
Abstract
Physical activity has been recently documented to play a fundamental physiological role in the regulation of autophagy in several tissues. It has also been reported that autophagy is required for exercise itself and for training-induced adaptations in glucose homeostasis. These autophagy-mediated metabolic improvements are thought to be largely dependent on the activation of the metabolic sensor PRKAA1/AMPK. However, it is unknown whether these important benefits stem from systemic adaptations or are due solely to alterations in skeletal muscle metabolism. To address this we utilized inducible, muscle-specific, atg7 knockout mice that we have recently generated. Our findings indicate that acute inhibition of autophagy in skeletal muscle just prior to exercise does not have an impact on physical performance, PRKAA1 activation, or glucose homeostasis. However, we reveal that autophagy is critical for the preservation of mitochondrial function during damaging muscle contraction. This effect appears to be gender specific affecting primarily females. We also establish that basal oxidative stress plays a crucial role in mitochondrial maintenance during normal physical activity. Therefore, autophagy is an adaptive response to exercise that ensures effective mitochondrial quality control during damaging physical activity.
Abbreviations:
- ACACA, acetyl-CoA carboxylase alpha
- ATG7, autophagy-related 7
- BNIP3, BCL2/adenovirus E1B 19 kDa interacting protein 3
- FDB, flexor digitorum brevis
- MAP1LC3A, microtubule-associated protein 1 light chain 3
- NAC, N-acetylcysteine
- PARK2, parkin RBR E3 ubiquitin protein ligase
- PRKAA1, protein kinase AMP-activated, alpha 1 catalytic subunit
- ROS, reactive oxygen species
- SQSTM1, sequestosome 1
- TA, tibialis anterior
- TMRM, tetramethylrhodamine, methyl ester
Introduction
Autophagy is an evolutionarily conserved system in which long-lived proteins, organelles, and fractions of cytoplasm are engulfed into double-membraned vesicles called autophagosomes.Citation1 The newly formed vesicles are subsequently delivered to lysosomes where they fuse leading to cargo degradation. Amino acids, and presumably lipids and sugars, are then released by the lysosomes to be recycled by the cell for the de novo synthesis of various molecules and organelles.Citation2-5 Autophagy plays a fundamental role in a variety of physiological and pathological processes ranging from cell proliferation, protein, and lipid homeostasis to neurodegeneration, muscle wasting, and cancer.Citation6 Although many facets of autophagy remain to be identified, it is now generally accepted that this process is crucial for health and longevity.Citation7
Autophagy is constantly ongoing at a low level in most tissues. However, under conditions of cellular stress, such as during limited nutrient availability, autophagy flux is elevated as a compensatory mechanism in an effort to sustain energetic demands and proper cellular function. Among the tissues with the highest basal autophagy fluxCitation8 are striated muscles, which also possess the capacity to dramatically enhance autophagosome biogenesis during catabolic conditions. Skeletal muscle is the most abundant tissue in mammals and plays an important role in locomotion and in the regulation of glucose and lipid metabolism. Indeed, skeletal muscle accounts for 80% of whole-body, insulin-mediated glucose utilization. Efficient autophagy is required for muscle mass maintenance, integrity, and proper turnover of nascent mitochondria.Citation9 Reactivation of deficient autophagy can also ameliorate the myopathic phenotype of several inherited muscle diseases including collagen VI deficiency and Duchenne muscular dystrophy.Citation10,11
Physical exercise is an important lifestyle practice that renders a multitude of beneficial adaptations to humans and rodents alike. Regular physical activity has been demonstrated to improve glucose and lipid homeostasis, maintain muscle mass, and delay aging.Citation12-17 For instance, 5 mo of exercise training are sufficient to completely reverse the premature aging phenotype of the mitochondrial DNA mutator mice, which possess a dysfunctional copy of the mitochondrial proofreading-exonuclease, POLG (poly[A] polymerase gamma).Citation18 Although the positive effects of exercise are undisputed, the underlying mechanisms are still under vigorous investigation.
We have previously revealed that an acute bout of exercise is sufficient to induce autophagy in skeletal muscle.Citation19 Others have further confirmed these findings reporting that physical activity-induced adaptations may be mediated by the activation of autophagy.Citation20,21 Mice with defective stress-induced autophagy, but proper basal autophagy, run significantly less on a treadmill than the wild types. Moreover, these mice do not obtain the same exercise-mediated benefits and are not protected from high fat diet-induced glucose intolerance. This supports the notion that exercise-induced metabolic rejuvenation occurs through the stimulation of autophagy. A look into the mechanisms behind these exercise-induced autophagy-mediated metabolic improvements, revealed PRKAA1 as the potential culprit.Citation20 PRKAA1 activation presumably leads to the upregulation of the glucose transporter, SLC2A4/GLUT4, at the muscle membrane, thus increasing the capacity for muscle glucose uptake. However, the signaling cascade responsible has not been illuminated yet. Moreover, these findings remain controversial as skeletal muscle–specific autophagy knockout mice show the exact opposite phenotype. These mice appear to have an improved metabolic profile and increased sensitivity to insulin, rendering them protected from diet-induced obesity.Citation21 These contrasting results may be due to the difference in tissue-specific vs. general autophagy disturbance and, therefore, highlight a potential cell autonomous regulation, which has yet to be investigated. Thus, whether it is whole body or muscle specific autophagy that is required to sustain contraction, maintain glucose homeostasis, and render exercise-induced benefits, remains unknown.
In order to address these important questions we used tamoxifen-inducible muscle-specific atg7 knockout mice.Citation9 In this model we deleted the atg7 gene acutely, just prior to exercise. We then monitored several parameters including physical performance, blood glucose levels, and related metabolites, as well as PRKAA1 activation and mitochondrial function. Our data indicate that autophagy inhibition does not have an impact on physical capability, PRKAA1 activation, or glucose homeostasis. However, we reveal autophagy as a key mechanism for muscle injury repair following damaging muscle contraction. Specifically, autophagy is necessary for the removal of mitochondria that are damaged by contraction. Moreover, we found that basal oxidative stress plays a crucial role in mitochondrial maintenance during normal physical activity.
Results
Autophagy is not required to sustain muscle contraction during physical activity
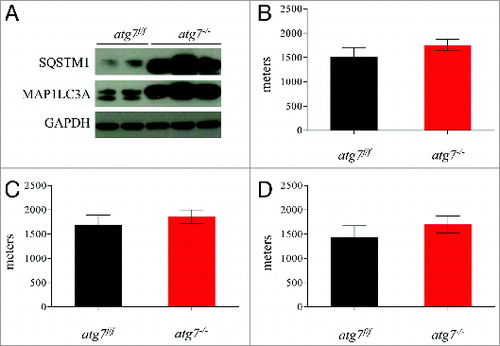
To address the role of skeletal muscle autophagy during physical activity we acutely deleted the atg7 gene in adult animals by treating inducible muscle-specific atg7 knockout mice with tamoxifen.Citation9 This inducible model was used in order to minimize the chance of any adaptations and compensations that occur with constitutive or conditional deletion of genes embryonically or at a very young age. The efficiency of atg7 deletion (atg7−/−) and of autophagy inhibition was confirmed by western blotting for MAP1LC3A/LC3 and SQSTM1/p62 (A). Tamoxifen-treated mice showed accumulation of MAP1LC3A-I and SQSTM1 protein (A) confirming that autophagosome formation and substrate delivery to lysosomes were efficiently inhibited. In order to investigate whether acute block of autophagy in muscle could affect exercise performance, atg7f/f and atg7−/− mice were exercised on a treadmill. We used a standard concentric exercise protocol while monitoring the maximum distance ran to exhaustion.Citation20 Surprisingly, we did not find any significant differences in running capacity between atg7f/f and atg7−/− (B). We then investigated whether gender had any effect on running performance, and once again no differences were found between the atg7f/f and atg7−/− mice (C and D). Morphological analysis did not show signs of inflammation or degeneration (data not shown). Thus, consistent with a recent report,Citation21 our findings further confirm that autophagy is not required to sustain muscle contraction during physical activity.
Figure 1. Autophagy is not required to sustain muscle contraction during physical activity. (A) Immunoblot for MAP1LC3A and SQSTM1 proteins on muscle extracts from inducible atg7f/f mice after tamoxifen treatment. (B) Histogram showing the mean maximum distance ran to exhaustion by atg7f/f and atg7−/− mice during acute concentric exercise (n = 16 each genotype). (C) Mean distance covered by females (n = 5 each genotype) and (D) males (n = 11 each genotype) after concentric exercise.
Autophagy is important during damaging contraction
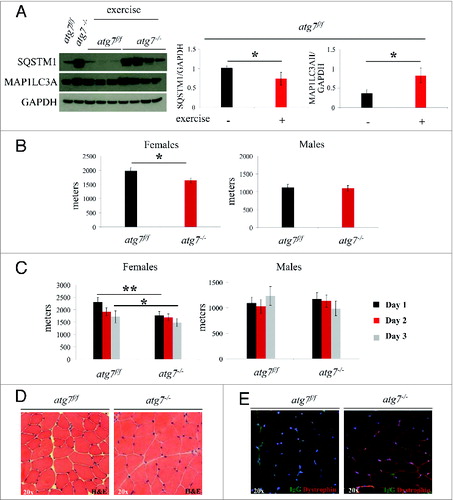
Since autophagy is important for effective protein and organelle turnover as well as for survival under cellular stress, we tested whether a damaging eccentric-type muscle contraction might unravel a novel role for autophagy during muscle repair postexercise. To address this, we performed a downhill running exercise to induce damaging eccentric contraction in atg7f/f and atg7−/− animals while recording maximal running distance achieved. We observed that autophagy was activated in response to eccentric exercise as indicated by the lipidation of MAP1LC3A and a decrease of SQSTM1 in the muscle of atg7f/f mice. Conversely, atg7−/− mice maintained their high levels of MAP1LC3A-I and SQSTM1 protein, confirming the efficient inhibition of autophagy (A). On average, autophagy-deficient mice ran less than wild types (Fig. S1). However, when we took gender into consideration we found that atg7-deficient females but not males ran less than their wild-type counterparts (B). Next, we investigated whether repeated bouts of eccentric exercise for 3 consecutive d would induce an additive detrimental effect on muscle performance, as a consequence of cumulative damage. Interestingly, only a trend for decreased physical performance was observed over time in both genotypes, suggesting that most of the damaging events took place during the first round of eccentric contraction (C). However, autophagy-deficient females continued to run less than their littermate controls after 3 d of exercise. Consistent with previous data, control males were not affected by the repetitive strain of eccentric contraction while autophagy-deficient males showed a trend for a decrease in performance over time. We next aimed to uncover some of the mechanisms responsible for this decrease in physical performance observed in females. Morphological analyses did not reveal any abnormalities, such as necrosis or inflammation, following exercise (D). To assess whether eccentric contraction caused damage to plasma membranes or altered electrolyte homeostasis, we stained myofibers for serum immunoglobulins. However, we did not find any positive staining inside myofibers, confirming that plasma membrane integrity was retained after eccentric contraction in both genotypes (E). Therefore, the impairment in performance of atg7−/− females is not due to major structural alterations.
Figure 2. Autophagy is required for eccentric exercise. (A) Representative immunoblots and quantification histograms for MAP1LC3A and SQSTM1 proteins on muscle extracts from inducible atg7f/f mice before and after eccentric exercise (n = 7 each conditions). (B and C) Histograms showing the mean maximum distance ran to exhaustion by atg7f/f and atg7−/− females and males (B) during an acute bout of eccentric exercise (n = 10 males each genotype and n = 10 females each genotype, *P < 0.05), (C) during 3 consecutive d of eccentric exercise (n = 10 males each genotype and n = 10 females each genotype, **P < 0.01; *P < 0.05). (D) Representative hematoxylin and eosin staining of tibialis anterior (TA) muscle cross sections from exercised atg7f/f and atg7−/− animals. No major morphological alterations such as inflammation, center-nucleated fibers are present in exercised muscles of both genotypes. (E) Representative images of IgG staining of cross-sections from exercised TA of atg7f/f and atg7−/− mice. No significant membrane permeabilization was found in either genotype after 3 d of eccentric exercise.
Autophagy is not required for PRKAA1 activation or for exercise-mediated glucose uptake
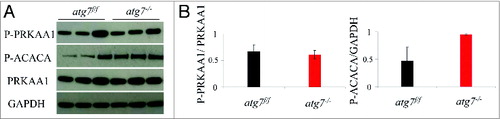
It has been reported that exercise-induced autophagy plays a critical role in PRKAA1 activation and glucose homeostasis.Citation20 It is therefore conceivable that the energy imbalance caused by exercise may explain the exercise intolerance observed in autophagy-deficient females. We first monitored the level of phospho-PRKAA1 and of its downstream target phospho-ACACA/ACC in exercised muscles, but no significant differences were observed between atg7f/f and atg7−/− mice (A and B).
Figure 3. Autophagy is not required for the phosphorylation of PRKAA1. (A) Representative immunoblots from exercised atg7f/f and atg7−/− females. (B) Histograms representing the densitometric quantification of immunoblots in (A). No significant differences in protein expression were observed (n = 3 each genotype).
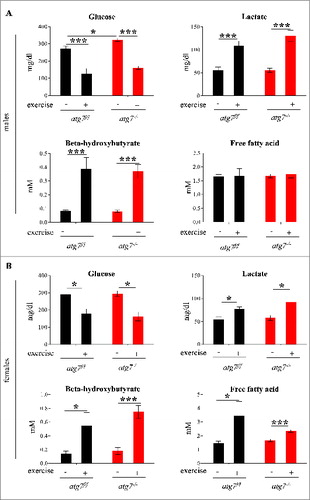
Next we compared the blood metabolic profiles of atg7f/f and atg7−/− mice both at rest and following exercise. Blood glucose was reduced following exercise along with a concomitant increase in blood lactate. Interestingly this metabolic profile was unaltered by the block in autophagy (). Indeed both males and females showed a decrease of glycemia and a concomitant increase of lactacidemia after exercise that did not differ between atg7f/f and atg7−/− animals (A and B). The only difference found was a slight elevation in blood glucose in atg7−/− males prior to exercise compared with controls. Both genders showed an increase in blood β-hydroxybutyrate after exercise, suggesting that ketones were produced and used during exhaustive contraction. However, there were no significant differences between wild-type and autophagy-deficient mice post exercise. Blood-free fatty acids were significantly increased after exercise in females but not in males. However, once again no differences were found between controls and knockout mice. Moreover, periodic acid-Schiff and Oil Red staining did not reveal any glycogen or lipid accumulation in atg7−/− mice (data not shown). Altogether, these data suggest that muscle autophagy is not required for metabolic regulation during exercise, as indicated by the lack of differences in PRKAA1 activation as well as glucose and lipid utilization.
Figure 4. Autophagy inhibition does not impair glucose homeostasis. (A and B) Histograms representing blood concentrations of metabolites in atg7f/f and atg7−/− animals. Analyses were done before and after 3 d of eccentric exercise in (A) males (n = 6 atg7f/f, n = 4 atg7−/−), and (B) females (n = 6 atg7f/f, n = 6 atg7−/−).
Autophagy is required to prevent the accumulation of dysfunctional mitochondria during damaging muscle contraction
Since autophagy is important for organelle quality control, we tested whether mitochondrial homeostasis was altered in wild-type and autophagy-deficient animals following exercise. Interestingly, flexor digitorum brevis (FDB) myofibers isolated from atg7−/− mice showed a significant increase in depolarized mitochondria following treatment with the F1F0-ATPase blocker, oligomycinCitation11 (A). While eccentric exercise did not affect mitochondrial membrane potential in wild-type animals, it exacerbated the percentage of depolarized fibers in atg7-deficient mice (B; Fig. S2). Males lacking autophagy also demonstrated fibers with depolarized mitochondria, however, to a much lower extent than females (pre exercise: 15% vs 35%; post exercise: 35% vs 54%, respectively) (Fig. S3). Next, we monitored whether, in the absence of autophagy, the depolarized fibers persist or continue to deteriorate. On the one hand, mitochondrial membrane potential remained normal 3 d after the last bout of eccentric exercise in fibers isolated from atg7f/f animals. On the other hand, the number of depolarized fibers continued to grow in autophagy-deficient mice (Fig. S4), indicating a deterioration in mitochondrial function.
Figure 5. Autophagy inhibition leads to accumulation of dysfunctional mitochondria and increase of oxidative stress during eccentric contraction. (A and B) Mitochondrial membrane potential as measured by TMRM fluorescence in isolated FDB muscle fibers from atg7f/f (top) and atg7−/− (bottom) female mice, (A) pre-exercise and (B) postexercise. Oligomycin (Olm) and the protonophore FCCP were added at the indicated time points. The percentage of depolarized fibers is shown on the bottom of the graphs. Fibers were considered depolarized if TMRM fluorescence decreased by 10% or more of the initial value following the addition of Olm. Each trace represents the TMRM fluorescence of a single fiber. (C) Overall protein carbonylation in exercised atg7f/f and atg7−/− muscles. Left panel: A representative immunoblot for carbonylated proteins. Right panel: Densitometric quantification of the carbonylated proteins. Postexercised atg7−/− mice show higher protein carbonylation than atg7f/f (n = 5 each genotype, *P < 0.05). (D) Mitochondrial ROS production. Mt-roGFP1 fluorescence was measured in single fibers of atg7f/f and atg7−/− (n = 3 each condition, P < 0.05).
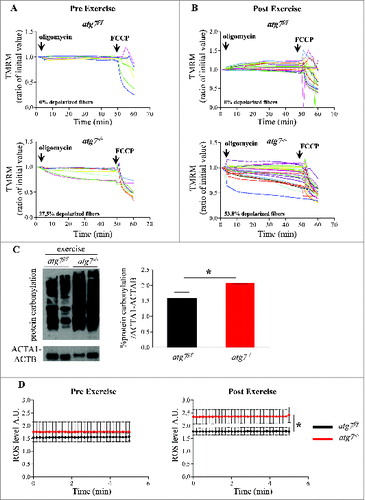
Because mitochondria are the main source and effectors of reactive oxygen species (ROS) in the cell, it is feasible that oxidative stress may play a role in the observed results. Therefore, we measured total protein carbonylation in exercised muscles. As expected, atg7 null muscles showed more carbonylated proteins than exercise-matched controls (C; Fig. S5). To determine whether mitochondria are the source of oxidative stress we transfected adult muscles with a mitochondrial targeted ROS sensor.Citation22,23 Under basal conditions, atg7 knockout mice tended to exhibit higher levels of ROS production when compared with controls. Following eccentric exercise, both genotypes generated more ROS, but atg7−/− mitochondria produced significantly higher levels than controls (D; Fig. S6).
Therefore, acute inhibition of autophagy in females led to the appearance of dysfunctional mitochondria, as indicated by an increase in depolarized fibers, higher oxidative stress, and reduced physical performance during eccentric contraction.
Antioxidant treatment does not improve physical performance of atg7 knockouts but instead blocks autophagy in controls, resulting in a deterioration of mitochondrial function and running capacity
Excessive oxidative stress has been documented to impair muscle function, which could potentially explain the reduced physical performance of atg7−/− mice. We therefore treated female atg7f/f and atg7−/− mice with the antioxidant N-acetylcysteine (NAC) for 6 wk, and then subjected them to eccentric exercise. The treatment successfully reduced protein carbonylation in atg7 knockout mice (Fig. S7). Surprisingly, NAC treatment severely impaired performance of atg7f/f but did not elicit any benefit in atg7−/− mice (). Indeed, the difference in running distance between atg7f/f and atg7−/− mice was abolished both at 1 and 3 d of exercise (A). To further investigate this unexpected effect in wild-type animals we monitored mitochondrial function in atg7f/f mice before and after exercise. Interestingly, NAC treatment resulted in impaired basal mitochondrial membrane potential in atg7f/f mice (B). Indeed, NAC-treated atg7f/f mice exhibited a percentage of depolarized fibers prior to exercise that was similar to that of exercised atg7−/− mice (compare B with B). Interestingly, exercise was able to slightly attenuate the number of depolarized fibers from 62% to 42%. As expected, NAC treatment elicited minor effects on atg7−/− mice, with the percentage of fibers with dysfunctional mitochondria being similar to untreated atg7 knockout mice (Fig. S8). To investigate whether the profound effect of NAC on physical performance was due to the prolonged inhibition of ROS, we treated mice with a ROS scavenger for just a few days before exercise. Moreover, to unravel the role of mitochondria-mediated ROS production we used Mito-TEMPO, an established mitochondria specific antioxidant.Citation24 Consistent with NAC treatment, acute inhibition of mitochondrial ROS resulted in lower performance in atg7f/f mice and did not improve the activity of atg7−/− mice (A). Similarly to NAC, Mito-TEMPO induced mitochondrial depolarization in control mice (B and C) and did not ameliorate the percentage of fibers with dysfunctional mitochondria in autophagy-deficient muscles (Fig. S9A and S9B).
Figure 6. NAC treatment impairs physical performance and mitochondria function in atg7f/f mice. (A) Mean maximal running distance after 1 (left) and 3(right) d of eccentric exercise with and without NAC treatment in atg7f/f and atg7−/− females (NAC-treated n = 6 atg7f/f, n = 4 atg7−/−). NAC treatment did not improve atg7−/− but rather worsened atg7f/f physical performance. (B) TMRM analysis of atg7f/f females pre-exercise (top) and postexercise (bottom) after NAC treatment. Mitochondrial ability to maintain membrane potential is compromised by prolonged antioxidant treatment (n > 15 per condition).
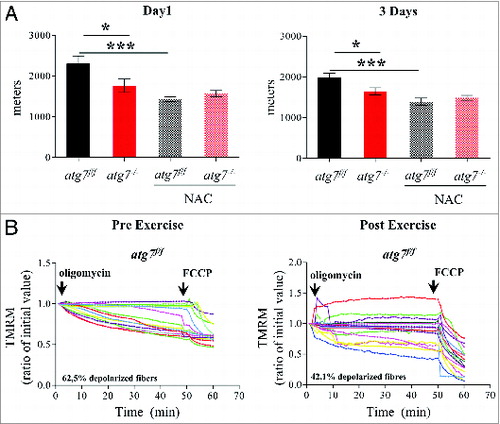
Figure 7. Mitochondria targeted antoxidant Mito-TEMPO impairs the physical performance and mitochondria function of atg7f/f mice. (A) Mean maximal running distance after 1 (left) and 3 (right) d of eccentric exercise with and without Mito-TEMPO treatment in atg7f/f and atg7−/− females (Mito-TEMPO-treated n = 3 atg7f/f, n = 3 atg7−/−). Mito-TEMPO treatment worsened atg7f/f physical performance. (B and C) TMRM analysis of atg7f/f females pre-exercise (B) and postexercise (C) after Mito-TEMPO treatment. Mitochondrial ability to maintain membrane potential is compromised by prolonged antioxidant treatment (n > 15 per condition). (D) Representative immunoblots for the activation of PRKAA1 and ACACA in control and exercised atg7f/f and atg7−/− females, following NAC treatment. (E) Histogram of the densitometric quantification of phospho-PRKAA1 corrected for its total content (n = 3 each condition, P < 0.05).
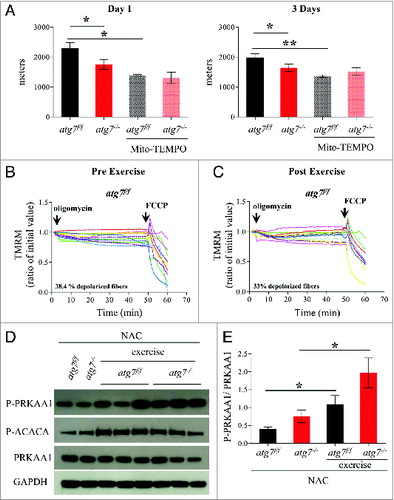
Next we investigated the potential signaling events triggered by the antioxidant treatment that may explain the mitochondrial dysfunction observed in control mice. We first monitored the activation of the metabolic master regulator PRKAA1 as it is heavily implicated in exercise autophagy and mitochondrial regulation and may act as a metabolic link between these events. Consistent with previous data, exercise triggered PRKAA1 and ACACA phosphorylation both in control and autophagy-deficient mice (D and E).
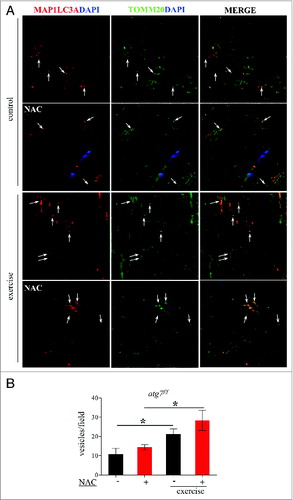
We therefore continued our efforts in search of the mechanisms responsible for the decrease in physical and mitochondrial performance in NAC-treated atg7f/f mice. When we monitored autophagy in control atg7f/f animals we found that antioxidant treatment led to a decrease of MAP1LC3A-II and an increase of SQSTM1 (A and B). MAP1LC3A lipidation after eccentric exercise was slightly induced in NAC-treated mice. Moreover, exercise resulted in a reduction of SQSTM1 in NAC-treated mice, suggesting that antioxidant treatment reduced basal autophagy but that exercise was able to partially reactivate autophagic signaling (A and C). To further address this point we purified mitochondria from muscles of exercised atg7f/f mice and monitored the presence of lipidated MAP1LC3A, SQSTM1, BNIP3, and PARK2/PARKIN in the enriched mitochondrial fraction. Both NAC and vehicle-treated exercised muscles showed a recruitment of MAP1LC3A-II, SQSTM1, and BNIP3 to the mitochondria (D). However, PARK2 showed minor changes after exercise only in the untreated control group. These data suggest that BNIP3- and SQSTM1-mediated mitophagy might be affected by both NAC treatment and exercise. To further confirm this point we immunostained longitudinal cryosections for TOMM20/TOM20, a mitochondrial protein, along with MAP1LC3A and quantified the colocalization of these markers (A), which would indicate mitophagy in progress. Exercise triggered mitophagy in both NAC-treated and untreated mice (B) which explains why exercise was able to partially restore the abnormal mitochondrial membrane potential in NAC-treated atg7f/f mice. However, exercise-induced mitophagy was not sufficient to clear the dysfunctional mitochondria that persisted during NAC treatment. Altogether, these findings underline the critical role of autophagy in the maintenance of proper mitochondrial function. Moreover, we reveal an important physiological role of oxidative stress for basal autophagy regulation in skeletal muscle.
Figure 8. NAC treatment reduces basal autophagy in atg7f/f mice. (A) Representative western blots for SQSTM1 and MAP1LC3A-I/MAP1LC3A-II pre-exercise and postexercise in atg7f/f mice in the presence or absence of NAC. (B and C) Histograms representing the densitometric quantification of (B) MAP1LC3A-II and (C) SQSTM1 (n = 5 each condition, P < 0.05). (D) Representative immunoblots showing the presence of SQSTM1, MAP1LC3A-II, BNIP3, PARK2, COX4I1/COXIV on isolated mitochondria from pre-exercised and postexercised atg7f/f muscles in the presence or absence of NAC. GAPDH immunoblot indicates the purity of the enriched mitochondrial fraction.
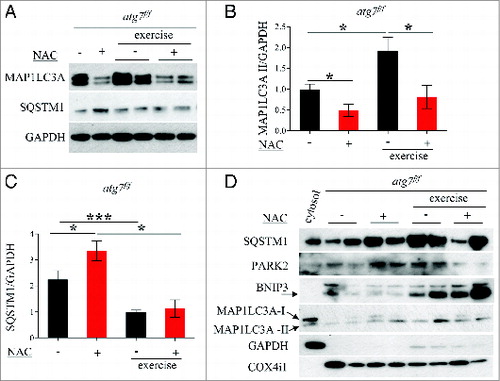
Figure 9. Exercise activates mitophagy in control and NAC-treated mice. (A) Representative confocal images of immunofluorescence staining for MAP1LC3A (red) and the mitochondrial protein TOMM20 (green) of longitudinal cryosections of pre-exercised and postexercised in atg7f/f animals with or without NAC treatment. (B) Quantification of MAP1LC3A and TOMM20 double-positive vesicles (n = 7 each condition, *P < 0.05).
Discussion
Autophagy is an important cellular recycling mechanism that is constitutively active and can be further induced by several stress stimuli. Exercise has been recently implicated in the regulation of autophagy in different tissues.Citation20,25 Indeed, we have previously demonstrated that acute exercise activates autophagosome formation in skeletal muscle. However, the mechanisms responsible for this activation have not been fully elucidated. Whether autophagy is required for contraction or is an indirect consequence of the metabolic changes that take place during physical activity is still under debate. Here, we employed a genetic approach to establish the role of autophagy in skeletal muscle during physical activity. Our data highlight a vital role for autophagy in preserving normal muscle function following damage induced by eccentric contraction. It has previously been reported that autophagy is required for proper energy provision and glucose homeostasis during muscle contraction potentially through PRKAA1 and its downstream targets.Citation20 However, another study reached the opposite conclusion indicating that autophagy inhibition results in an improved blood glucose profile.Citation21 Therefore, no general consensus exists as to the metabolic impact of autophagy during exercise and whether this effect is cell-autonomous.
In order to address these issues, we used the tamoxifen-inducible muscle-specific atg7 knockout miceCitation9,26 that we have recently generated. With this model we were able to acutely delete atg7 specifically in skeletal muscles just prior to exercise. In our hands, acute inhibition of autophagy in skeletal muscle did not have an impact on physical performance, glucose homeostasis, or PRKAA1 signaling. In accordance with our data, a recent report where mice heterozygous for BECN1/BECLIN1 were subjected to exercise also found no differences in running capacity,Citation27 providing further evidence that autophagy is not required during an acute bout of exercise. The same study also demonstrated that the absence of exercise-induced autophagy results in a lack of exercise training-induced adaptations.Citation27 The conclusions of that study were that autophagy is required for chronic exercise-induced muscular adaptations which are mediated by mitochondrial turnover and remodeling.Citation27 Here we demonstrate that indeed autophagy is critical for mitochondrial quality control and its absence results in reduced ability of the mitochondria to maintain membrane potential. Interestingly, this effect is gender specific where females are more affected than males. These data are in line with the concept that females are more prone to muscle wasting during catabolic conditions. Alterations in the pathways controlling protein and organelle turnover may manifest more rapidly in females than in males. It is plausible that different exercise regimens or a more chronic autophagy inhibition may unravel alterations in muscle performance in males as well. Further research into gender difference is required in order to discern the mechanism responsible for male protection and female predisposition.
Another interesting observation is that the reactive oxygen species produced by mitochondria are important for mitochondrial quality control. The notion that antioxidants are detrimental for exercise-induced benefits has already been reported in humans, although the mechanisms remain unclear.Citation28 In our hands, prolonged NAC treatment led to autophagy inhibition, which is in line with the lack of exercise-induced adaptations observed with antioxidant treatment in humans. Inhibition of autophagy caused appearance of dysfunctional mitochondria culminating in decreased muscle performance. However, this effect was not due to increased oxidative stress in the autophagy-deficient mice as NAC treatment did not improve the impaired muscle function in these animals. Moreover, since PRKAA1 signaling and blood glucose and lipid levels did not correlate with reduced autophagy, other factors must be involved in mediating impaired performance in knockout females. One possibility is mitochondria-derived metabolites, which play an important role in signaling and could therefore impinge on cellular adaptations to stress. For instance, α-ketoglutarate-derived glutamine inhibits MTOR activity and activates autophagy.Citation29,30 Similarly, accumulation of fumarate and succinate as a consequence of alterations in the Krebs cycle are linked to cancer.Citation31-33 Indeed, impaired mitochondrial function in muscle-specific atg7 knockouts is protective from diet-induced obesity through the induction of a mitochondrial stress mitokine, known as FGF21.Citation21 Therefore, it is feasible that dysfunctional mitochondria generate metabolites or myokines that communicate with pathways important for myofiber function during exhausting and damaging muscle contraction. In this scenario autophagy is critical for mitochondrial quality control and for the regulation of proper metabolite turnover. In conclusion, our findings highlight the crucial role of autophagy in the maintenance of mitochondrial function but not in PRKAA1 activation, exercise-dependent glucose homeostasis, or energy provision to fuel muscle contraction. Moreover, the detrimental effects of antioxidants on physical performance underline the important role of mild oxidative stress in regulating autophagy and mitochondrial network in skeletal muscles.
Materials and Methods
Muscle-specific atg7 HSA knockout mice, antioxidant treatment, and exercise
In this study 6-mo-old, tamoxifen-inducible muscle-specific atg7 null (atg7−/−) mice were used; their generation was previously described.Citation9 Mice performed concentric exercise on a treadmill (Biological Instruments, LE 8710 Panlab Technology 2B), with 10° incline, according to the protocol of acute exercise previously described.Citation20 The eccentric training protocol consisted of 3 d of treadmill running to exhaustion, with a 10° decline. Total running distance was recorded for each mouse. All procedures are specified in the projects approved by the Italian Ministero Salute, Ufficio VI (authorization numbers C65).
A group of females was treated with NAC (Sigma, A9165), for 6 wk. We used 1% NAC drinking water for 5 wk and 2% NAC drinking water for the last week. The treatment was also maintained during exercise training. A second group of females was treated with Mito-TEMPO (Enzo Life Science, ALX-430-150-M005) at a dose of 1.4 mg/kg, and was administered through an intraperitoneal injection every day for 7 d.
Immunoblotting
Cryosections of frozen tibialis anterior (TA) muscles were lysed in a buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 10 mM MgCl2, 0.5 mM DTT, 1 mM EDTA, 10% glycerol, 2% SDS, 1% Triton X-100, Complete protease inhibitor cocktail (Roche, 11-836-145001) and phosphatase inhibitor cocktail (Sigma, P5726). The samples were immunoblotted as previously describedCitation34 and visualized with SuperSignal west pico chemiluminescent substrate (Pierce, 34080). The following primary antibodies were used: rabbit anti-MAP1LC3A (Sigma, L7543), anti-SQSTM1 (Sigma, P0067), anti-phospho-PRKAA1(Thr 172) (Cell Signaling Technology, 2535), anti-PRKAA1 (Cell Signaling Technology, 4182), anti-phospho-ACACA (Cell Signaling Technology, 3661), anti-PARK2 (Santa Cruz Biotechnology, sc-30130), anti-BNIP3 (Cell Signaling Technology, 3769), anti-COX4Il (Abcam, ab14744), anti-GAPDH (Abcam, ab8245), anti-ACTA1/ACTB (Sigma, A4700). Mouse and rabbit HRP-conjugated antibody were from Bio-Rad (170-6516, 170-6515). Quantification analyses were performed with ImageJ Software and all values were normalized for the signal of the housekeeping protein GAPDH.
Histology and fluorescence microscopy
Cryosections of TA were stained for hematoxylin and eosin (H&E). Immunofluorescence staining was performed on cryosections as previously describedCitation9 and then monitored with a fluorescence microscope. The following primary antibodies were used: anti-DMD/DYSTROPHIN (Abcam, ab15277), rabbit anti-MAP1LC3A (Cell Signaling Technology, 27755), anti-TOMM20 (Santa Cruz Biotechnology, sc-11415). Mouse-Alexa 488 was from Life Technologies (A11001) and rabbit-Cy3 was from Jackson Immunoresearch (111-165-003).
Blood metabolites quantification
Blood samples were collected at 3 time points: before exercise and immediately after the last bout of exercise. Blood was collected from the orbital sinus in heparin-coated Pasteur pipettes and centrifuged immediately after collection. Plasma samples were kept at −20°C until dosing.
Free fatty acids (FFAs) and β-hydroxybutyrate were dosed using an automated spectrophotometer Cobas Fara II (Roche) according to the manufacturer's instruction.
Blood glucose and lactate levels were measured with an YSI 2300 STAT Plus™ glucose and lactate analyzer (YSI Life Sciences, Yellow Springs, OH) according to the manufacturer's instruction.
Single fibers mitochondrial membrane potential analyses
Mitochondrial membrane potential was measured in single fibers isolated from flexor digitorum brevis muscles. Mitochondrial membrane potential was measured by epifluorescence microscopy based on the accumulation of TMRM fluorescence as previously described.Citation35
Protein carbonyls detection
Carbonylation of muscle proteins were detected by using the OxyBlot protein oxidation detection kit (Millipore, s7150).Citation9 Quantification analysis was performed with ImageJ Software and all values were normalized for the housekeeping proteins ACTA1-ACTB/PAN ACTIN.
Mitochondrial oxidative stress measurement
Mt-roGFP1, which measures the thiol/disulfide equilibrium in the mitochondrial matrix, was used as an indicator of mitochondrial redox status.Citation22,23 Adult FDB muscles were transfected by electroporation with mt-roGFP1 plasmid. After 8 d of transfection single muscle fibers were isolated from control and exercised mice. Mt-roGFP1 fluorescence (excitation: 405 and 480 nm, emission: 535 nm, 20× objective) was measured for 5 min every 10 s. The ratio of fluorescence intensities (exc 405/480) was determined by ImageJ Software.
Mitochondrial isolation
Mitochondria were isolated from quadriceps using differential centrifugation, as previously described.Citation36 Briefly, muscles were minced on ice and homogenized using a Teflon pestle and mortar, and suspended in mitochondrial isolation buffer (MIB) (250 mM Sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl, 1 mM EDTA, 1 mM EGTA) supplemented with protease and phosphatase inhibitors. The homogenate was then centrifuged at 1,000 g for 10 min at 4°C to pellet the nuclei. The supernatant fraction was recentrifuged at 16,000 g for 20 min at 4°C to pellet the mitochondria. The mitochondrial pellet was washed twice and finally resuspended in a one-fold dilution of MIB. Mitochondria were subsequently sonicated to yield the enriched mitochondrial fraction. Protein concentrations within the samples were determined using the Bradford method (Bio-Rad, 500-0006).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental data for this article can be accessed on the publisher's website.
Supplementary Material.pdf
Download PDF (340.3 KB)Acknowledgments
We thank Mattia Albiero, Elisabetta Iori, and Maria Cristina Marescotti for assistance with the metabolites analyses and Rubén Quintana Cabrera for mt-roGFP construct. This work was supported from Telethon-Italy (TCP04009), from ERC (282310-MyoPHAGY), from the European Union (MYOAGE, contract: 223576 of FP7), from Leducq Foundation and from Italian Ministry of Education (MiUR) (PRIN 2010/2011).
References
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/10.1016/j.cell.2011.10.026
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011; 334:678-83; PMID:22053050; http://dx.doi.org/10.1126/science.1207056
- Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ 2013; 20:3-11; PMID:22595754; http://dx.doi.org/10.1038/cdd.2012.63
- Nascimbeni AC, Fanin M, Masiero E, Angelini C, Sandri M. Impaired autophagy contributes to muscle atrophy in glycogen storage disease type II patients. Autophagy 2012; 8:1697-700; PMID:22940840; http://dx.doi.org/10.4161/auto.21691
- Nascimbeni AC, Fanin M, Masiero E, Angelini C, Sandri M. The role of autophagy in the pathogenesis of glycogen storage disease type II (GSDII). Cell Death Differ 2012; 19:1698-708; PMID:22595755; http://dx.doi.org/10.1038/cdd.2012.52
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/10.1016/j.cell.2007.12.018
- Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell 2011; 146:682-95; PMID:21884931; http://dx.doi.org/10.1016/j.cell.2011.07.030
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11; PMID:14699058; http://dx.doi.org/10.1091/mbc.E03-09-0704
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab 2009; 10:507-15; PMID:19945408; http://dx.doi.org/10.1016/j.cmet.2009.10.008
- De Palma C, Morisi F, Cheli S, Pambianco S, Cappello V, Vezzoli M, Rovere-Querini P, Moggio M, Ripolone M, Francolini M, et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis 2012; 3:e418; PMID:23152054; http://dx.doi.org/10.1038/cddis.2012.159
- Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 2010; 16:1313-20; PMID:21037586; http://dx.doi.org/10.1038/nm.2247
- Melov S, Tarnopolsky MA, Beckman K, Felkey K, Hubbard A. Resistance exercise reverses aging in human skeletal muscle. PLoS One 2007; 2:e465; PMID:17520024; http://dx.doi.org/10.1371/journal.pone.0000465
- Fontana L, Klein S, Holloszy JO. Effects of long-term calorie restriction and endurance exercise on glucose tolerance, insulin action, and adipokine production. Age (Dordr) 2010; 32:97-108; PMID:19904628; http://dx.doi.org/10.1007/s11357-009-9118-z
- Sandri M, Barberi L, Bijlsma AY, Blaauw B, Dyar KA, Milan G, Mammucari C, Meskers CG, Pallafacchina G, Paoli A, et al. Signalling pathways regulating muscle mass in ageing skeletal muscle: the role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology 2013; 14:303-23; PMID:23686362; http://dx.doi.org/10.1007/s10522-013-9432-9
- Schiaffino S, Dyar KA, Ciciliot S, Blaauw B, Sandri M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J 2013; 280:4294-314; PMID:23517348; http://dx.doi.org/10.1111/febs.12253
- Coen PM, Jubrias SA, Distefano G, Amati F, Mackey DC, Glynn NW, Manini TM, Wohlgemuth SE, Leeuwenburgh C, Cummings SR, et al. Skeletal muscle mitochondrial energetics are associated with maximal aerobic capacity and walking speed in older adults. J Gerontol A Biol Sci Med Sci 2013; 68:447-55; PMID:23051977; http://dx.doi.org/10.1093/gerona/gls196
- Toledo FG, Goodpaster BH. The role of weight loss and exercise in correcting skeletal muscle mitochondrial abnormalities in obesity, diabetes and aging. Mol Cell Endocrinol 2013; 379:30-4; PMID:23792186; http://dx.doi.org/10.1016/j.mce.2013.06.018
- Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, et al. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A 2011; 108:4135-40; PMID:21368114; http://dx.doi.org/10.1073/pnas.1019581108
- Grumati P, Coletto L, Schiavinato A, Castagnaro S, Bertaggia E, Sandri M, Bonaldo P. Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy 2011; 7:1415-23; PMID:22024752; http://dx.doi.org/10.4161/auto.7.12.17877
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012; 481:511-5; PMID:22258505; http://dx.doi.org/10.1038/nature10758
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim H, Hur KY, Kim HK, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 2013; 19:83-92; PMID:23202295; http://dx.doi.org/10.1038/nm.3014
- Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem 2004; 279:22284-93; PMID:14985369; http://dx.doi.org/10.1074/jbc.M312847200
- Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem 2004; 279:13044-53; PMID:14722062; http://dx.doi.org/10.1074/jbc.M312846200
- Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 2010; 107:106-16; PMID:20448215; http://dx.doi.org/10.1161/CIRCRESAHA.109.214601
- He C, Sumpter R Jr., Levine B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 2012; 8:1548-51; PMID:22892563; http://dx.doi.org/10.4161/auto.21327
- Masiero E, Sandri M. Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy 2010; 6:307-9; PMID:20104028; http://dx.doi.org/10.4161/auto.6.2.11137
- Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS, Hoehn KL, Yan Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J 2013; 27:4184-93; PMID:23825228; http://dx.doi.org/10.1096/fj.13-228486
- Ristow M, Zarse K, Oberbach A, Klöting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Blüher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A 2009; 106:8665-70; PMID:19433800; http://dx.doi.org/10.1073/pnas.0903485106
- Sandri M. FOXOphagy path to inducing stress resistance and cell survival. Nat Cell Biol 2012; 14:786-8; PMID:22854812; http://dx.doi.org/10.1038/ncb2550
- van der Vos KE, Eliasson P, Proikas-Cezanne T, Vervoort SJ, van Boxtel R, Putker M, van Zutphen IJ, Mauthe M, Zellmer S, Pals C, et al. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat Cell Biol 2012; 14:829-37; PMID:22820375; http://dx.doi.org/10.1038/ncb2536
- Frezza C, Zheng L, Folger O, Rajagopalan KN, MacKenzie ED, Jerby L, Micaroni M, Chaneton B, Adam J, Hedley A, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011; 477:225-8; PMID:21849978; http://dx.doi.org/10.1038/nature10363
- Sciacovelli M, Guzzo G, Morello V, Frezza C, Zheng L, Nannini N, Calabrese F, Laudiero G, Esposito F, Landriscina M, et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab 2013; 17:988-99; PMID:23747254; http://dx.doi.org/10.1016/j.cmet.2013.04.019
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013; 496:238-42; PMID:23535595; http://dx.doi.org/10.1038/nature11986
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004; 117:399-412; PMID:15109499; http://dx.doi.org/10.1016/S0092-8674(04)00400-3
- Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J 2010; 29:1774-85; PMID:20400940; http://dx.doi.org/10.1038/emboj.2010.60
- Vainshtein A, Kazak L, Hood DA. Effects of endurance training on apoptotic susceptibility in striated muscle. J Appl Physiol (1985) 2011; 110:1638-45; PMID:21474699; http://dx.doi.org/10.1152/japplphysiol.00020.2011