics and pharmacodynamics
We performed population pharmacokinetic (PK) analysis using 117 nonbaseline blood samples from 34 patients collected over a period up to 135 d. The population model PK parameters do not specifically represent steady-state values, as they were determined from multiple repeated single doses taken by individual patients during their period of participation in the study. The model that best described the disposition of HCQ blood concentrations was a 2-compartment model with first-order absorption and a lag time. No covariate interactions were identified that significantly improved the model. A nondiagonal fit was superior to a diagonal fit based on -2(LL). The final model was as follows: first order absorption rate constant (Ka) = typical value (tv)Ka * exp(nKa); apparent volume of distribution in central compartment (V/F) = tvV * exp(nV)/F; apparent volume of distribution in peripheral compartment (V2/F) = tvV2 * exp(nV2)/F; apparent oral clearance (Cl/F) = tvCl * exp(nCl)/F; intercompartmental clearance (Q) = tvQ * exp(nQ); lag time (tLag) = tvTlag = exp(nTlag). The residual error was supported by an additive error model, as described by: observed concentration = C + zero mean normally distributed random variable, where C is the predicted concentration. shows the individual predicted concentrations vs. the observed concentrations from the population PK model. HCQ population PK parameters are presented in .
Figure 5. Pharmacokinetic analysis of HCQ in patients receiving temsirolimus and HCQ. (A) Observed vs. individually predicted concentrations of HCQ based on the population PK model. (B) Estimated peak concentrations (Cmax). (C) Estimated trough concentrations (Cmin). (D) Estimated average concentrations (Cavg).
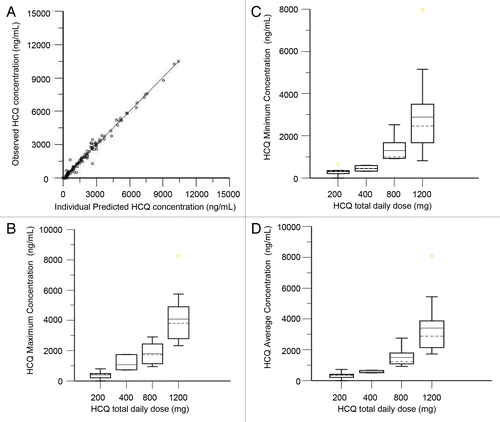
Table 4. HCQ population pharmacokinetic parameters
The final PK model developed was used to simulate HCQ blood concentrations for individual patients at steady-state, which was achieved on average after 350 h (15 d). Individual PK parameter estimates derived from the population were most variable for central volume of distribution (Vc/F) and intercompartmental clearance (Q). Blood HCQ concentration relationships for area under the concentration-time curve, Cmax, Cmin, and Cavg were proportional to daily HCQ dose (). For patients that had both complete PK and PD data available for correlation, 18/20 patients had an increase in AV after 5 wk of TEM + HCQ compared with baseline. The effect of the threshold value for Cmax on the change in AV after 4 wk of combined TEM + HCQ compared with pretreatment measurements in PBMC was then investigated. Patients with Cmax below 1348 ng/mL (n = 9) produced a median AV change of 0.280 (inter-quartile range: 0.005, 1.029), while patients with Cmax above 1348 ng/mL (n = 18) produced a median AV change of 1.380 (IQR: 0.421, 1.820) (Kruskal-Wallis P = 0.0742; Kolmogorov-Smirnov P = 0.3639). Neither the Kruskal-Wallis test for comparing median values nor the Kolmogorov-Smirnov 2-sample test identified any significant shift in the distribution. It is likely that due to this nearly uniform therapy-associated increase in AV from baseline, and the small sample size, classification and regression trees analysis analysis could not identify a threshold of HCQ exposure that significantly predicted AV increase for this combination.
Discussion
Despite the fact that the phosphoinositide-3 kinase (PI3K) pathway is one of the most frequently dysregulated pathways in cancer,Citation13 targeting PI3K-AKT-MTOR signaling has not yielded responses or even prolonged stable disease in most malignancies. While there are many contributing factors to this observation, one mechanism of resistance to inhibition of this signaling pathway is through the induction of autophagy. The direct link between growth factor signaling and autophagy is through MTORC1 kinase activity.Citation4 Therefore, there is considerable rationale and preclinical evidenceCitation8 that rapamycin analogs induce autophagy, and combined MTOR and autophagy inhibition could significantly enhance the efficacy of rapamycin analogs in multiple diseases. This phase I trial of TEM and HCQ has demonstrated the safety of this approach in patients with advanced solid tumors. The combination of standard doses of TEM with the highest doses of HCQ used in clinical practice was safe and tolerable. As with the combination of dose-intense temozolomide with HCQ (Rangwala et al., this issue)Citation14, and bortezomib and HCQ (Vogl et al., this issue)Citation15, an MTD was not defined in this trial, but frequent grade 2 or 3 nausea, anorexia, and fatigue lead to frequent dose reductions after 2 or 3 mo of treatment. Therefore further dose escalation of HCQ is not likely to be tolerable for prolonged periods of time, but future studies could consider a higher loading dose strategy to expedite time to peak concentrations and achievement of steady-state concentrations.
Assessments of autophagy modulation by EM, and tumor metabolism by FDG-PET/CT imaging in this study demonstrated that high-dose HCQ can modulate autophagy in surrogate tissues and tumor tissue. The PK-PD correlation analysis did not find a statistically significant correlation between HCQ exposure, or peak concentration and AV accumulation, but this is likely due to the small sample size and the fact that almost all of the patients had a significant increase in AV in PBMC after 4 wk of combined therapy compared with baseline. This consistent increase in AV was not seen in other HCQ combinations where significant PK-PD correlations were made (Rosenfeld, et al. this issueCitation16, Rangwala et al., this issueCitation14), suggesting that at 4 wk of combined therapy, the contribution of TEM to AV accumulation may be more significant than the contribution of temozolomide, for instance. However, as in other HCQ clinical studies (Vogl et al., this issueCitation15; Mahalingam et al., this issueCitation17, Rangwala et al., this issueCitation14), the magnitude of autophagy modulation may need to be maximized further to elicit tumor responses, either through higher doses of HCQ being tested, or by using more potent autophagy inhibitors that currently under development.Citation18 Larger studies in melanoma patients with biopsiable disease will determine if HCQ in fact is concentrated in tumor tissue as demonstrated by Barnard et al. (this issueCitation19) in dog lymphomas, and whether or not the magnitude of autophagy inhibition is in fact much higher than in PBMC with combined MTOR and autophagy inhibition.
In patients with stage IV melanoma TEM and HCQ produced a 74% stable disease rate. While there were no responses in this clinical trial this rate of stable disease is in striking contrast to a previous phase II trial of temsirolimus in advanced melanoma, where 1/33 patients had a partial response but 0/33 patients had stable disease.Citation20 This degree of stable disease is also superior to the stable disease rate observed in a phase I trial of TEM and sorafenib,Citation21 which was associated with more significant toxicity than TEM and HCQ. The high rate of stable disease coupled with the median progression-free survival of 3.5 mo observed in this clinical trial is a signal of activity that warrants further study in a melanoma population. The safety of this regimen opens up the possibility of further combination of TEM and HCQ with other rational targeted therapies, or chemotherapy in multiple malignancies.
One of the known resistance mechanisms to pure MTORC1 inhibition is feedback activation of AKT. This study of TEM and HCQ did not address this resistance mechanism, and there is preclinical evidence that autophagy inhibition in combination with a dual PI3K-MTOR inhibitor produces more synergistic antitumor activity in vivo in multiple preclinical models than rapamycin analogs in combination with autophagy inhibitors.Citation22,Citation23 Preclinical studies combining multitargeted PI3K pathway inhibitors with more potent autophagy inhibitors are currently underway.
Patients and Methods
Patient population
For the dose-escalation portion of this study, patients who had histologically or cytologically confirmed malignancies that were metastatic or unresectable, and for which standard curative or palliative therapies did not exist or were not considered effective were eligible. Patients had to have an Eastern Cooperative Oncology Group performance status of 0 or 1, be ≥ 18 y of age, have measurable disease by RECIST 1.0,Citation11 have adequate bone marrow, hepatic, and renal function as defined by absolute neutrophil count ≥ 1500/mm3, platelets ≥ 100,000/mm3, creatinine ≤ 2 times, total bilirubin ≤ 1.5 mg/dl, alanine aminotransferase and aspartate aminotransferase ≤ 5 times the upper limits of the institutional norm. Any number and type of prior therapy except prior MTOR inhibitors were allowed. Patients must have discontinued active immunotherapy, chemotherapy, or targeted therapies at least 4 wk prior to entering the study. Patients with treated brain metastases that had been stable for 3 mo were eligible. Patients with active, clinically significant and/or uncontrolled medical conditions were excluded, including patients with uncontrolled psoriasis. In addition, patients with human immunodeficiency virus and porphyria were excluded, the latter due to the risk of disease exacerbation. Patients receiving P450 enzyme-inducing anticonvulsants were ineligible. The study protocol was approved by the Institutional Review Board at the University of Pennsylvania; written informed consent was mandatory and obtained from all enrolled patients. For the melanoma-expansion cohort, the same eligibility criteria were used except patients with treated brain metastases that were proven stable for ≥ 1 mo were eligible.
Study design and HCQ dose escalation
This was a phase I, open label, single institution, 3 + 3 dose escalation studyCitation24 of oral HCQ with intravenous, weekly TEM in patients with advanced solid tumors. Due to significant antitumor activity during the dose escalation portion of the study, an expansion at the maximal tolerated or maximal administered dose was planned in 12 patients with advanced melanoma. Treatment consisted of intravenous TEM 25 mg monotherapy and oral HCQ 200 to 1200 mg daily. All patients were treated with a 1-wk run-in of single agent TEM followed by combination therapy with daily HCQ to enable correlative studies. The starting dose for HCQ was 200 mg. The planned dose escalation schema is as per Table S1: HCQ daily dose cohorts: 1) 200 mg, 2) 400 mg, 3) 800 mg, and 4) 1200 mg. Cycle length was 4 wk. Dose-limiting toxicities (DLTs) were evaluated initially during the first 6 wk, and after the HCQ 400 mg cohort was amended to 5 wk. DLTs were defined as Common Terminology Criteria for Adverse Events version 3.0 ≥ grade 3 nonhematologic toxicity. Hematological DLTs consisted of febrile neutropenia, grade 4 neutropenia > 7 d, or platelet count less than 10,000/mm3. Any signs or symptoms of peripheral retinal toxicity led to immediate discontinuation of HCQ and prompt ophthalmologic evaluation. Any DLT that led to a dose delay of > 28 consecutive d of HCQ resulted in the patient being taken off treatment. Patients were evaluable for toxicity if they had taken at least 1 dose of HCQ. Patients were evaluable for dose escalation decisions, and response if they completed at least 2 wk of concurrent HCQ and TEM. Nonevaluable patients were replaced. The maximum tolerated dose (MTD) was defined as the highest dose level at which ≤ 1 of 6 patients experienced DLT during the first 6 wk of the study. No intrapatient dose escalation of HCQ was allowed. If a significant adverse event was observed at any dose level that was not related to study drugs, but was serious or life threatening in way, 4 to 10 additional patients were enrolled at that dose level to explore safety. Dose escalation beyond 1200 mg HCQ/d was not pursued due because this is the highest administered dose typically used in other disorders such as rheumatoid arthritis.Citation25 If no MTD was established, 1200 mg dose level would be the recommended phase II dose.
Treatment, monitoring, and dose modifications
TEM was provided by Wyeth/Pfizer, and HCQ was obtained for patients through prescriptions filled at general outpatient pharmacy. HCQ tablets were manufactured by a number of generic pharmaceutical companies. Oral HCQ doses greater than 200 mg daily were taken by patients in divided doses at least 6 h apart. Intravenous TEM was infused over 30 min. Prophylactic intravenous diphenhydramine 25–50 mg was administered approximately 30 min prior to temsirolimus infusion. If antiemetic therapy was needed, phenothiazines or ondansetron were prescribed. Treatment was administered until disease progression as defined by a greater than 20% increase in measurable disease, or the appearance of a new lesion; treatment delay due to toxicity for ≥ grade 3 attributed as possibly, probably or definitely related to HCQ resulted in the dose being held until the adverse event (AE) has resolved to ≤ grade 1 or baseline. If the AE resolved, reinstitution of treatment could occur but at a reduced dose of HCQ (reduced by 200 mg daily from the previous dose). If the AE recurred at the reduced dose, treatment was held until the AE had resolved to ≤ grade 1 and when resolved treatment could be reinstituted at the next lower dose level. No more than 2 dose reductions were allowed. Toxicities that were attributed to HCQ included nausea, vomiting, diarrhea, rash, and visual field deficit. If any of these AEs occur at grade ≤ 2, HCQ may be continued and the AE managed with supportive care. Within 3 d prior to TEM dosing patients must have had an absolute neutrophil count > 1.0 × 109/L and platelet count > 75 × 109/L. All nonhematological toxicity grade 3 or 4 (except for alopecia, nausea, and vomiting) must have resolved to grade ≤ 2. If toxicity persisted, treatment was delayed by 1 wk for up to 3 consecutive wk. If after 3 wk of delay all toxicity has still not resolved then any further treatment with TEM was stopped. HCQ could have been continued after TEM was stopped. Known TEM toxicities including hematological and metabolic (e.g., hyperglycemia) toxicities were only attributed to TEM and resulted in the following TEM dose modifications: TEM 20 mg IV, and TEM 15 mg. For patients who required greater than 2 dose reductions TEM was stopped.
Safety and efficacy assessments
Clinical and laboratory assessments were obtained at baseline. Blood counts were obtained weekly for the first 5 wk and every 2 wk thereafter; liver and renal function were assessed every 2 wk for the first cycle and monthly thereafter. Safety assessments included an EKG obtained on wk 4 of combined therapy; a lipid panel was obtained every 2 wk for the first cycle and monthly thereafter. Adverse events were assessed at every visit. Responses were assessed every 2 mo or as clinically indicated, using RECIST version 1.0.Citation11
Pharmacokinetic and pharmacodynamic analysis
All patients enrolled had whole blood drawn for PK analysis at the following time points: HCQ concentrations were determined from whole blood samples (collected after the TEM run-in, and at 4 wk, 8 wk, 12 wk, and 6 mo of combination therapy). Blood was collected in tubes containing sodium heparin, and stored at −70 °C until analysis.
Whole blood concentrations of HCQ were measured using high-performance liquid chromatography with tandem mass spectrometry detection. Sample aliquots containing 500 ng of internal standard (IS) (d4-HCQ) were vortexed with acetonitrile/methanol, then centrifuged. An aliquot of the supernatant was withdrawn, dried under nitrogen gas, then reconstituted with mobile phase and 10 uL injected onto a Kinetex 50 × 3 mm 2.6 um 100A HPLC column (Phenomenex, Torrance, CA). Samples were eluted with a gradient mobile phase of 0.1% formic acid in acetonitrile and water using a 1200 Series Agilent HPLC system with an API 4000™ (AB SCIEX, Foster City, CA) mass spectrometer and electrospray interface operated in positive mode with multiple reaction monitoring detection. The capillary voltage was 4000 V with a source temperature of 500 °C. Mass spectrometer parameters were adjusted to maximize the intensity of the [M + H]+ ions in quadrupole 1 and the m/z transition ions of HCQ (337.275 → 248.152) and IS (341.150 → 252.035) in quadropole 3.
The mass spectrometers were controlled by AB SCIEX Analyst® software (Version 1.6.1) and data collection and analyses were conducted with the same software. Standard curves were constructed by plotting the analyte to IS ratio vs. the known concentration of HCQ (x) in each sample. Standard curves were fit by linear regression with weighting by 1/x. Samples were assayed in duplicate; samples for which the percent difference exceeded 15% were reanalyzed and samples for which concentrations exceeded the range values for the calibration curve were diluted appropriately and reanalyzed. The calibration curve was linear from 1 to 5000 ng/mL with correlation coefficients ranging from 0.9990- to 0.9999. The lower limit of quantitation was 1.0 ng/mL. The correlation coefficients for both inter- and intra-day variability were < 5.6% for each concentration (15 ng/mL, 150 ng/mL, and 1500 ng/mL) studied. The mean accuracy for inter- and intra-day evaluations was between 97.2 and 102%.
Whole blood HCQ concentration data were analyzed by nonlinear mixed-effect modeling using Phoenix™ NLME 1.2 (Pharsight, Cary, NC). Initial estimates for a base population pharmacokinetic model were derived from a naïve-pooled data analysis of individual patient blood concentration time data. One and 2-compartment models with first-order absorption and elimination, with and without a lag time, were evaluated as the potential pharmacokinetic base structural model. Inter-individual variability of population pharmacokinetic parameters was considered to be log-normally distributed with mean of 0 and variance of ω2. Visual inspection of standard goodness of fit/diagnostic plots and numerical diagnostics were used to determine optimal model fits. The first-order conditional maximum likelihood estimation, Lindstrom-Bates method was used for the modeling process. Diagnostic scatter plots (individual and population predicted values vs. observed concentrations, conditional weighted residuals vs. time and vs. observed concentrations), Akaike information criteria, and the likelihood ratio test, were used to select the base model. Conditional weighted residuals vs. time and predicted concentration time plots helped confirm that the chosen residual error model was appropriate.
Visual inspections of scatter and box plots for eta (random effect) values were used to explore potential continuous (age, weight), and categorical (sex, dose cohort) covariates. Covariates were centered on their median values. A stepwise covariate selection process was performed to build the full model. Model building criteria were based on covariate models associated with an increase in objection function value greater than 3.84 with one degree of freedom (P < 0.05) using the likelihood ratio test. A visual predictive check with 200 replicates was performed to assess the model performance. A total of 1000 bootstrap runs were performed to provide estimates of the precision of parameter estimates and the 95% confidence intervals for the pharmacokinetic parameters.
Individual pharmacokinetic parameters for each patient were derived from the final population model and used to simulate time-concentration profiles using WinNonlin® 6.2 (Pharsight Corporation, Cary, NC). The simulated blood HCQ concentrations were compared with observed concentrations to determine the predictive performance of the model. HCQ pharmacokinetic parameter estimates (peak blood concentration, Cmax; trough blood concentration, Cmin, average blood concentration, Cavg, area under the concentration-time curve) from these simulations were used to explore pharmacokinetic-pharmacodynamic (PK-PD) relationships.
Measurement of autophagic vacuole accumulation as a surrogate for autophagic flux was assessed in peripheral blood mononuclear cells (PBMCs) and serial tumor biopsies. Venous blood samples were collected in 2 BD Vacutainer® CPT tubes at the following timepoints: dose cohort 1 and 2: 1) predose, 2) after 1 wk (after single agent temsirolimus prior to dosing of infusion #2), and 3) after 4 wk of combination therapy (prior to dosing). In later cohorts, the time point for collection of sample #2 was changed to 24 h after the first dose of combined HCQ and TEM. Manufacturer’s instructions were followed to collect PBMC in 2 cell pellets. Cells obtained from PBMC pellet 1 were immediately fixed with 2% glutaraldehyde (EM fixative; company, catalog number) and stored at 4 °C until embedding. Embedding and image capture were performed as previously described.Citation26 Tumor biopsies were obtained by punch biopsies of cutaneous melanoma metastases. Tumor was dissected on ice and immediately fixed in EM fixative. For quantification of AV in PBMC using electron microscopy, high-powered micrographs (10000–12,000×) of 20–25 mononuclear cells from multiple distinct low-powered fields in each sample were obtained. AVs were scored by 2 independent investigators who were blinded to treatment time points. Morphological criteria for AV included 1) circularity, 2) contrast with structures that were white or lighter than the cytoplasm, 3) vesicles with contents, 4) vesicles > 200 nm in size and, 5) vesicles > 200 nm interior to the plasma membrane. Vesicular structures with cristae characteristic of mitochondria in cross section were excluded. The average of 2 counts assessed by investigators, are presented as mean ± standard error of the mean. PBMC pellet 2 was immediately frozen. The PK-PD relationship between HCQ and AV accumulation was first investigated by using an exploratory classification tree (Salford Predictive Modeler Builder v6.6), which identified a threshold effect of the peak blood concentration Cmax. The classification and regression trees analysis analysis was conducted using PK and PD data of 20 patients, with the target variable defined as any positive change in number of AV from pre to TEM + HCQ and Cmin, Cmax, and area under the curve as predictors. The effect of the threshold value for Cmax on the change in AV was then investigated using Kruskal-Wallis test for comparing median values and Kolmogorov-Smirnov 2-sample test to identify any significant shift in the distribution.
PET/CT imaging
Twelve patients with metastatic melanoma were imaged by whole body FDG-PET/CT imaging at the following timepoints: before treatment, 48 to 72 h after the first temsirolimus dose prior to HCQ administration, and 48 to 72 h after the 6th temsirolimus + HCQ dose. Fingerstick serum glucose levels were measured to ensure ≤ 150 mg/dL prior to administration. Subjects had fasted for at least 4 h prior to FDG administration. Study subjects then received ~555 MBq of intravenous FDG and stayed at rest for ~1 h. Subsequently, subjects were placed on a 16 detector-row PET/CT scanner with time-of-flight capabilities (Gemini TF, Philips Healthcare, Best, the Netherlands) to undergo whole body (skull vertex to toes) PET/CT imaging.
PET/CT data sets were initially assessed qualitatively for presence of metabolically active sites of tumor, locations of tumor lesions, and number of tumor lesions. Subsequently, through use of available image analysis software that utilizes an automatic adaptive thresholding method (ROVER, ABX GmbH, Radeberg, Germany),Citation27,Citation28 3D masks were placed about tumor lesions on PET images for automatic lesion delineation and subsequent measurement of lesional standardized uptake value (SUV), in order to assess for changes in tumor metabolism following therapy. In addition, lesional metabolically active tumor volume (MAV) and total MAV (defined as the sum of lesional MAV among all lesions in the body) were quantified and used to calculate lesional metabolic volumetric product (MVP) (defined as the product of lesional MAV and lesional SUV) and total MVP (defined as the sum of lesional MVP among all lesions in the body). Tumor MAV and MVP based on FDG-PET/CT have been utilized in the study of a variety of cancers, as they can be useful for pretreatment planning, patient selection for clinical trials, prognostication of patient outcome prior to treatment, and response assessment.Citation29 For example, Oh et al. showed that pretreatment tumor MAV is an independent predictor of overall survival and progression-free survival in 106 patients with small cell lung cancer, and provides a more detailed prediction of prognosis compared with tumor staging alone.Citation30
Statistical analysis
The primary objective of this phase I study was to determine the MTD of HCQ when given in combination with weekly temsirolimus in patients with refractory solid tumors. Secondary endpoints included response rates, toxicity rates, and pharmacodynamic and pharmacokinetic correlative endpoints. The target DLT rate was 33% using a traditional 3 + 3 design. The MTD was defined as a) the dose producing DLT in 2 out of 6 patients, or b) the dose level below the dose which produced DLT in ≥ 2 out of 3 patients, or in ≥ 3 out of 6 patients. Significance testing was conducted by the Student t test, or the Mann-Whitney test using Graphpad software.
| Abbreviations: | ||
| AE | = | adverse event |
| AV | = | autophagic vacuole |
| C | = | predicted concentration |
| Cl/F | = | apparent oral clearance |
| CQ | = | chloroquine |
| CT | = | computed tomography |
| DLT | = | dose-limiting toxicity |
| EM | = | electron microscopy |
| FDG-PET | = | [18]-fluorodeoxyglucose-positron emission tomography |
| HCQ | = | hydroxychloroquine |
| Ka | = | first order absorption rate constant |
| MTORC | = | mechanistic target of rapamycin complex |
| MTD | = | maximal tolerated dose |
| MVP | = | metabolic volumetric product |
| PBMC | = | peripheral blood mononuclear cells |
| PD | = | pharmacodynamics |
| PK | = | pharmacokinetics |
| Q | = | intercompartmental clearance |
| RECIST | = | response evaluation criteria in solid tumors |
| SUVmax | = | standardized uptake value |
| TEM | = | temsirolimus |
| tv | = | typical value |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
| V/F | = | apparent volume of distribution in central compartment |
| V2/F | = | apparent volume of distribution in peripheral compartment |
Additional material
Download Zip (446.6 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was partially funded by a clinical trial agreement between University of Pennsylvania and Wyeth/Pfizer incorporated. The study was also supported by the Abramson Cancer Center and NCI 1K23CA120862 (RKA). The authors would like to thank Ray Meade in the UPENN EM facility. We would like to thank Kurt D’Andrea, Richard Letrero, and Christopher Watt for providing genotyping analysis for patients on the study.
References
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709 - 30; http://dx.doi.org/10.1038/nrd3802; PMID: 22935804
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12:401 - 10; http://dx.doi.org/10.1038/nrc3262; PMID: 22534666
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17:654 - 66; http://dx.doi.org/10.1158/1078-0432.CCR-10-2634; PMID: 21325294
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992 - 2003; http://dx.doi.org/10.1091/mbc.E08-12-1249; PMID: 19225151
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274 - 93; http://dx.doi.org/10.1016/j.cell.2012.03.017; PMID: 22500797
- Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov 2011; 10:868 - 80; http://dx.doi.org/10.1038/nrd3531; PMID: 22037041
- Xie X, White EP, Mehnert JM. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS One 2013; 8:e55096; http://dx.doi.org/10.1371/journal.pone.0055096; PMID: 23383069
- Bray K, Mathew R, Lau A, Kamphorst JJ, Fan J, Chen J, Chen HY, Ghavami A, Stein M, DiPaola RS, et al. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS One 2012; 7:e41831; http://dx.doi.org/10.1371/journal.pone.0041831; PMID: 22848625
- Bukowski RM. Temsirolimus: a safety and efficacy review. Expert Opin Drug Saf 2012; 11:861 - 79; http://dx.doi.org/10.1517/14740338.2012.713344; PMID: 22861825
- Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, et al, Global ARCC Trial. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007; 356:2271 - 81; http://dx.doi.org/10.1056/NEJMoa066838; PMID: 17538086
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000; 92:205 - 16; http://dx.doi.org/10.1093/jnci/92.3.205; PMID: 10655437
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51 - 64; http://dx.doi.org/10.1016/j.ccr.2006.06.001; PMID: 16843265
- Sheppard K, Kinross KM, Solomon B, Pearson RB, Phillips WA. Targeting PI3 kinase/AKT/mTOR signaling in cancer. Crit Rev Oncog 2012; 17:69 - 95; http://dx.doi.org/10.1615/CritRevOncog.v17.i1.60; PMID: 22471665
- Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M, et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1369 - 79; http://dx.doi.org/10.4161/auto.29118; PMID: 24991839
- Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, Rangwala R, Piao S, Chang YC, Scott EC, et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014; 10:1380 - 90; http://dx.doi.org/10.4161/auto.29264; PMID: 24991834
- Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014; 10:1359 - 68; http://dx.doi.org/10.4161/auto.28984; PMID: 24991840
- Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, Mita AC, Curiel TJ, Espitia CM, Nawrocki ST, et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014; 10:1403 - 14; http://dx.doi.org/10.4161/auto.29231; PMID: 24991835
- McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, Lynch JP, Uehara T, Sepulveda AR, Davis LE, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A 2012; 109:8253 - 8; http://dx.doi.org/10.1073/pnas.1118193109; PMID: 22566612
- Barnard RA, Wittenburg LA, Amaravadi RK, Gustafson DL, Thorburn A, Thamm DH. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy 2014; 10:1415 - 25; http://dx.doi.org/10.4161/auto.29165; PMID: 24991836
- Margolin K, Longmate J, Baratta T, Synold T, Christensen S, Weber J, Gajewski T, Quirt I, Doroshow JH. CCI-779 in metastatic melanoma: a phase II trial of the California Cancer Consortium. Cancer 2005; 104:1045 - 8; http://dx.doi.org/10.1002/cncr.21265; PMID: 16007689
- Davies MA, Fox PS, Papadopoulos NE, Bedikian AY, Hwu WJ, Lazar AJ, Prieto VG, Culotta KS, Madden TL, Xu Q, et al. Phase I study of the combination of sorafenib and temsirolimus in patients with metastatic melanoma. Clin Cancer Res 2012; 18:1120 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-11-2436; PMID: 22223528
- Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, Haas-Kogan D, James CD, Oakes SA, Debnath J, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal 2010; 3:ra81; http://dx.doi.org/10.1126/scisignal.2001017; PMID: 21062993
- Mirzoeva OK, Hann B, Hom YK, Debnath J, Aftab D, Shokat K, Korn WM. Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3K-mTOR pathway in pancreatic adenocarcinoma. J Mol Med (Berl) 2011; 89:877 - 89; http://dx.doi.org/10.1007/s00109-011-0774-y; PMID: 21678117
- Lin Y, Shih WJ. Statistical properties of the traditional algorithm-based designs for phase I cancer clinical trials. Biostatistics 2001; 2:203 - 15; http://dx.doi.org/10.1093/biostatistics/2.2.203; PMID: 12933550
- Munster T, Gibbs JP, Shen D, Baethge BA, Botstein GR, Caldwell J, Dietz F, Ettlinger R, Golden HE, Lindsley H, et al. Hydroxychloroquine concentration-response relationships in patients with rheumatoid arthritis. Arthritis Rheum 2002; 46:1460 - 9; http://dx.doi.org/10.1002/art.10307; PMID: 12115175
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117:326 - 36; http://dx.doi.org/10.1172/JCI28833; PMID: 17235397
- Torigian DA, Lopez RF, Alapati S, Bodapati G, Hofheinz F, van den Hoff J, Saboury B, Alavi A. Feasibility and performance of novel software to quantify metabolically active volumes and 3D partial volume corrected SUV and metabolic volumetric products of spinal bone marrow metastases on 18F-FDG-PET/CT. Hell J Nucl Med 2011; 14:8 - 14; PMID: 21512658
- Hofheinz F, Pötzsch C, Oehme L, Beuthien-Baumann B, Steinbach J, Kotzerke J, van den Hoff J. Automatic volume delineation in oncological PET. Evaluation of a dedicated software tool and comparison with manual delineation in clinical data sets. Nuklearmedizin 2012; 51:9 - 16; http://dx.doi.org/10.3413/Nukmed-0419-11-07; PMID: 22027997
- Kwee TC, Torigian DA, Alavi A. Overview of positron emission tomography, hybrid positron emission tomography instrumentation, and positron emission tomography quantification. J Thorac Imaging 2013; 28:4 - 10; http://dx.doi.org/10.1097/RTI.0b013e31827882d9; PMID: 23249967
- Oh JR, Seo JH, Chong A, Min JJ, Song HC, Kim YC, Bom HS. Whole-body metabolic tumour volume of 18F-FDG PET/CT improves the prediction of prognosis in small cell lung cancer. Eur J Nucl Med Mol Imaging 2012; 39:925 - 35; http://dx.doi.org/10.1007/s00259-011-2059-7; PMID: 22270509