
Abstract
CHDH (choline dehydrogenase) is an enzyme catalyzing the dehydrogenation of choline to betaine aldehyde in mitochondria. Apart from this well-known activity, we report here a pivotal role of CHDH in mitophagy. Knockdown of CHDH expression impairs CCCP-induced mitophagy and PARK2/parkin-mediated clearance of mitochondria in mammalian cells, including HeLa cells and SN4741 dopaminergic neuronal cells. Conversely, overexpression of CHDH accelerates PARK2-mediated mitophagy. CHDH is found on both the outer and inner membranes of mitochondria in resting cells. Interestingly, upon induction of mitophagy, CHDH accumulates on the outer membrane in a mitochondrial potential-dependent manner. We found that CHDH is not a substrate of PARK2 but interacts with SQSTM1 independently of PARK2 to recruit SQSTM1 into depolarized mitochondria. The FB1 domain of CHDH is exposed to the cytosol and is required for the interaction with SQSTM1, and overexpression of the FB1 domain only in cytosol reduces CCCP-induced mitochondrial degradation via competitive interaction with SQSTM1. In addition, CHDH, but not the CHDH FB1 deletion mutant, forms a ternary protein complex with SQSTM1 and MAP1LC3 (LC3), leading to loading of LC3 onto the damaged mitochondria via SQSTM1. Further, CHDH is crucial to the mitophagy induced by MPP+ in SN4741 cells. Overall, our results suggest that CHDH is required for PARK2-mediated mitophagy for the recruitment of SQSTM1 and LC3 onto the mitochondria for cargo recognition.
Abbreviations:
- ANT, adenine nucleotide translocator
- Baf, bafilomycin A1
- CCCP, carbonyl cyanide m-chlorophenylhydrazone
- CHX, cycloheximide
- FB1, FAD/NAD (P)-binding domain 1
- FB2, FAD/NAD (P)-binding domain 2
- IM, inner membrane
- IMS, inter-membrane space
- Mat, matrix
- MPP+, 1-methyl-4-phenylpyridinium
- MTS, mitochondrial targeting sequence
- OM, outer membrane
- PB1, Phox and Bem 1 domain
- PD, Parkinson disease
- PK, proteinase K
- RD, FAD-linked reductase domain
Introduction
CHDH is an enzyme catalyzing the dehydrogenation of choline to betaine aldehyde in the mitochondria.Citation1 Betaine aldehyde is subsequently converted to betaine, an important methyl donor for the synthesis of methionine from homocysteine.Citation2,3 CHDH has been highlighted by Ma et al., who report that CHDH is overexpressed in patients with a favorable outcome associated with tamoxifen monotherapy in a cohort of early-stage estrogen receptor-positive breast cancer patients.Citation4 In addition, a population-based study shows that CHDH expression correlates with ESR (estrogen receptor) and HER2 status and is regulated by estrogen in an ESR-dependent manner.Citation5 A recent study demonstrates that CHDH plays a pivotal role in mitochondrial function in several tissues, with the most striking effects in sperm of chdh−/− mice.Citation6 Several single nucleotide polymorphisms (SNPs) of CHDH are common in humans. Between 38% and 42% of the population is carrying one allele of the rs12676 SNP and they show liver and muscle dysfunction when fed a choline-deficient diet.Citation7,8 Moreover, this SNP affects the motility and mitochondrial morphology of human sperm.Citation9
Mitochondrial quality control is performed within certain steps along with the level of damage.Citation10 Cells must eliminate mitochondria when they fail to repair their components using proteolytic systems or ROS regulation. Autophagic machineries are employed in this type of organellar-level mitochondrial degradation, termed ‘mitophagy,’ which is critical for the maintenance of cellular homeostasis and survival.Citation11 Autophagy is responsible for the initial flux of mitophagy, and the 2 processes share the same pathway, including ULK1, ATG13,Citation12,13 MAP1LC3B/LC3B (refer to LC3 hereafter), and BECN1/Beclin1Citation14 with a controversy in case of BECN1.Citation15 Inhibition of MTORC1 is also required for autophagy and mitophagy.Citation16 However, it remains unclear which critical factor targets autophagic machineries to the mitochondria. In recent years, PINK1/PARK6 (PTEN-induced putative kinase 1) and PARK2/parkin (parkin RBR E3 ubiquitin protein ligase) have emerged as a pivotal paradigm of mitophagic process. These genes are mutated in autosomal recessive Parkinson disease (PD)Citation17,18 and contribute to regulation of mitochondrial integrity, including mitophagy.Citation14 PINK1 has a short half-life and is rapidly degraded under steady-state conditions by proteases located in the inner membrane (IM) of mitochondria. However, once the mitochondrial membrane potential has dissipated, it becomes stable and recruits PARK2.Citation19,20 The ubiquitin-ligase activity of PARK2 is then activatedCitation21 and promotes ubiquitination of mitochondrial proteins, driving mitophagy. Although the mechanism of mitophagy has been intensively studied,Citation22-24 the mitophagy-specific factors remain unclear.
In this study, we assess the role of CHDH in PARK2-mediated mitophagy. Beyond its well-known role in betaine production, we demonstrate that CHDH is required for mitophagy in which CHDH interacts with SQSTM1, a mitophagic adaptor molecule, and subsequently facilitates the recruitment of LC3 into the mitochondria.
Results
CHDH is required for PARK2-mediated mitophagy
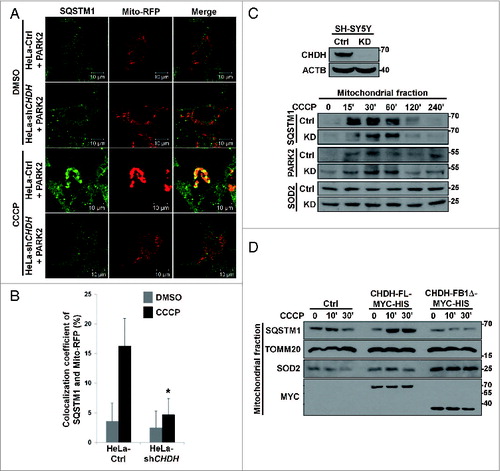
First, we examined the necessity of CHDH for mitophagy induced by carbonyl cyanide m-chlorophenylhydrazone (CCCP), a mitochondrial uncoupler that triggers mitochondrial depolarization and mitophagy.Citation25 By using CHDH shRNA, we generated stable HeLa cells that showed reduced expression of CHDH (HeLa-shCHDH cells) (B, upper). As has been previously reported,Citation26 immunofluorescence analysis revealed that CCCP treatment induced the degradation of TOMM20-positive mitochondria in the presence of PARK2 in control HeLa cells (A, left), which contain no endogenous PARK2. However, knockdown of CHDH expression impeded the degradation of mitochondria (A, right). Mitochondrial degradation did not occur in the absence of PARK2, consistent with the previous report.Citation11,27 When flow cytometry analysis was employed to measure the total fluorescence intensity of Mito-RFP, the results of CCCP exposure showed that clearance of Mito-RFP-positive mitochondria was also retarded in HeLa-shCHDH cells (C). Similarly, quantification of the degradation of mitochondrial DNA and proteins revealed that quantities of MT-CYB/cytochrome b DNA and mitochondrial proteins, such as SOD2/MnSOD and TOMM20, were less reduced in HeLa-shCHDH cells than in control cells during mitophagy (D and E). These results indicate that CHDH is required for the proper functioning of PARK2-mediated mitophagy in HeLa cells.
Figure 1. CHDH is required for CCCP-induced and PARK2-mediated mitophagy. (A and B) HeLa-Control (Ctrl) and HeLa-CHDH knockdown (HeLa-shCHDH) stable cells were transfected with FLAG-Ctrl or FLAG-PARK2 and then exposed to DMSO or 10 μM CCCP for 24 h. After staining with TOMM20 antibody and Hoechst 33258 dye, cells were examined under a confocal microscope. Scale bar: 10 μm (A). Expression of CHDH in HeLa-Ctrl and HeLa-shCHDH cells was assessed using western blot analysis (B, upper). The signal intensity of TOMM20 in HeLa-Ctrl and HeLa-shCHDH cells (n > 50) exposed to CCCP and expressing PARK2 in (A) were quantified using the ImageJ program and are represented as bars with the mean ± SD, *P < 0.05 (B, lower). (C) HeLa-Ctrl and HeLa-shCHDH cells were transfected with PARK2 and Mito-RFP and treated with DMSO or 10 μM CCCP. After 24 h, cells were fixed and subjected to flow cytometry analysis, as described in Materials and Methods. (D and E) HeLa-Ctrl and HeLa-shCHDH cells were transfected with PARK2 and then left untreated or exposed to 10 μM CCCP for 24 h. Mitochondrial DNA and proteins were extracted and subjected to PCR analysis using synthetic primers for cytochrome b (MT-CYB) and ACTB (D), and western blot analysis using SOD2 and TOMM20 antibodies (E), respectively. (F and G) SN4741 cells were transiently transfected with Mito-RFP and either pSuper (Ctrl) or CHDH shRNA and then exposed to DMSO or 20 μM CCCP. After 24 h, cells were stained with Hoechst 33258 dye and examined under a confocal microscope. Scale bar: 10 μm (F). The signal intensities of Mito-RFP in CCCP-treated control and CHDH knockdown cells were quantified as in (B) and are represented as% of cells (n > 50) with cleared mitochondria. Bars represent the mean ± SD, *P < 0.001 (G). TUBA, tubulin, α.
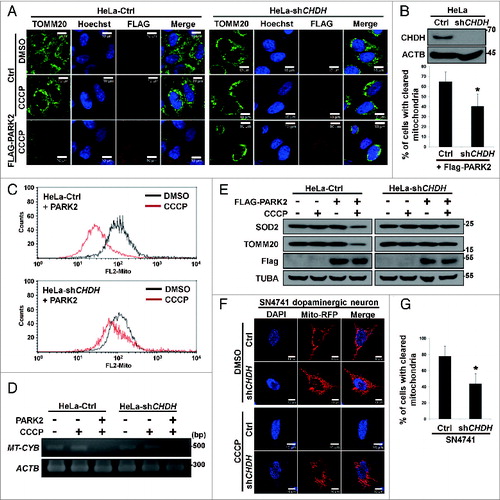
Given that PARK2-mediated mitophagy is related to PD, we also examined the role of CHDH in mitophagy of SN4741 neuronal cells, a clonal substantia nigra dopaminergic neuronal murine cell line which expresses PARK2.Citation28 Using western blot analysis, we confirmed the expression of PARK2 in SN4741 cells (data not shown). Exposure of SN4741 cells to CCCP resulted in clearance of Mito-RFP-positive mitochondria (F and G). On the contrary, knockdown of CHDH expression significantly impaired the clearance of Mito-RFP-positive mitochondria in SN4741 cells (F and G), suggesting that CHDH is required for PARK2-mediated mitophagy not only in HeLa cells but also in SN4741 dopaminergic neuronal cells.
We also examined the effect of CHDH overexpression on mitophagy. When HeLa cells expressing PARK2 were exposed to CCCP, colocalization of GFP-LC3 and Mito-RFP was enhanced by CHDH overexpression (A). Interestingly, CHDH overexpression followed by CCCP treatment also increased the colocalization of mitochondria and LC3. When we analyzed mitochondrial DNA and protein quantities following CCCP treatment, we found that clearance of MT-CYB DNA and mitochondrial COX4I1/COX-IV protein was accelerated by CHDH overexpression (B and C). Consistent with this result, the fluorescence intensity of Mito-GFP was rapidly dissipated by CHDH overexpression in HEK293T cells during mitophagy, which is almost equivalent to that by PINK1 overexpression (D). These results indicate that CHDH overexpression enhances CCCP-induced clearance of mitochondria. However, expression level of CHDH did not affect the stability of PINK1 protein, although CCCP treatment stabilized PINK1 in mitochondria as previously reported (Fig. S1A and S1B).Citation29,30 In addition, PINK1 knockdown attenuated CCCP-induced mitophagy in both control cells and cells overexpressing CHDH. However, the overexpression of CHDH still enhanced mitophagy in PINK1 knockdown cells (Fig. S1C).
Figure 2. Overexpression of CHDH accelerates mitochondrial clearance independent of its enzymatic activity. (A) HeLa-Ctrl and HeLa-CHDH cells were cotransfected with GFP-LC3, Mito-RFP and either GFP control vector (Ctrl) or PARK2 and then incubated with 10 μM CCCP. After 2 h, cells were analyzed under a confocal microscope; the colocalization coefficient (%) of GFP-LC3 and Mito-RFP is shown with bars representing the mean ± SD, *P < 0.001. (B and C) HeLa-Ctrl and HeLa-CHDH cells were left untreated or exposed to 10 μM CCCP for 30 h (B) or for the indicated times (C). Mitochondrial DNA (MT-CYB) and proteins (CHDH, COX4I1) were extracted and analyzed by PCR (B) and western blotting (C), respectively. (D) HEK293T cells were cotransfected with Mito-GFP and pcDNA (Ctrl), CHDH + PARK2 or PINK1 + PARK2. Following treatment with DMSO or 100 μM CCCP for 2 h, the fluorescence of cells was measured, as described in Materials and Methods. The signal of Mito-GFP in control extracts is fixed as 100 and the relative ratio to the control is indicated as the mean ± SD, *P < 0.05. (E) Schematic of CHDH full-length (FL) and deletion mutants (FB1Δ, RDΔ, and FB2Δ). (F) The colocalization coefficient (%) of GFP-LC3 and Mito-RFP in the presence of CHDH-FL or deletion mutants was analyzed in the transfected cells under a confocal microscope and is represented as a bar graph with the mean ± SD. Cells were treated with 10 μM CCCP for 4 h. *P < 0.001. (G) HEK293T cells were transfected with CHDH or a series of deletion mutants and then subjected to LC-MS for measurement of enzyme activities, as described in Materials and Methods.
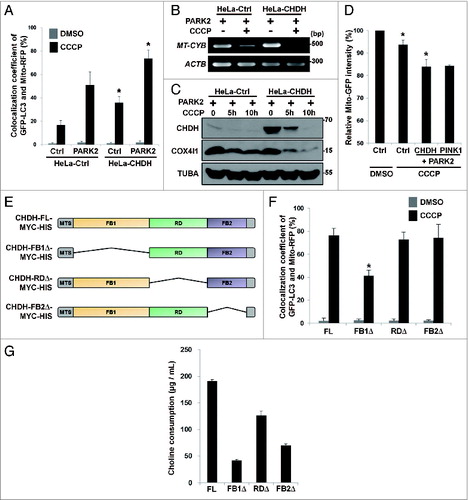
Mitophagic activity of CHDH is independent of enzyme activity
We next examined whether this mitophagic activity of CHDH is related to its enzymatic activity that converts choline to betaine aldehyde. We constructed a series of CHDH deletion mutants based on bioinformatic analysis (materials and methods). CHDH appears to have a mitochondria-targeting sequence at its N-terminus (residues 1 to 38) and 3 functional domains, named FAD/NAD(P)-binding domain 1 (FB1, residues 39 to 326), FAD-linked reductase domain (RD, residues 333 to 515) and FAD/NAD(P)-binding domain 2 (FB2, residues 511 to 574) (E). Expression of these constructs was confirmed by western blot analysis (Fig. S2A). Overexpression of the CHDH-RDΔ or CHDH-FB2Δ mutants induced colocalization of GFP-LC3 with Mito-RFP as effectively as wild-type CHDH, but the CHDH-FB1Δ mutant failed to do so (Fig. S2B; F), indicating that the FB1 domain of CHDH is critical for its mitophagy-stimulating activity. However, enzyme activity assays using these mutants illustrated that all of these CHDH mutants exhibited impaired activity of the enzyme that generates betaine aldehyde; the FB1 and FB2 domains were crucial for this activity, as was the RD domain, which was less important but still required (G). Further, when we tested the mitophagic activity of betaine, the product of CHDH and betaine aldehyde dehydrogenase, which is involved in many biochemical pathways,Citation31 we discovered that betaine did not affect the colocalization of LC3 with mitochondria (Fig. S3). Overall, it appears that the mitophagy-stimulating function of CHDH is independent of its well-known enzymatic activity and enzymatic product.
CHDH accumulates on the outer membrane following mitochondrial damage
We next investigated the subcellular location of CHDH. Consistent with the previous reports,Citation32,33 we found that CHDH was present in mitochondria (Fig. S4A). We further assessed the submitochondrial location of CHDH using a submitochondrial fractionation assay (A). CHDH and the outer membrane (OM)-located TOMM20 were detected in the OM fraction, as well as in the IM fraction, in which COX4I1 was also located (A). However, CHDH was not detected in the cytosol and matrix fractions. To confirm this result, we purified mitochondria from HeLa and HEK293T cells and performed a proteinase K (PK) degradation assay. Incubation of the purified mitochondria with the increased concentrations of PK resulted in gradual degradation of CHDH and TOMM20, but it did not affect the levels of SOD2 in the matrix or IM-localized COX4I1 (B). Whereas the previous study proposed that CHDH is located on the IM of mitochondria, these results indicate that CHDH is present on both the OM and IM of mitochondria. According to the western blot analysis showing that CHDH was retained in the membrane-associated fractions after the treatment with high salt (Fig. S4B), we believe that CHDH is present as a transmembrane protein in the mitochondria.
Figure 3. CHDH resides on the IM and OM of mitochondria and is enriched in the OM following CCCP treatment. (A) HEK293T cells were subjected to submitochondrial fractionation analysis, as described in Materials and Methods, and the fractions were verified using western blot analysis using the indicated antibodies. (B) Mitochondria isolated from HeLa and HEK293T cells were treated with the increasing concentrations (0.25, 0.5, 0.75, 1, and 2 μg/mL) of proteinase K, and the reaction products were analyzed with western blotting. (C–E) Following treatment of HEK293T cells with 0.1 μM CCCP for 30 min, mitochondria were purified and treated with Proteinase K (1 μg/mL), and the reaction products were analyzed using western blotting (C). HEK293T cells were pretreated with cycloheximide (CHX, 20 μg/mL) for 3 h and then exposed to 0.1 μM CCCP for 30 min, followed by submitochondrial fractionation and western blot analysis (D). The normalized relative ratio of CHDH signals detected in the OM and IM fractions on the blots in the (D) control group was determined by densitometric analysis using the ImageJ program (E). (F) HeLa cells were left untreated (Ctrl) or transfected with HA-CHDH-MYC-HIS. After permeabilization with 0.001% digitonin or 0.05% Triton X-100, cells were coimmunostained with TOMM20 (mouse/red) and SOD2 (rabbit/green) antibodies to confirm partial permeabilization (Ctrl) or with HA (mouse/red) and HIS (rabbit/green) antibodies (HA-CHDH-MYC-HIS) and then visualized under a confocal microscope. TUBA, tubulin, α. Scale bar: 10 μm
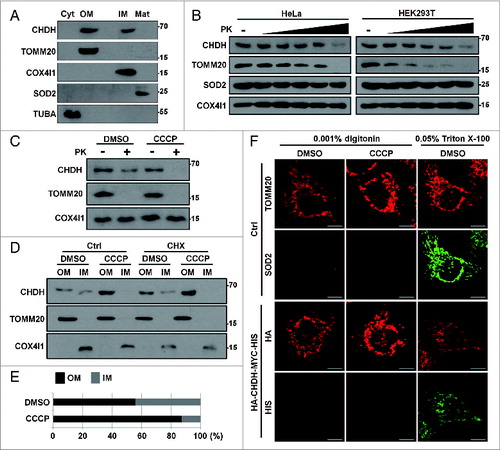
Next, we compared the quantity of CHDH on the OM and IM of mitochondria following CCCP treatment. Instead of using the conventional concentration (10 to 20 μM) of CCCP, which induces mitochondrial membrane potential dissipation but may interfere with membrane integrity and thus result in marker protein contamination in PK and fractionation assays, we used a lower concentration of CCCP that would induce mild mitochondrial depolarization but not affect membrane integrity during the assays. To determine the required concentration, we assessed different concentrations of CCCP, ranging from 0.01 to 10 μM of CCCP, in pilot experiments and found that 0.1 μM CCCP was sufficient to induce significant depolarization of the mitochondrial membrane (Fig. S4C). Using this concentration of CCCP, we found that in mitochondria isolated from CCCP-treated cells, CHDH was more sensitive to degradation by PK than it was in mitochondria isolated from untreated control cells (C). We thus hypothesized that CHDH accumulates on the OM of mitochondria following the loss of the mitochondrial membrane potential. A subsequent fractionation assay also showed that CCCP treatment induced the accumulation of CHDH in the OM fraction, with a concomitant reduction of CHDH in the IM fraction (D and E). We also performed an assay using cycloheximide to block protein translation, which showed that CHDH accumulated on the OM of mitochondria in the presence of cycloheximide (D), indicating that the accumulation of CHDH on the OM of mitochondria does not result from de novo protein synthesis.
When we assessed the topology of CHDH in the mitochondrial membrane, a PK assay following the expression of N-terminal-tagged CHDH (HA-CHDH) revealed that the N terminus of CHDH may be exposed to the cytosol in control cells (Fig. S4D). We confirmed this result with immunocytochemical analysis. In a control analysis, an immunofluorescence assay following differential permeabilization of cells revealed that the immunofluorescent signal of TOMM20, but not SOD2, was detectable by mild permeabilization using 0.001% digitonin. In contrast, immunofluorescence of both TOMM20 and SOD2 were observed after permeabilization with 0.05% Triton X-100, which permeabilizes both the plasma membrane and the mitochondrial membrane (F, upper). Identical experiments were then performed following the expression of dual-tagged HA-CHDH-MYC-HIS containing HA at the N terminus and MYC-HIS at the C terminus. In contrast to the N-terminal HA, immunofluorescence of the C-terminal HIS was not observed after permeabilization with 0.001% digitonin but observed with Triton X-100 (F, lower). These results suggest that the N terminus of OM-located CHDH is exposed to the cytosolic side, whereas the C terminus is located inside mitochondria in both control and CCCP-treated cells. Further, western blot analysis using CHDH antibody recognizing the C terminus revealed that approximately 45-kDa cleavage product of CHDH appeared in a PK assay of purified mitochondria and its amount was increased by CCCP treatment in SH-SY5Y cells (Fig. S4E). Thus, approximately a 20-kDa N-terminal region of CHDH is exposed to the cytosol.
To further assess the mechanism by which CHDH translocates across the mitochondrial membranes, we hypothesized that the mitochondrial OM–IM contact site was a candidate site for the CHDH path. Because the VDAC1-adenine nucleotide translocator (ANT) complex is one of the contact sites,Citation34 we tested whether VDAC1 functions as a path for CHDH during mitophagy. Interestingly, we found that CHDH interacted with VDAC1 following CCCP treatment (Fig. S5A). Moreover, compared with controls, overexpression of VDAC1 increased PK-sensitive cleavage of CHDH during mitophagy (Fig. S5B), indicating that the accumulation of CHDH on the OM is enhanced by VDAC1. Thus, the VDAC1-ANT complex may function to regulate the accumulation of CHDH in the OM of mitochondria during mitophagy. On the other hand, this accumulation was not affected by the overexpression of PLSCR3 (phospholipid scramblase 3) (Fig. S5C), which plays a role in the externalization of cardiolipin to the OM of mitochondria during mitophagy.Citation35 Also, total amount of CHDH protein was not changed during mitophagy or by PINK1 knockdown (Fig. S5D).
CHDH interacts with SQSTM1 independently of PARK2 during mitophagy
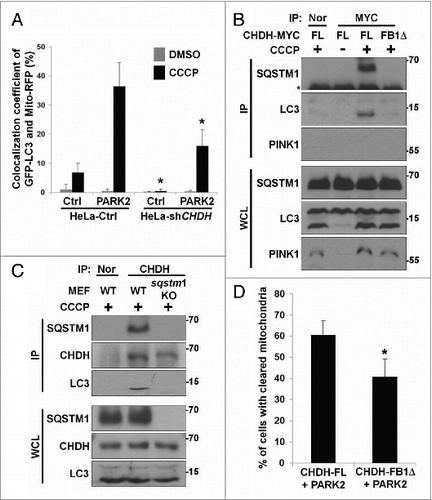
Because knockdown of CHDH expression resulted in reduced colocalization of LC3 with mitochondria (Fig. S3), we hypothesized that CHDH may affect the cargo recognition step in mitophagy via SQSTM1, an important adaptor that connects damaged cargo to LC3.Citation36,37 Using immunoprecipitation analysis, we observed that CHDH interacted with SQSTM1 in the transfected cells (A). Interestingly, this interaction remarkably increased in cells following CCCP treatment. Similarly, endogenous CHDH bound to SQSTM1 in SH-SY5Y neuronal cells and this interaction also increased following exposure to CCCP; the interaction reached a maximum level at 30 min and remained high up to 240 min (B). However, CHDH did not interact with other mitochondrial proteins, such as TOMM20 and COX4I1. We confirmed the endogenous interaction using a reciprocal immunoprecipitation assay (C).
Figure 4. For figure legend, see page 1913.Figure 4 (See previous page). CHDH binds to SQSTM1, which requires the CHDH FB1 and SQSTM1 PB1 domains. (A) HEK293T cells were transfected with pcDNA (Ctrl) or HA-CHDH and then left untreated or treated with 10 μM CCCP for 30 min. Cell extracts were prepared and subjected to immunoprecipitation (IP) analysis using a CHDH antibody. The immunoprecipitates and whole cell lysates (WCL) were analyzed by western blotting. (B and C) SH-SY5Y cells were treated with 20 μM CCCP for the indicated times (B) or for 30 min (C). Cell extracts were subjected to immunoprecipitation (IP) analysis using preimmune normal (Nor) and CHDH antibody (B) or SQSTM1 antibody (C). TOMM20, COX4I1, and SOD2 were used as negative controls in western blotting (B). (D) HeLa-Ctrl and HeLa-shCHDH cells were transfected with FLAG-Ctrl or FLAG-PARK2 and then left untreated or exposed to 20 μM CCCP for 30 min. Cell extracts were subjected to immunoprecipitation (IP) analysis using a CHDH antibody. The asterisk indicates a heavy chain that is weakly bound across species. (E) HeLa cells were transfected with HA-VDAC1, CHDH and either control vector or PARK2 as indicated, and then treated with 10 μM CCCP for 3 h in the presence or absence of 5 μM MG132 or 20 nM bafilomycin A1 (Baf). (F) A schematic representation of SQSTM1 full-length (FL) and the PB1-deletion mutant (PB1Δ). (G and H) HEK293T cells were transfected with SQSTM1-FL-MYC-HIS or SQSTM1-PB1Δ-MYC-HIS and then treated with 20 μM CCCP for 30 min, after which cell lysates were subjected to immunoprecipitation (IP) analysis using CHDH antibody (G, upper) or MYC antibody (H, upper). The immunoprecipitates and whole cell lysates (WCL) were analyzed by western blotting (lower). (I) HEK293T cells were transfected with CHDH-MYC and its deletion mutants and then treated with 10 μM CCCP for 30 min, after which cell lysates were subjected to IP analysis using a MYC antibody.
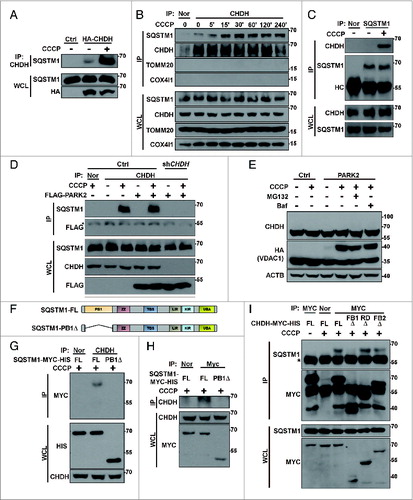
We then addressed whether this interaction was dependent on PARK2 in HeLa cells. Although PARK2 expression is absent in HeLa cells, a basal level of the interaction between CHDH and SQSTM1 was also detected in HeLa cells, and this interaction increased following CCCP treatment (Fig. S6A). However, the interaction was impaired in HeLa-shCHDH cells compared with control HeLa cells. Moreover, the overexpression of PARK2 in HeLa cells did not affect the interaction (D), indicating that the binding of CHDH to SQSTM1 is independent of PARK2. Further, when we examined PARK2-mediated ubiquitination of mitochondrial proteins during mitophagy, we observed the ubiquitination of VDAC1, a substrate of PARK2,Citation26 but not of CHDH (E), indicating that CHDH is not a substrate of PARK2.
Next, we determined the domains responsible for the interaction between CHDH and SQSTM1 using deletion mutants (E; F). The results of an immunoprecipitation assay showed that, unlike full-length SQSTM1, a SQSTM1-PB1Δ mutant lacking the PB1 domain, which is responsible for its interaction with certain aggregate-prone mutant proteins,Citation38,39 failed to interact with CHDH in HEK293T cells (G and H). We also determined the binding region of CHDH and observed that the CHDH-FB1Δ mutant lacking an FB1 domain did not interact with SQSTM1 (I). These results suggest that the PB1 domain of SQSTM1 and the FB1 domain of CHDH are required for their interaction. We confirmed these results using a colocalization assay under a confocal microscope. Unlike wild-type CHDH, mutant CHDH-FB1Δ did not colocalize with SQSTM1 in cells before or after CCCP treatment (Fig. S6B). Expression of CHDH or CHDH-FB1Δ did not affect the function of PARK2, as examined by VDAC1 ubiquitination (Fig. S6C). SNPs of CHDH, especially rs12676 (G233T; R78L), which is located in the FB1 domain, are known to be responsible for mitochondrial function and morphology in mice and humans, especially in sperm.Citation6,9 When this SNP was examined for its ability to interact with SQSTM1, there was no significant difference between CHDH wild-type and rs12676 (Fig. S6D).
The interaction of CHDH with SQSTM1 brings LC3 to damaged mitochondria for cargo recognition during mitophagy
To investigate the functional significance of the interaction between CHDH and SQSTM1 during mitophagy, we examined knockdown effects of CHDH expression on SQSTM1 recruitment into impaired mitochondria. Compared with HeLa control cells, colocalization between Mito-RFP and SQSTM1 was abrogated in HeLa-CHDH knockdown cells during mitophagy (A and B). In addition, a mitochondrial fractionation assay illustrated that SQSTM1 was dramatically recruited into the mitochondrial fraction as early as 15 min after CCCP treatment in SH-SY5Y cells and resided in mitochondria for up to 60 min (C). Strikingly, this recruitment was much reduced in SH-SY5Y-CHDH knockdown cells. However, CHDH did not affect the recruitment of PARK2 into mitochondria. To further investigate the recruitment of SQSTM1 to damaged mitochondria, we performed a fractionation assay. Compared with untreated control cells, CCCP treatment increased the quantity of mitochondrial fraction-localized SQSTM1 in HEK293T cells overexpressing CHDH, but not in cells expressing the SQSTM1 binding-defective CHDH-FB1Δ mutant (D). We also found that the degradation of mitochondrial proteins during mitophagy was partially but significantly impaired by SQSTM1 knockdown in HEK293T cells (Fig. S6E). These observations suggest that CHDH is required for the recruitment of SQSTM1 into damaged mitochondria for degradation.
Figure 5. CHDH recruits SQSTM1 onto mitochondria during mitophagy. (A and B) HeLa-Ctrl and HeLa-shCHDH cells were transfected with Mito-RFP and FLAG-PARK2 and treated with 10 μM CCCP for 4 h. Following immunostaining with a SQSTM1 antibody, cells were examined under a confocal microscope (A) and the colocalization coefficient (%) of SQSTM1 and Mito-RFP was determined. Bars represent the mean ± SD, *P < 0.0001 (B). (C) SH-SY5Y-Ctrl and SH-SY5Y-shCHDH stable cells were treated with 20 μM CCCP for the indicated times and mitochondria were purified and analyzed by western blotting using the indicated antibodies. (D) HEK293T cells were transfected with CHDH-FL or CHDH-FB1Δ and then treated with 10 μM CCCP for the indicated times. Cell extracts were fractionated to isolate mitochondria followed by western blot analysis.
Further, confocal imaging analysis revealed that the colocalization of GFP-LC3 and Mito-RFP was decreased by 2-fold in HeLa-shCHDH cells compared with HeLa control cells (A). This reduction in the colocalization was also observed in HeLa control cells lacking PARK2 (A, lanes 2 and 6). It thus appears that CHDH recruits LC3 independently of PARK2, as was observed in the SQSTM1-binding pattern of CHDH. Given that SQSTM1 directly links damaged and ubiquitinated mitochondria to LC3,Citation36 we examined protein complex formation among CHDH, SQSTM1, and LC3. The results of an immunoprecipitation assay showed that CHDH, but not CHDH-FB1Δ, formed a protein complex with SQSTM1 and LC3 in HeLa cells following CCCP treatment (B). Under identical conditions, knockout of SQSTM1 expression prevented formation of the ternary protein complex containing LC3 and CHDH (C). Because an LC3-containing autophagosome fused with a lysosome is destined for degradation, we then examined the relative effects of CHDH and CHDH-FB1Δ on PARK2-dependent degradation of mitochondria. Immunofluorescence analysis revealed that CHDH-FB1Δ was less effective than CHDH to clear TOMM20-positive mitochondria following CCCP treatment (40.8% vs. 60.5%) (D). These results suggest that CHDH binds to SQSTM1 to bring LC3 to the damaged mitochondria for the degradation of mitochondria and is indispensable for PARK2-mediated mitophagy at the cargo-recognition step.
Figure 6. Interaction of CHDH with SQSTM1 increases the recruitment of LC3 into mitochondria to process mitophagy. (A) HeLa-Ctrl and HeLa-shCHDH cells were cotransfected with GFP-LC3, Mito-RFP, and either control vector (Ctrl) or PARK2. Following treatment with DMSO or 10 μM CCCP for 4 h, cells were fixed and observed under a confocal microscope. The colocalization coefficient (%) of GFP-LC3 and Mito-RFP was determined and bars represent the mean ± SD, *P < 0.005. (B) After HeLa cells were transfected with CHDH-FL-MYC-HIS or CHDH-FB1Δ-MYC-HIS and treated with 10 μM CCCP for 30 min, samples were subjected to an immunoprecipitation (IP) assay using preimmune (Nor) or a MYC antibody. The asterisk indicates the heavy chain of the MYC antibody. (C) sqstm1 KO MEF cells were exposed to 10 μM CCCP for 30 min. Cell extracts were subjected to an immunoprecipitation (IP) assay using a CHDH antibody and the immunoprecipitates and whole cell lysates (WCL) were analyzed by western blot. (D) HeLa cells were cotransfected with PARK2-GFP and either CHDH or CHDH-FB1Δ, treated with 10 μM CCCP for 24 h and then immunostained with a TOMM20 antibody. The percentage of cells showing reduced immunoreactivity against TOMM20 was determined using a confocal microscope and is represented as bars ± SD, *P < 0.001.
Based on the domain-mapping results for CHDH, we asked whether the CHDH FB1 domain is sufficient for LC3 recruitment into mitochondria. We generated a TOMM20-FB1-GFP chimera, a mitochondrial OM-targeting chimeric construct containing TOMM20 fused to CHDH FB1-GFP (A, upper), and examined its ability to recruit LC3 into the mitochondria under a confocal microscope. When these constructs were overexpressed in HeLa cells, no difference was observed between TOMM20-GFP and TOMM20-FB1-GFP in the recruitment of mRFP-LC3 into mitochondria. However, when cells were exposed to CCCP, TOMM20-FB1-GFP increased colocalization of mRFP-LC3 with the mitochondria by 2-fold, compared with TOMM20-GFP (A, lower). These observations suggest that the CHDH FB1 domain is necessary for LC3 recruitment to damaged mitochondria.
Figure 7. For figure legend, see page 1917.Figure 7 (See previous page). Inhibition of the interaction between CHDH and SQSTM1 hampers mitochondrial degradation. (A) Schematic diagram of TOMM20-FB1-GFP chimera (upper). HeLa cells were transfected with mRFP-LC3 and either TOMM20-GFP or TOMM20-FB1-GFP and exposed to 10 μM CCCP for 1 h. Cells were analyzed under a confocal microscope and the colocalization coefficient was determined. Bars represent the mean ± SD, *P < 0.0001 (lower). (B) HEK293T cells were transfected with different concentrations of GFP (Ctrl-GFP) or FB1-GFP (+: 2 μg, ++: 5 μg) and then treated with 10 μM CCCP for 30 min. Cell lysates were analyzed by immunoprecipitation (IP) and western blotting. (C and D) HeLa cells were transfected with PARK2 and either Ctrl-GFP or FB1-GFP. Following 24 h treatment with 10 μM CCCP, cells were subjected to immunocytochemical analysis using the indicated antibodies and then visualized under a confocal microscope. Scale bar: 10 μm. Statistical values for (C) are represented as bars with the mean ± SD, *P < 0.05 (D). (E) Stable SN4741-Ctrl or SN4741-shCHDH cells were transfected with GFP-LC3 and Mito-RFP and then treated with 100 μM MPP+ for 4 h. Cells were examined under a confocal microscope. The expression level of CHDH was determined using western blot analysis (upper) and the colocalization coefficient (%) between GFP-LC3 and Mito-RFP was determined (lower). Bars represent the mean ± SD, *P < 0.0001. (F) Proposed role of CHDH in mitophagy. CHDH resides on both the OM and IM of mitochondria and exposes its FB1 domain-containing N terminus to the cytosol on the OM. Following mitochondrial depolarization, CHDH accumulates on the OM probably through VDAC1 and interacts with SQSTM1 to recruit LC3 to the damaged mitochondria for mitophagy.
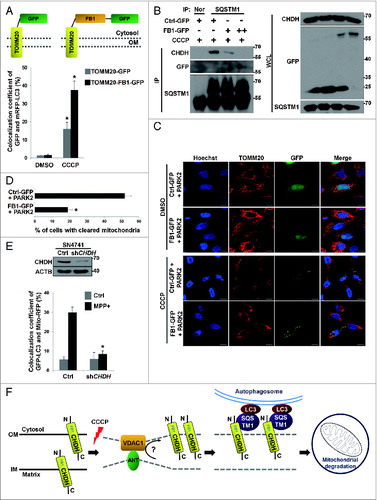
In addition, we addressed whether the interaction between CHDH and SQSTM1 is critical for mitophagy using a competition assay. We found that FB1-GFP could bind to SQSTM1 in the transfected cells during mitophagy, although the interaction appears to be weak (B). Moreover, ectopic expression of FB1-GFP weakened the interaction between CHDH and SQSTM1 in a dose-dependent manner. We then assessed the effect of FB1-GFP on mitochondrial clearance during mitophagy. Immunofluorescence analysis revealed that ectopic expression of FB1-GFP significantly inhibited PARK2-dependent clearance of TOMM20-positive mitochondria during CCCP-induced mitophagy (from 51.8% to 19.0%) (C and D). Consistently, FB1-GFP colocalized with SQSTM1-MYC in the transfected cells (Fig. S6F). These results suggest that the interaction between CHDH and SQSTM1 is important for mitophagy to proceed.
CHDH is implicated in MPP+-induced mitophagy in SN4741 dopaminergic cells
The failure of appropriate recognition of damaged mitochondria causes accumulation of defective mitochondriaCitation11,26 and this failure is closely associated with the pathogenesis of PD.Citation40 In SN4741 dopaminergic cells, we thus addressed the role of CHDH during mitophagy triggered by 1-methyl-4-phenylpyridinium (MPP+), a Parkinsonism-causing reagent.Citation41 Like CCCP, treatment with MPP+ induced a significant level (30.0%) of colocalization of GFP-LC3 with mitochondria in SN4741 cells (E). In contrast, downregulation of CHDH expression abolished the colocalization (8.5%). These results indicate that CHDH is essential in the mitophagy of SN4741 dopaminergic neurons following exposure to MPP+.
Discussion
Although CHDH has been proposed to localize to the IM of mitochondria,Citation1,32,33 it is surprising to note that no report has clearly demonstrated the presence of CHDH in the IM of mitochondria. Our results indicate that CHDH resides on both the OM and IM of mitochondria. More interestingly, CHDH accumulates on the OM during mitochondrial depolarization. Thus, it appears that CHDH plays a dual role in choline conversion and mitophagy in the IM and OM, respectively, of mitochondria. Concordant with this hypothesis, we found no correlation between the enzymatic activity and mitophagic function of CHDH, in that enzyme activity-dead mutant of CHDH were functional in mitophagy and the CHDH FB1 domain was crucial only for mitophagy. In addition, our observation that betaine, the enzymatic product of CHDH, cannot rescue the impaired mitophagy caused by CHDH deficiency further distinguishes CHDH's mitophagic activity from its enzymatic activity.
Given this newly discovered role in mitophagy, the location of CHDH within mitochondria becomes more important. PINK1 also resides on both the OM and IM of mitochondria, and is likewise involved in mitophagy. Although PINK1 is normally degraded in the IM of mitochondria following cleavage by PARL proteases,Citation19 it accumulates on the mitochondrial surface to recruit PARK2 during mitophagy.Citation20,42 In contrast to PINK1, however, western blot analysis did not show any cleavage or posttranslational modification, such as ubiquitination, of CHDH during mitophagy, suggesting that the accumulation of CHDH on the OM may not be accompanied by such modifications. Cardiolipin, a phospholipid located on the mitochondrial IM, is externalized to the OM by PLSCR3 following mitochondrial injury, where it interacts with LC3.Citation35 However, we observed no significant effect of PLSCR3 on the accumulation of CHDH in the OM. Instead, we found that the accumulation of CHDH on the OM depends on the mitochondrial potential and may use the VDAC1-containing mitochondrial membrane contact site as a path.
It appears that the accumulation of CHDH on the OM of depolarized mitochondria is critical to facilitate interaction with cytosolic SQSTM1. In particular, the CHDH N-terminal region, which is approximately 20 kDa in size and exposed to the cytosol in the FB1 domain, seems to be important for this interaction. Although the CHDH FB1 domain is required for this interaction and ectopic expression of the CHDH FB1 domain interrupts the interaction of CHDH with SQSTM1, it remains unclear whether the CHDH FB1 domain alone is sufficient to recruit SQSTM1 onto damaged mitochondria. Our observation that TOMM20-FB1-GFP functioned in recruiting LC3 to depolarized mitochondria in the presence of CCCP does not exclude a mechanism in which other mitophagic component(s) or machinery, such as PARK2 substrates, operate in parallel during mitophagy, in addition to the presence of CHDH on the OM.
Although the ubiquitination of mitochondrial substrates by PARK2 is also crucial to recruit SQSTM1 into impaired mitochondria,Citation26,43,44 it has previously been reported that SQSTM1 interacts with nonubiquitinated substrates via its PB1 domain.Citation38,39 The PB1 domain is responsible for forming SQSTM1 aggregates through self- or hetero-oligomerizationCitation45,46 and is required for mitochondrial aggregation and localization of SQSTM1 to mitochondria during CCCP conditions.Citation44 Here, we elucidate the mechanism by which SQSTM1 may be recruited into impaired mitochondria, involving an interaction between SQSTM1 and nonubiquitinated CHDH on the OM of mitochondria. Although we observed some interaction of SQSTM1 with CHDH at the steady-state level required for basal mitophagy, the interaction drastically increased upon mitophagy induction. Despite some debate on the necessity and sufficiency of SQSTM1 in mitochondrial clearance,Citation11,26,44,47 the results from our assays show that SQSTM1 is necessary for the degradation of depolarized mitochondria. It remains possible that SQSTM1 and other adaptor proteins may work together in mitophagy. While SQSTM1 is crucial in mediating mitophagy in several cell lines, it does not act alone, because knockdown of CHDH expression suppressed CCCP-induced LC3 recruitment into the mitochondria by just over 50%, despite complete inhibition of the colocalization of SQSTM1 with mitochondria. Thus, in the absence of SQSTM1, other adaptors, such as HDAC6, may compensate for SQSTM1 deficiency.Citation43,48
The effect of CHDH on mitophagy was displayed largely with PINK1 and PARK2. In the absence of PINK1 and PARK2, however, CHDH overexpression could still stimulate mitophagy, while absolute levels of mitophagy were reduced. Similarly, PARK2-mediated mitophagy was significantly impaired in the absence of CHDH. The function of CHDH appears to be independent of PINK1 and PARK2, since neither the recruitment of PARK2 to depolarized mitochondria nor the stabilization of PINK1 was affected by CHDH. Further, CHDH is apparently not a substrate of PARK2 and its interaction with SQSTM1 is PARK2-independent. Our current hypothesis is that CHDH functions in parallel with PINK1/PARK2 pathway in mitophagy. Thus, CHDH is also important for the recruitment of LC3 into mitochondria in PARK2-mediated mitophagy. In addition to PINK1 and PARK2, BNIP3L/NIX and FUNDC1 have recently been reported to function in hypoxial mitophagy.Citation49,50 While it seems that CHDH may not function during hypoxial mitophagy (data not shown), we now add CHDH to the list of mitophagy coordinators. In conclusion, CHDH accumulates on the OM of mitochondria following mitochondrial damage and interacts there with SQSTM1 to recruit LC3 into the impaired mitochondria for mitophagic clearance (F). This mitophagic function of CHDH is independent of its conventional enzymatic activity but is necessary for PARK2-mediated mitophagy. Finally, the implications of CHDH malfunction in mitophagy-associated pathology are now widely open and remain to be further addressed.
Materials and Methods
Cell culture and transfection
HeLa, HEK293T, and SH-SY5Y cells were grown in DMEM (Hyclone, SH30243.01) supplemented with 10% fetal bovine serum (Hyclone, SH30919.03), 50 U/mL penicillin and 50 μg/mL streptomycin at 37 °C under 5% CO2. SN4741 dopaminergic neuronal cells were cultured as described previously.Citation28 Stable cell lines were generated after selection with G418 (1 mg/mL; Invitrogen, 10131027) for 2 wk following DNA transfection and confirmed by western blot analysis. sqstm1 MEFs were gifted by Dr J Shin (Sungkyunkwan University, Korea). DNA, shRNA and siRNA transfections were performed with Polyfect reagent (Qiagen, 1015586) and Lipofectamine 2000 reagent (Invitrogen, 11668019) for 24 and 48 h, respectively, according to the manufacturer's instructions.
Plasmids and siRNA
CHDH shRNA constructs were generated in pSUPER-neo (OligoEngine, VEC-PBS-0004) using the following target sequence for human CHDH: 5′-CCACATTCAG TCAGATAAA-3′ and for mouse Chdh: 5′-GGTAATGATT GCAGAGAAA-3′. Serial deletion (Δ) mutants of CHDH were generated by excising the FB1 domain (115 to 978) (CHDH-FB1Δ), RD domain (997 to 1545) (CHDH-RDΔ) and FB2 domain (1531 to 1722) (CHDH-FB2Δ) based on the bioinformatic domain prediction using MITOPROT (http://ihg.gsf.de/ihg/mitoprot.html) and SUPERFAMILY Sequence Search (http://supfam.org/SUPERFAMILY/hmm.html). The HA-CHDH and CHDH-MYC-HIS were produced by subcloning CHDH into pcDNA3-HA and pEF1/MYC-HIS (Invitrogen, V92120), respectively. HA-CHDH-MYC-HIS was constructed by subcloning CHDH-MYC-HIS into pcDNA3-HA for HA tagging at the N terminus and MYC-HIS tagging at the C terminus. The SQSTM1-FL-MYC-HIS, SQSTM1-PB1Δ-MYC-HIS were gifts from Dr M Tanaka (Kyoto Prefectural University, Japan) and PINK1-GFP was from Dr J Chung (Seoul National University, Korea). TOMM20-GFP, TOMM20-FB1-GFP and FB1-GFP were constructed by subcloning of TOMM20 and/or the FB1 domain cDNA into pEGFP-N1 (Invitrogen, 6085-1). Rs12676 SNP mutant of CHDH (R78L) was generated by site-directed mutagenesis using PCR: The following primers were used. Fwd, 5′-GCCCAAGGAC GTGCTCGCGG GGAGCAAGCG-3′ and Rev, 5′-CGCTTGCTCC CCGCGAGCAC GTCCTTGGGC-3′. The SQSTM1 siRNA was kindly provided by Dr J Shin (Sungkyunkwan University, Korea).
Measurement of Mito-GFP intensities
After transfection with Mito-GFP, the fluorescence of cells was measured with EnVision multilabel plate reader (PerkinElmer, 940 Winter Street, Waltham, 02451, MA USA).
Measurement of enzyme activity and LC-MS
Mitochondria were purified from cells using differential centrifugation as described previously.Citation20 After resuspension in reaction buffer (10 mM sodium phosphate, 10 mM Tris-Cl, pH 8.0), mitochondria were burst by sonication and then mixed with 10 mM choline chloride and 2 mM phenazine methosulfate. The reaction was incubated at 37°C for 1 h in the dark and samples were subjected to ultracentrifugation at 100,000 g for 30 min to precipitate mitochondrial debris; the supernatant fraction was kept at −70°C until analysis. For LC-MS analysis, a Thermo-Finnigan Surveyor instrument (Thermo Scientific, USA), equipped with autosampler and PDA-UV detector and Thermo-Finnigan LCQ Deca XP plus ion trap mass spectrometer with electrospray ionization interface was used. ZoRBAX 300SB-C8 P/No. 865750-906, size 2.1 × 50 mm, 3.5 μm, S/No. USACR01562 was used for chromatographic separations. The injection volume was 10 μL and the mobile phase consisted of 0.1% formic acid in distilled water (A) and in acetonitrile (B). Gradient elution at a flow rate of 150 μL/min was as following: 95% (A) and 5% (B) from 0 to 20 min, 5% (A) and 95% (B) from 20 min to 25 min, and returned to the initial condition from 20 min to 30 min. Total running time was 30 min. Ionization of analytes was performed using electrospray ionization. The capillary temperature was maintained at 275°C. The ion source voltage and the nebulizer gas were set at 5 kV and 30 units, respectively. The capillary voltage was at 46 V in positive and −15 V in negative ionization mode. The average scan time was 0.01 min while the average time for changing polarity was 0.02 min. The collision energy was generally chosen in order to maintain about 35% abundance of the precursor ion.
Immunoprecipitation, western blot, and antibodies
Immunoprecipitation and western blot analysis were performed as reported previously.Citation51 Briefly, cells were lysed in lysis buffer (20 mM Tris-Cl, pH 7.6, 150 mM NaCl, 1% Triton X-100) and clarified by centrifugation. Cell lysates were dichotomized for immunoprecipitation and whole cell lysate quantification. For immunoprecipitaion, primary antibody was added overnight at 4°C and pulled down by protein G Sepharose beads (GE Healthcare, 17-0618-01) at 4°C for 1 h. Both samples were boiled in 1× SDS sample buffer, separated by SDS-PAGE and transferred onto nitrocellulose membrane (Pall Corporation, 66485). After blocking with 5% skim milk in TBST, the membrane was incubated with the following antibodies: CHDH (Santa Cruz Biotechnology, sc-102442), TOMM20 (sc-17764), GFP (sc-8334), MYC (sc-40), TUBA/tubulin, α (sc-23948) and ACTB (sc-47778) SOD2 (Upstate, 06-984), COX4I1 (Abcam, ab14744-100), SQSTM1 (Abnova, H00008878-M01 and Santa Cruz Biotechnology, sc-25575), PARK2 (Abcam, ab77924 and Santa Cruz Biotechnology, sc-32282), LC3 (Novus, NB100-2220), FLAG (Sigma, F3165) and TIMM23 (BD Bioscience, 611222).
Immunofluorescence and colocalization coefficient
Cells were fixed with 4% paraformaldehyde and permeabilized with 0.005% digitonin in PBS (Gibco, 70011-044). After blocking with 10% fetal bovine serum, the fixed cells were incubated with antibodies at 1:100 to 1:250 dilution ratio, followed by incubation with Alexa Fluor secondary antibodies (Molecular Probes, A11005, A11001, A11008, and A11012). Samples were visualized under a confocal laser scanning microscope (Carl Zeiss, LSM700, Carl-Zeiss-Promenade 10, 07745, Jena Germany) and colocalization coefficient was analyzed using ZEN software (Carl Zeiss).
Flow cytometry
Cells were harvested and monocellized with Trypsin-EDTA, followed by fixation with 70% ethanol with voltexing and then subjected to analysis using a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ USA) according to the manufacturer's instructions.
Mitochondrial DNA quantification
Mitochondrial DNA was extracted using Exprep kit (GeneAll, 101-102) and subjected to PCR analysis using MT-CYB primers: Fwd, 5′-TTCTGAGGGG CCACAGTAAT TACA AACTTA-3′, Rev, 5′-ATGGAGGATG GGGATTATTG CTAGGATGAG-3′. Prior to mitochondrial DNA extraction, whole cell DNA was also extracted for quantification as described previouslyCitation52 and subjected to PCR analysis using ACTB primers: Fwd, 5′-CGTTGGCATC CACGAAACTA-3′, Rev, 5′-AGTACTTGCG CTCAGGAGGA-3′.
Mitochondria fractionation
Cells were resuspended in PBS and subjected to sonication twice at 30% amplitude for 3 s with an Ultrasonic Processor 130 W (Sonics and Materials, Newtown, CT USA). Unbroken cells and nuclei were removed by centrifugation at 1,000 g for 10 min. The postnuclear supernatant fraction was pelleted by further centrifugation at 10,000 g for 15 min to obtain the mitochondria pellet. For submitochondria fractionation, the pelleted mitochondria were rinsed with swelling buffer (10 mM KH2PO4, pH 7.4) once and resuspended in swelling buffer for 20 min with gentle mixing, followed by addition of an equal volume of shrinking buffer (10 mM HEPES, pH 7.4, 32% sucrose, 30% glycerol, 10 mM MgCl2) for 15 min. Subsequent centrifugation at 10,000 g for 15 min yielded the pelleted mitoplast and suspended OM and intermembrane space (IMS) fraction which was kept for further separation. The mitoplast was rinsed twice with washing buffer (250 mM sucrose, 1 mM EGTA, 10 mM HEPES, pH 7.4), resuspended in swelling buffer and burst 5 times by sonication at 50% amplitude for 3 s. this suspension was centrifuged at 12,000 g for 15 min to discard the unbroken mitoplast. Finally, samples were processed by ultracentrifugation at 100,000 g for 40 min to separate OM and IMS fractions, and the IM matrix fractions. Every procedure was performed at 4°C.
Proteinase K degradation assay
For proteinase K degradation assay, the purified mitochondria were resuspended in PK assay buffer (20 mM HEPES, pH 7.4, 250 mM sucrose, 80 mM potassium acetate, 5 mM magnesium acetate) and added with proteinase K (GenDepot, P2170-005) or control BSA in ice. After 10 min, the digestion was stopped with addition of 1 mM PMSF.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental data for this article can be accessed on the publisher's website.
Supplementary Material.pdf
Download PDF (679.3 KB)Acknowledgments
Mr S Park and SM Yoo were in part supported by BK21 program.
Funding
This work was supported by the grants from the Global Research Laboratory Program (GRL, NRF-2010-00341) and a CRI grant (NRF-2013R1A2A1A01016896) funded by the Ministry of Education, Science and Technology (MEST) in Korea.
References
- De Ridder JJ, van Dam K. The efflux of betaine from rat-liver mitochondria, a possible regulating step in choline oxidation. Biochim Biophys Acta 1973; 291:557-63; PMID:4690868; http://dx.doi.org/10.1016/0005-2736(73)90507-5
- Garrow TA. Purification, kinetic properties, and cDNA cloning of mammalian betaine-homocysteine methyltransferase. J Biol Chem 1996; 271:22831-8; PMID:8798461
- Olthof MR, Verhoef P. Effects of betaine intake on plasma homocysteine concentrations and consequences for health. Curr Drug Metab 2005; 6:15-22; PMID:15720203; http://dx.doi.org/10.2174/1389200052997366
- Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, Fuller A, Muir B, Mohapatra G, Salunga R, Tuggle JT, et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell 2004; 5:607-16; PMID:15193263; http://dx.doi.org/10.1016/j.ccr.2004.05.015
- Wang Z, Dahiya S, Provencher H, Muir B, Carney E, Coser K, Shioda T, Ma XJ, Sgroi DC. The prognostic biomarkers HOXB13, IL17BR, and CHDH are regulated by estrogen in breast cancer. Clin Cancer Res 2007; 13:6327-34; PMID:17975144; http://dx.doi.org/10.1158/1078-0432.CCR-07-0310
- Johnson AR, Craciunescu CN, Guo Z, Teng YW, Thresher RJ, Blusztajn JK, Zeisel SH. Deletion of murine choline dehydrogenase results in diminished sperm motility. FASEB J 2010; 24:2752-61; PMID:20371614; http://dx.doi.org/10.1096/fj.09-153718
- da Costa KA, Kozyreva OG, Song J, Galanko JA, Fischer LM, Zeisel SH. Common genetic polymorphisms affect the human requirement for the nutrient choline. FASEB J 2006; 20:1336-44; PMID:16816108; http://dx.doi.org/10.1096/fj.06-5734com
- Xu X, Gammon MD, Zeisel SH, Lee YL, Wetmur JG, Teitelbaum SL, Bradshaw PT, Neugut AI, Santella RM, Chen J. Choline metabolism and risk of breast cancer in a population-based study. FASEB J 2008; 22:2045-52; PMID:18230680; http://dx.doi.org/10.1096/fj.07-101279
- Johnson AR, Lao S, Wang T, Galanko JA, Zeisel SH. Choline dehydrogenase polymorphism rs12676 is a functional variation and is associated with changes in human sperm cell function. PLoS One 2012; 7:e36047; PMID:22558321; http://dx.doi.org/10.1371/journal.pone.0036047
- Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J 2008; 27:306-14; PMID:18216873; http://dx.doi.org/10.1038/sj.emboj.7601972
- Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 2010; 285:27879-90; PMID:20573959; http://dx.doi.org/10.1074/jbc.M110.119537
- Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci 2012; 125:1488-99; PMID:22275429; http://dx.doi.org/10.1242/jcs.094110
- Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K, Zhang J, Iyengar R, Jung CH, Suen DF, et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell 2011; 43:572-85; PMID:21855797; http://dx.doi.org/10.1016/j.molcel.2011.06.018
- Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 2013; 20:31-42; PMID:22743996; http://dx.doi.org/10.1038/cdd.2012.81
- Chu CT, Zhu J, Dagda R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy 2007; 3:663-6; PMID:17622797; http://dx.doi.org/10.4161/auto.4625
- Gilkerson RW, De Vries RL, Lebot P, Wikstrom JD, Torgyekes E, Shirihai OS, Przedborski S, Schon EA. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum Mol Genet 2012; 21:978-90; PMID:22080835; http://dx.doi.org/10.1093/hmg/ddr529
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998; 392:605-8; PMID:9560156; http://dx.doi.org/10.1038/33416
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004; 304:1158-60; PMID:15087508; http://dx.doi.org/10.1126/science.1096284
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 2012; 13:378-85; PMID:22354088; http://dx.doi.org/10.1038/embor.2012.14
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 2010; 191:933-42; PMID:21115803; http://dx.doi.org/10.1083/jcb.201008084
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 2010; 189:211-21; PMID:20404107; http://dx.doi.org/10.1083/jcb.200910140
- Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 2009; 17:98-109; PMID:19619495; http://dx.doi.org/10.1016/j.devcel.2009.06.014
- Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 2009; 17:87-97; PMID:19619494; http://dx.doi.org/10.1016/j.devcel.2009.06.013
- Orvedahl A, Sumpter R Jr., Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011; 480:113-7; PMID:22020285; http://dx.doi.org/10.1038/nature10546
- Cai Q, Zakaria HM, Simone A, Sheng ZH. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol 2012; 22:545-52; PMID:22342752; http://dx.doi.org/10.1016/j.cub.2012.02.005
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119-31; PMID:20098416; http://dx.doi.org/10.1038/ncb2012
- Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP, et al. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 2013; 78:65-80; PMID:23498974; http://dx.doi.org/10.1016/j.neuron.2013.02.029
- Son JH, Chun HS, Joh TH, Cho S, Conti B, Lee JW. Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J Neurosci 1999; 19:10-20; PMID:9870933
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010; 8:e1000298; PMID:20126261; http://dx.doi.org/10.1371/journal.pbio.1000298
- Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 2012; 22:320-33; PMID:22280891; http://dx.doi.org/10.1016/j.devcel.2011.12.014
- Lever M, Slow S. The clinical significance of betaine, an osmolyte with a key role in methyl group metabolism. Clin Biochem 2010; 43:732-44; PMID:20346934; http://dx.doi.org/10.1016/j.clinbiochem.2010.03.009
- Lin CS, Wu RD. Choline Oxidation and Choline Dehydrogenase. J Protein Chem 1986; 5:193-200; http://dx.doi.org/10.1007/BF01025488
- Huang S, Lin Q. Functional expression and processing of rat choline dehydrogenase precursor. Biochem Biophys Res Commun 2003; 309:344-50; PMID:12951056; http://dx.doi.org/10.1016/j.bbrc.2003.08.010
- Reichert AS, Neupert W. Contact sites between the outer and inner membrane of mitochondria-role in protein transport. Biochim Biophys Acta 2002; 1592:41-9; PMID:12191767; http://dx.doi.org/10.1016/S0167-4889(02)00263-X
- Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 2013; 15:1197-205; PMID:24036476; http://dx.doi.org/10.1038/ncb2837
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/10.1074/jbc.M702824200
- Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci 2012; 125:795-9; PMID:22448035; http://dx.doi.org/10.1242/jcs.093849
- Watanabe Y, Tanaka M. p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J Cell Sci 2011; 124:2692-701; PMID:21771882; http://dx.doi.org/10.1242/jcs.081232
- Gal J, Ström AL, Kwinter DM, Kilty R, Zhang J, Shi P, Fu W, Wooten MW, Zhu H. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem 2009; 111:1062-73; PMID:19765191; http://dx.doi.org/10.1111/j.1471-4159.2009.06388.x
- Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. EMBO J 2012; 31:3038-62; PMID:22735187; http://dx.doi.org/10.1038/emboj.2012.170
- Mandemakers W, Morais VA, De Strooper B. A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J Cell Sci 2007; 120:1707-16; PMID:17502481; http://dx.doi.org/10.1242/jcs.03443
- Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 2011; 20:867-79; PMID:21138942; http://dx.doi.org/10.1093/hmg/ddq526
- Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol 2010; 189:671-9; PMID:20457763; http://dx.doi.org/10.1083/jcb.201001039
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010; 6:1090-106; PMID:20890124; http://dx.doi.org/10.4161/auto.6.8.13426
- Wilson MI, Gill DJ, Perisic O, Quinn MT, Williams RL. PB1 domain-mediated heterodimerization in NADPH oxidase and signaling complexes of atypical protein kinase C with Par6 and p62. Mol Cell 2003; 12:39-50; PMID:12887891; http://dx.doi.org/10.1016/S1097-2765(03)00246-6
- Lamark T, Perander M, Outzen H, Kristiansen K, Øvervatn A, Michaelsen E, Bjørkøy G, Johansen T. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem 2003; 278:34568-81; PMID:12813044; http://dx.doi.org/10.1074/jbc.M303221200
- Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 2010; 15:887-900; PMID:20604804
- Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 2010; 29:969-80; PMID:20075865; http://dx.doi.org/10.1038/emboj.2009.405
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 2012; 14:177-85; PMID:22267086; http://dx.doi.org/10.1038/ncb2422
- Novak I, Dikic I. Autophagy receptors in developmental clearance of mitochondria. Autophagy 2011; 7:301-3; PMID:21206218; http://dx.doi.org/10.4161/auto.7.3.14509
- Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, Jung S, Jung YK. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun 2013; 4:2300; PMID:23939249; http://dx.doi.org/10.1038/ncomms3300
- Park S, Lee KM, Ju JH, Kim J, Noh DY, Lee T, Shin I. Protein expression profiling of primary mammary epithelial cells derived from MMTV-neu mice revealed that HER2/NEU-driven changes in protein expression are functionally clustered. IUBMB Life 2010; 62:41-50; PMID:19960538