Abstract
Autophagy is a highly conserved, closely regulated homeostatic cellular activity that allows for the bulk degradation of long-lived proteins and cytoplasmic organelles. Its roles in cancer initiation and progression and in determining the response of tumor cells to anticancer therapy are complicated, and only limited investigation has been conducted on the potential significance of autophagy in the pathogenesis and therapeutic response of acute myeloid leukemia. Here we demonstrate that the inducible or transfected expression of the acute promyelocytic leukemia (APL)-specific PML-RARα, but not PLZF-RARα or NPM-RARα, fusion protein upregulates constitutive autophagy activation in leukemic and nonleukemic cells, as evaluated by hallmarks for autophagy including transmission electron microscopy. The significant increase in autophagic activity is also found in the leukemic cells-infiltrated bone marrow and spleen from PML-RARα-transplanted leukemic mice. The autophagy inhibitor 3-methyladenine significantly abrogates the autophagic events upregulated by PML-RARα, while the autophagic flux assay reveals that the fusion protein induces autophagy by increasing the on-rate of autophagic sequestration. Furthermore, this modulation of autophagy by PML-RARα is possibly mediated by a decreased activation of the Akt/mTOR pathway. Finally, we also show that autophagy contributes to the anti-apoptotic function of the PML-RARα protein. Given the critical role of the PML-RARα oncoprotein in APL pathogenesis, this study suggests an important role of autophagy in the development and treatment of this disease.
Introduction
Autophagy or “self-eating” is a highly conserved, homeostatic process that allows for the bulk degradation of long-lived proteins and cytoplasmic organelles in eukaryotes.Citation1 During autophagy, portions of the cytoplasm including organelles are first engulfed within a double-membrane vesicle called the autophagosome, which then fuses with lysosomes to degrade the sequestered materials through the actions of various lysosomal hydrolytic enzymes, followed by the regeneration of amino acids that are recycled for macromolecular synthesis and energy production.Citation2 In the past decade, the molecular mechanism of autophagy was widely investigated in yeast and mammalian cells.Citation3,Citation4 More than 30 autophagy-related (Atg) genes, such as beclin 1 and human vacuolar protein sorting 34 (hVPS34), the homolog of class III phosphatidylinositol-3-kinase (PtdIns3KC3) in yeast, have been identified to participate in the autophagic process.Citation2,Citation5–Citation7 Among these molecules, microtubule-associated protein 1 light chain 3 type I (LC3-I), originally identified as a small subunit of microtubule-associated protein 1A and 1B from rat brain,Citation8 is processed into LC3-II (that is, LC3 conjugated to phosphotidylethanolamine) by a unique protein activation/conjugation system similar to ubiquitination to form an autophagosomal membrane during autophagy. The conversion of LC3-I into LC3-II is regarded to be indispensable for both the membrane association of LC3 itself and elongation of the autophagosomal membrane.Citation9,Citation10
Autophagy is important in cell differentiation, development and response to environmental stimuli. This cellular process also plays an important role in numerous diseases such as bacterial and viral infections, neurodegenerative disorders and cardiovascular diseases.Citation2,Citation11 Recent studies suggest that autophagy may also be essential in the regulation of cancer initiation and progression and in determining the response of tumor cells to anticancer therapy. However, the role of autophagy in these cancer-related processes is complicated and has diametrically opposite consequences for tumor cells.Citation12–Citation14 To our understanding, the potential roles of autophagy in the pathogenesis of acute myeloid leukemia (AML), a heterogeneous group of hematopoietic malignancies, have had limited investigation. Acute promyelocytic leukemia (APL), a unique subtype of AML, is characterized by the block of terminal differentiation at the promyelocytic stage of neutrophil maturation. Almost all cases of APL express an oncogenic fusion protein involving the retinoic acid receptor-α (RARα) gene due to chromosome translocations. More than 95% of APL patients express the PML (for promyelocytic leukemia)-RARα fusion protein that is generated as a result of a chromosome translocation 15;17, and this protein is essential for APL pathogenesis.Citation15–Citation18 More importantly, PML-RARα is directly targeted and degraded by two effective therapeutic agents for APL, all-trans retinoic acid (ATRA) and arsenic trioxide (As2O3.Citation19–Citation22 In addition to PML-RARα, at least five other APL-related RARα-carrying fusion proteins (PLZF-RARα, NPM-RARα, NuMA-RARα, STAT5b-RARα and PRKAR1A-RARα) have been identified and characterized at the molecular level.Citation23,Citation24 More recently, it is reported that both ATRA and As2O3 induce autophagy via the mammalian target of rapamycin (mTOR) pathway in APL cells, and autophagic degradation significantly contributes to the basal turnover as well as the therapy-induced proteolysis of the PML-RARα protein.Citation25,Citation26 Here, we provide the first demonstration that the expression of the PML-RARα, but not PLZF-RARα and NPM-RARα, fusion protein can significantly increase constitutive autophagic activity in leukemic and nonleukemic cell lines, as well as APL cells in vivo, possibly through a decreased activation of Akt/mTOR.
Results
PML-RARα fusion protein triggers constitutive autophagic activation in leukemic and nonleukemic cells.
To assess the possible involvement of the PML-RARα fusion protein in autophagy, the U937/PR9 cell line, a subclone of the monocytic leukemia cell line U937 stably transfected with the PML-RARα cDNA under the control of a zinc-inducible promoter,Citation27,Citation28 was treated with 100 µM ZnSO4 for different times together with its parental U937 cell line and EBSS incubation as controls. In the presence of Zn2+ , the PML-RARα protein was induced in U937/PR9 cells () but not in U937 cells (data not shown). LC3-II was regarded as a faithful marker of autophagic activity that can be distinguished from LC3-I by SDS-PAGE as a result of the slightly increased mobility of LC3-II that occurs as a consequence of increased hydrophobicity caused by lipidation.Citation29 The substitution of growth medium with the nutrient-free EBSS medium, the latter being a powerful inducer of autophagy,Citation30 increased LC3-II protein in U937/PR9 and U937 cells (> and Fig. S1A). More intriguingly, induction of the PML-RARα protein by Zn2+ significantly increased the endogenous LC3-II protein in U937/PR9 cells and this effect intensified with increasing expression level of this fusion protein (). PML-RARα expression also increased the expression of Beclin 1 (the mammalian ortholog of yeast Atg6) (), another indicator for autophagy initiation that participates in autophagosome formation by interacting with hVps34,Citation31 while it decreased the expression of p62 protein (), which is usually degraded through autophagy.Citation32
The most reliable and conventional technique for the visualization of autophagic vacuolization is transmission electron microscopy,Citation33 as some of the other detection methods involve numerous intrinsic pitfalls. With this in mind, U937/PR9 and U937 were observed by TEM after treatment with or without 100 µM Zn2+ for 4 h, together with a 1 h EBSS treatment as the positive control. Autophagic vacuoles (AVs) including double-membrane vesicles or single-membrane vesicles with intracellular contents (cytosol and organelles), namely autophagosomes and autolysosomes, could be clearly captured in the EBSS-treated cells (). Compared with the untreated U937/PR9 cells with less AVs and normal cellular organelles (such as mitochondria), abundant AV-like structures were observed in the cytoplasm of the Zn2+ -treated U937/PR9 cells (). As summarized in , the percentage of AV+ cells was significantly increased in the Zn2+-treated U937/PR9 cells (, left). Moreover, among the AV+ cells, the number of AVs per cell in PML-RARα-expressing U937/PR9 cells was also greater than those in the control cells (right part, ). However, Zn2+ addition did not produce such an effect in the parental U937 cells (Fig. S1A and B). All these data supported the hypothesis that PML-RARα expression induces constitutive autophagy activation in the leukemic U937 cells.
To address whether the above phenomenon is cell type-dependent, the PML-RARα fusion gene-expressing plasmid was transfected into the human osteosarcoma cell line, U2OS and embryonic kidney cell line, HEK293T. The results revealed that the ectopic expression of PML-RARα also induced autophagy in these cells, as judged by increases in the expression levels of LC3-II and Beclin 1 (Fig. S3), as well as the structures and numbers of AVs under TEM (Fig. S4 for HEK293T cells and data not shown for U2OS cells). Notably, the expression of the LC3-I protein was also enhanced in parallel with LC3-II accumulation following the expression of PML-RARα. Therefore, the expression of LC3 mRNA was examined by real-time RT-PCR. Unexpectedly, inducible or transfected expression of PML-RARα did not alter the LC3 mRNA level (Fig. S3A and B and data not shown). Furthermore, by monitoring the distribution of the fluorescent protein-tagged LC3 fusion protein, one could visually track autophagic responses by fluorescence microscopy when the cytoplasmically and diffusely distributed LC3-I is converted into the punctate LC3-II, which is targeted directly to the preautophagosomal and autophagosomal membranes.Citation29 Thus, GFP-tagged human LC3 plasmid together with DsRed, DsRed-PML-RARα or DsRed-wild type PML expression vector were transiently transfected into U2OS cells. Twenty-four hours later, the cells transfected with GFP-LC3 only were subsequently incubated with EBSS for 1 h or with 0.5 µM rapamycin (another widely used autophagy inducerCitation33) for 6 h as positive controls. The GFP-LC3+ cells incubated with EBSS or rapamycin showed dramatic transition from the diffuse cytoplasmic pattern to the punctate membrane pattern as assessed by calculating the percentages of punctate GFP-LC3+ cells (10.3 ± 0.2% for control; 52.0 ± 0.7% for EBSS; 60.2 ± 3.1% for rapamycin). Compared with the cells transfected with the DsRed vector (13.7 ± 3.1%), PML expression did not cause GFP-LC3 aggregation (10.4 ± 1.2%). More intriguingly, there was a significantly higher percentage of cells with the GFP-LC3 aggregation (40.7 ± 2.4%) in cells transfected with DsRed-PML-RARα, which presented a PML-RARα expression-specific microspeckled localization in the nucleus due to the disruption of the PML nuclear body ().Citation23 The statistical analysis of GFP-LC3 or endogenous LC3 dots per cell was also consistent with this observation (Figs. S1D and S5). The results suggested that the overexpression of PML-RARα protein, but not the wild-type PML, induces constitutive autophagy activation in a cell type-independent manner. It should be pointed out that, following the overexpression of PML protein, either ectopically expressed GFP-LC3 () or endogenous LC3 (Fig. S5) was partially co-localized within PML nuclear bodies.
The constitutive autophagic activity also exists in leukemic cells from PML-RARα-transplanted leukemic mice.
To evaluate the in vivo effect of PML-RARα on autophagy, leukemic cells from hMRP8-PML-RARα transgenic mice were intravenously injected into the syngenic FVB/N mice.Citation34 Based on our previous experience,Citation35,Citation36 we successfully generated transplant leukemic mice at about 29 d after injection of 3 × 105 cells per mouse, as evidenced by the accumulation of strictly monomorphic and immature promyelocyte-like cells in peripheral blood, BM and spleen (). We compared LC3 and p62 protein levels in leukemic cell-infiltrated organs between the normal and leukemic mice. The results revealed that the LC3-II protein significantly increased while p62 decreased in BM and spleen from leukemic mice with PML-RARα expression, compared with those from normal mice (). Furthermore, TEM observation demonstrated that large numbers of AVs were accumulated in the cytosol of the immature promyelocytes from the BM of leukemic mice, compared with ones from normal mice ( and D). These results indicated the presence of the increased constitutive autophagic activity in leukemic cells from an in vivo source.
Increased autophagic activity cannot be seen with the APL-specific PLZF-RARα and NPM-RARα fusion proteins.
Other rare chromosomal translocations in certain cases of APL involve fusion proteins disrupting the RARα locus on chromosome 17, such as PLZF-RARα and NPM-RARα.Citation23,Citation37 To test whether these variant fusion proteins had autophagy-modulating capabilities, U2OS cells were transiently co-transfected with GFP-LC3 along with a HcRed-PLZF-RARα or DsRed-PML-RARα expression plasmid. To exclude possible disturbance caused by the overlap of the CFP (CFP-NPM-RARα) and GFP (GFP-LC3) channels, we co-transfected a Myc-LC3 plasmid with CFP-NPM-RARα. Different from the PML-RARα-induced GFP-LC3+ punctate structures, HcRed-PLZF-RARα or CFP-NPM-RARα expression did not significantly alter GFP-LC3 or Myc-LC3 localization from the diffuse pattern into the punctate pattern ( and B). Similarly, the overexpression of NPM-RARα or PLZF-RARα did not increase the level of LC3-II in transfected U2OS cells () and in the U937 cells with Zn2+-inducible expression of PLZF-RARα (). Consistently, the increased endogenous LC3 aggregation was only found in PML-RARα-expressing cells (Fig. S5).
hVps34 inhibitor prevents PML-RARα-induced constitutive autophagy activation in the manner of on-rate increase.
Although the membrane-conjugated LC3-II found in autophagosomes is typically used to estimate the abundance of autophagosomes before they are degraded through fusion with lysosomes, LC3-II itself is also degraded by lysosomal proteases in the late phase of autophagy. Therefore, the accumulation of autophagosomes induced by PML-RARα could involve an enhanced autophagic sequestration (on-rate increase) or a reduced degradation of autophagic material (off-rate decrease). To distinguish between these possibilities, we performed an “autophagic flux” assay and assessed PML-RARα-induced vacuolization by monitoring the localization of GFP-LC3 and LC3 conversion, either in the presence or absence of the cysteine protease inhibitor E64d and the aspartic protease inhibitor pepstatin A. Consistent with previous reports,Citation29,Citation33 the addition of these two protease inhibitors to the starvation medium could effectively inhibit the degradation of lysosomal protein, resulting in further accumulation of LC3-II and the increase of GFP-LC3 puncta (Fig. S6A–C). Intriguingly, the inhibitors also increased the accumulation of GFP-LC3 puncta ( and B) and LC3-II protein (), suggesting that PML-RARα enhances autophagic activity in the manner of on-rate increase of AV formation.
Furthermore, distinct classes of PtdIns3Ks are involved in the signaling pathways that control autophagy in mammalian cells. Initiation of the autophagy process requires hVps34 and its complex formation with Beclin 1 and protein kinase 150.Citation38 The addition of 3-MA, which prevents autophagy at the sequestration step,Citation39 could significantly reduce the EBSS-induced accumulation of GFP-LC3+ cells and LC3 conversion (Fig. S6A, B and D). Thus, U2OS cells were transiently transfected with DsRed-PML-RARα for 24 h, followed by treatment with or without 10 mM 3-MA for an additional 4 h. As shown in , B and D, 3-MA treatment significantly suppressed the increase of the punctate GFP-LC3+ cells and LC3-II protein accumulation induced by PML-RARα expression. Similar effects on PML-RARα-induced autophagy activation were also seen in U937/PR9 cells treated with 3-MA and two lysosome inhibitors (data not shown). Of note, our results showed that the constitutive autophagy activation induced by the fusion protein did not accelerate the degradation of ectopic expression of PML-RARα ().
PML-RARα-induced autophagic activation is possibly mediated by a decrease in Akt/mTOR activation in PR9 cells.
To ask whether the PML-RARα-induced constitutive autophagy activation involves the Akt-mTOR signaling pathway, we examined the phosphorylation state of Akt and the downstream substrates of mTOR. The results showed that Akt activation, as determined by phosphorylation at Thr308 and Ser473, was significantly inhibited in PML-RARα-expressing cells. After the induction of PML-RARα protein in Zn+ -treated U937/PR9 cells, the mTOR protein level remained unchanged but its activation was significantly reduced in U937/PR9 cells, as assessed by a decrease in the phosphorylation of p70-S6K1 (, left part), the direct substrate of mTOR.Citation40 However, these alterations did not occur in the Zn+-treated U937 (, right part) and U937/PLZF-RARα cells ().
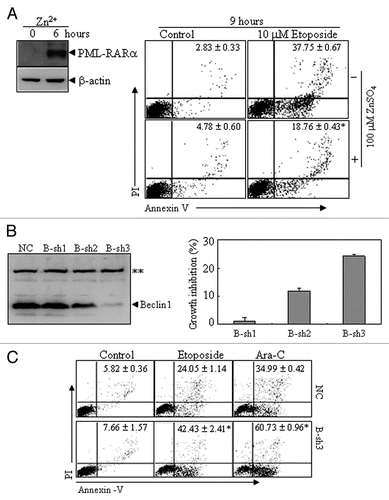
Autophagy contributes to the anti-apoptotic function of PML-RARα.
To explore the potential role of PML-RARα-induced constitutive autophagy activation, U937/PR9 cells were induced with or without Zn2+ for 6 h, followed by treatment with or without 10 µM etoposide for an additional 9 h. As assessed by the percentage of annexin-V+ cells, etoposide-induced apoptosis was significantly reduced in the presence of the inducible expression of PML-RARα compared with that in cells without Zn2+ incubation (), which was consistent with the previous notion that PML-RARα presents anti-apoptotic function.Citation41 To investigate the potential role of increasing constitutive autophagy in anti-apoptotic effects of the PML-RARα protein, we transfected shRNAs against beclin 1 (named B-Sh1,2,3) into the APL cell line NB4. As shown in , B-sh2 and B-sh3, but not B-sh1, partially or completely suppressed beclin 1 expression. Interestingly, the silencing of beclin 1 expression inhibited the growth of these leukemic cells and this effect was consistent with the degree of silencing. Furthermore, the NB4 cells expressing B-sh3 were found to be more prone to apoptosis induction through treatment with etoposide or Ara-C than NC cells (). Collectively, these results indicated that autophagy contributes to the growth advantage and anti-apoptotic properties induced by PML-RARα.
Discussion
Autophagy, which occurs at low basal levels in virtually all cells to perform homeostatic functions, such as protein and organelle turnover, is rapidly upregulated when cells need to generate intracellular nutrients and energy during starvation, growth factor withdrawal, or in conditions of high bioenergetic demand. Such upregulation of autophagy also occurs when cells are preparing to undergo structural remodeling, such as during developmental transitions, or when cells need to eliminate damaging cytoplasmic components in some poor environmental conditions, such as oxidative stress, infection or protein aggregate accumulation.Citation42 The regulation of autophagy overlaps closely with signaling pathways that regulate tumorigenesis.Citation12–Citation14,Citation42 In the present study, we demonstrated that the induction of the PML-RARα protein could significantly upregulate constitutive autophagic activation in leukemic U937 cells, as evidenced by the increase of LC3-positive punctate structures, the elevation of LC3-II and Beclin 1 protein levels, the decrease of p62 protein, and the accumulation of cytoplasmic AV-like structures based on TEM. This phenomenon could also be seen in two other nonleukemic cell lines upon the transient expression of PML-RARα. These results suggest that the upregulating effect of PML-RARα fusion protein on autophagy was independent of the cellular context. Besides the PML-RARα fusion protein, rare APL cases also express other fusion proteins due to different chromosome translocations, which always involve the RARα gene in chromosome 17. The in vitro and transgenic mice analysis showed that all these fusion proteins are critical for the pathogenesis of APL.Citation23,Citation43 Therefore, we also investigated the potential effect of the two other APL-related fusion proteins, PLZF-RARα and NPM-RARα, on autophagy. Unlike PML-RARα, unexpectedly, these two fusion proteins have no significant effect on constitutive autophagy formation, as seen in multiple assays for autophagy. We then investigated whether the enhancing effect of PML-RARα on autophagy activation is restricted to the in vitro conditions by using leukemic cells derived from the MRP8 PML-RARα transgenic mice. Transplanted APL-like cells that had infiltrated into the BM and spleen also showed increased constitutive autophagic activation. All these data supported the hypothesis that PML-RARα enhances autophagy generation.
Autophagy involves the delivery of the cytoplasmic cargo sequestered inside double-membrane vesicles to the lysosome, where the captured material, together with the inner membrane, is degraded.Citation42 Our results demonstrated that the lysosomal protease inhibitors E64d and pepstatin A further increased PML-RARα-induced LC3-II accumulation and GFP-LC3 foci formation, indicating that the upregulation of autophagy by PML-RARα was not due to the alterations of an autolysosomal event. On the other hand, the inhibition of autophagy by 3-MA at the sequestration step could significantly reduce the increase of autophagy induced by PML-RARα, indicating that PML-RARα induces autophagy through the increase of autophagosome formation.
The observation that PML-RARα expression triggers mammalian cells to undergo autophagy led to the speculation that the alteration of autophagy-related signaling molecules possibly contributed to PML-RARα-enhanced autophagic activity. As widely reviewed in reference Citation5 and Citation6, autophagy is regulated by a series of signaling molecules such as Atg7 and Vps34. Our results showed that there was no significant difference in the expression of Atg7 and Vps34 in the presence and absence of PML-RARα (data not shown). More recently, it has been demonstrated that an endoplasmic reticulum stress-related autophagic modulator, Grp78, is upregulated in more than 10% of AML patients with a favorable prognosis while Grp78va, a variant form of Grp78, is increased in several human leukemic cells and leukemia patients.Citation44,Citation45 However, Grp78 expression was also not significantly affected by the transient expression of PML-RARα in U2OS and HEK293T cells (data not shown). On the other hand, PML-RARα expression simultaneously increased the accumulation of LC3-I and LC3-II protein, but had no effect on its mRNA, implying the modulation of PML-RARα on LC3 is independent of its transcriptional level. But the exact mechanism of how PML-RARα increases LC3-I protein level remains to be further investigated.
Considering that the promotion of autophagic activity was only specifically produced by PML-RARα, but not by PLZF-RARα or NPM-RARα, we extrapolated that the PML-RARα-induced autophagic activity involves its PML motif. Previous studies show that PML interacts with mTOR and inhibits its activity by negatively regulating its association with Rheb, thus further sequestering mTOR in the nucleus under normal, and especially in hypoxic, conditions.Citation46 Moreover, PML has also been found to interact with the Akt phosphatase, PP2a, thereby causing the dephosphorylation of Akt, which further inhibits mTOR activity.Citation47 However, we found that the transient expression of PML did not influence autophagic activities ( and Figs. S1C and S5), but it appeared to sequester part of the LC3 population within PML nuclear bodies, which deserves further investigation for its significance. The mTOR protein belongs to the phosphatidylinositol kinase-related kinase (PIKK) family and binds several proteins to form two distinct protein complexes, mTORC1 (mTOR complex 1) and mTORC2. Emerging evidence suggests that the inhibition of mTORC1 activity, which is targeted and regulated by several pathways such as nutrient starvation and reduced growth factor, is a crucial step for autophagy induction in eukaryotes.Citation48 The growth factor signaling that regulates mTORC1 mainly involves the insulin/insulin-like growth factor-PI3K-Akt pathway, which negatively regulates autophagy induction. Indeed, our data showed that PML-RARα induction also reduced the activation of Akt with decreased phosphorylation of the direct downstream substrate of mTORC1. Although it remains to be further explored how PML-RARα inhibits Akt/mTOR activation, our results supported the notion that PML-RARα-induced constitutive autophagy activation may involve the Akt-mTOR signaling pathway.
All of the RARα-involving fusion proteins found so far can trigger leukemia with similar or distinct phenotypes in transgenic mice.Citation23,Citation49 However, they present different responses to ATRA and As2O3 in mouse models and human APL patients.Citation49 For example, leukemia involving PML-RARα, but not PLZF-RARα, is responsive to ATRA or As2O3 treatment, which induces the proteolysis of PML-RARα, but not PLZF-RARα. PML-RARα destruction is essential for the induction of terminal differentiation and/or the eradication of leukemia-initiating cells of APL.Citation18 Recently, Isakson et al. reported that both ATRA- and As2O3-induced autophagy contributes significantly to both the basal turnover and the therapy-induced proteolysis of PML-RARα in APL cells. They also showed a correlation between autophagy and therapy-induced differentiation of APL cells. However, here we demonstrated that the autophagy induced by PML-RARα was not accompanied by the acceleration of fusion protein degradation. We extrapolated that the discrepancy was possibly due to difference between the overexpressed and endogenous expression of PML-RARα in APL cells. However, we did find the easy decay of this fusion protein in U937/PR9 cells following induced expression for a longer period of time (>24 h) as previously reported in reference Citation43.
Coincidentally, the studies from two other groups also reported that autophagy is a critical mechanism for As2O3-induced anti-leukemic effect and ATRA-induced cell differentiation by accelerating the degradation of PML-RARα and its interaction with p62.Citation50,Citation51 Although the suppression of beclin 1 expression did not compromise ATRA-induced leukemic cell differentiation (Fig. S7), we found the fusion protein-induced autophagy was responsible for leukemic cell growth and apoptotic resistance when insulting with apoptotic inducers. Taken together, given the critical role of the PML-RARα oncoprotein in APL pathogenesis, this study extends our understanding of the important role of autophagy in the development and treatment of this disease.
Materials and Methods
Cell lines and reagents.
Human acute leukemic cell line NB4, U937 and its subclone U937/PR9 as well as ZnSO4 (Zn2+)-inducible expression of PLZF-RARα leukemic cells (U937/PLZF-RARα),Citation52 were cultured in RPMI-1640 medium (Gibco, 31800-022), supplemented with 10% fetal bovine serum (FBS, Gibco, 26140). HEK293T and U2OS cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco, 12100-046) with 10% FBS. All cell lines were incubated in a 5% CO2/95% air humidified atmosphere at 37°C. For respective induction of PML-RARα and PLZF-RARα in U937/PR9 and U937/PLZF-RARα cells, 100 µM ZnSO4 was added to the culture medium and the expression of fusion proteins was confirmed by protein gel blot. For induction of autophagy as a positive control, cells were incubated in Earle's balanced salt solution (EBSS), which was made according to the media formulations from Invitrogen (14155-063). 3-methyladenine (3-MA, M9281) and pepstatin A (77170) were purchased from Sigma-Aldrich and E64d powder was obtained from Calbiochem (330005). 3-MA was dissolved in ultrapure water to form 200 mM stock solutions with heating. Pepstatin A and E64d were respectively dissolved in DMSO and ethanol as 10 mg/ml stock solutions. Rapamycin obtained from Tocris Bioscience (1292) was dissolved in DMSO as a 1 mM stock solution.
Plasmids and transfection.
Human LC3B cDNA was amplified from leukemic U937 cells by reverse transcription (RT)-PCR and then cloned into pEGFP-C1 expression vector to construct the GFP-LC3 plasmid. The sequence of the cDNA insert was confirmed by sequencing. pSG5-PML-RARα, GFP-PLZF-RARα and CFP-NPM-RARα plasmids were generous gifts from Dr. Shuo Dong of Baylor College of Medicine. DsRed-PML-RARα and HcRed-PLZF-RARα were respectively made by a swap of PML-RARα and PLZF-RARα cDNA into pDsRed-express-C1 (Clontech, 632430) and pHcRed-C1 (Clontech, 6364-1) vectors from pSG5-PML-RARα and GFP-PLZF-RARα. Flag-PML-RARα plasmid was generated with the similar strategy into pFlag-CMV4 expression vector (Sigma-Aldrich, E7158). Myc-LC3 was obtained as a gift from Dr. Qing Zhong of University of California at Berkeley. Transient transfection was performed with Fugene HD transfection reagents according to the manufacturer's procedures (Roche Applied Science, 04709705001).
Leukemic syngenic transplanted mice and histological analyses.
Leukemic blasts from leukemic hMRP8-PML-RARα transgenic mice were collected from bone marrow (BM) and spleen by flushing RPMI1640 medium through long bones and dissociated spleens. To generate leukemic mice, 3 × 105 leukemic blasts were injected into the tail veins of 6- to 8-week-old syngenic FVB/N mice (Shanghai Laboratory of Animal Center, Chinese Academy of Sciences, Shanghai, China) that had received a sub-lethal irradiation with a total of 3.5 Gy. Peripheral blood was obtained from the retro-orbital venous plexus. BM cells were extracted by flushing PBS through mouse long bones. Blood and BM smears were prepared according to standard hematological techniques and stained with Wright's Giemsa stain. Specimens were cut into small pieces and fixed in 10% neutral buffered formalin, paraffin embedded and stained with hematoxylin-eosin (H&E). Animal handling was approved by the committee for humane treatment of animals at SJTU-SM.
Transmission electron microscopy (TEM).
Fresh BM cells and the transfected cells were washed in cold PBS, pelleted and subsequently fixed in pre-cooling 2% glutaraldehyde for 2 h. Cells were then collected by centrifugation and cell pellets were post-fixed in 1% osmium tetroxide. The samples were then rinsed with phosphate buffer followed by an increasing gradient dehydration step using ethanol and then embedding in Epon 812. Ultrathin sections obtained by LEICA Ultracut R were stained with uranyl acetate and lead citrate and electron micrographs were taken with a Philip-CM120 transmission electron microscope. Data quantification was determined in three independent experiments. For counting AVs per cell, 50 cells were randomly observed.
Confocal microscopy and indirect immunofluorescence.
U2OS cells were grown on 13-mm glass cover-slips (VWR, 631-0149) in 12-well plates and then transfected with the indicated plasmids using Fugene HD reagent. Twenty-four hours after transfection, cells were fixed with 4% paraformaldehyde in PBS at room temperature and processed for fluorescence microscopy analysis using confocal microscopy (LEICA TCS SP5). For Myc-LC3 immunostaining process, the fixed cells were stained with rabbit anti-Myc tag antibody (Cell signaling, 2272) and then with anti-rabbit IgG-TR (Santa Cruz, sc-2780). To investigate the distribution of endogenous LC3, cells with transient transfection were fixed and processed with immunostaining by using monoclonal anti-LC3 antibody (MBL, M152-3B) followed by goat anti-mouse IgG-FITC (Santa Cruz, sc-2010) or IgG-TR (Santa Cruz, sc-2781). GFP-LC3-positive autophagosomes per cell were counted and cells with 8 or more green dots were regarded as GFP-LC3 puncta-positive cells. For each experiment, 200 GFP+ cells were calculated.
Protein gel blot.
The whole cell lysates and tissue lysates were extracted in High Salt Buffer (20 mM HEPES, pH 7.9, 20 mM NaF, 1 mM Na3VO4, 1 mM Na4P2O7, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 420 mM NaCl, 0.5 mM PMSF, 20% glycerol, protease and phosphatase inhibitor cocktails) plus 2x SDS, equally loaded on 8–15% SDS-polyacrylamide gels, and subsequently transferred to nitrocellulose membrane (Bio-Rad, 162-0115) and stained with 0.2% Ponceau S red (Sigma-Aldrich, P7170) to ensure equal protein loading. After blocking with 5% nonfat milk in PBS for 1 h at room temperature, the membranes were incubated at 4°C overnight with various specific primary antibodies. This was followed by incubation for 1 h at room temperature with appropriate horseradish peroxidase (HRP)-linked secondary antibodies (Cell Signaling, 7074). The signals were detected by chemiluminescence phototope-HRP kit (Millipore, WBKLS0500) according to the manufacturer's instructions. Polyclonal anti-Beclin 1 (NB110-87318) and anti-Vps34 (NB110-87320) antibodies were purchased from Novus Biologicals. Antibodies against LC3 (L8918), RARα (sc551) and Atg7 (3615) antibodies were purchased respectively from Sigma-Aldrich, Santa Cruz Biotechnology and ProSci Inc. Anti-Grp78 (ab32618) was from Abcam (Cambridge, UK). Antibodies against mTOR (2983), phospho-p70 S6 Kinase (Thr389, 9234), phospho-Akt (Thr308, 2965), phospho-Akt (Ser473, 4058) and Akt (9272) were purchased from Cell Signaling. p70S6 kinase α antibody (sc-9027) was obtained from Santa Cruz Biotechnology. p62 antibody (PM045) was obtained from Medical and Biological Laboratories.
Real-time quantitative reverse transcription polymerase chain reaction.
Total RNA from the cells was extracted by TRIzol reagent (Invitrogen, 15596018), followed by treatment with RNase-free DNase (Promega, M610A). RT-PCR was performed with Promega RNA PCR kit following the manufacturer's instructions. Real-time PCR was performed and data were analyzed as described previously in reference Citation53. cDNA encoding LC3 and β-actin was amplified by the primers: 5′-GTA AGA TCT ATG CCG TCG GAG AAG ACC-3′ (forward) and 5′-GCG AAT TCT TAC ACT GAC AAT TTC ATC CCG-3′ (reverse) for human LC3B and 5′-CAT CCT CAC CCT GAA GTA CCC-3′ (forward) and 5′-AGC CTG GAT AGC AAC GTA CAT G-3′ (reverse) for β-actin.
ShRNA design and transfection.
Three pairs of complementary oligonucleotides specifically against beclin 1 (respectively named B-sh1 to 3) were synthesized, annealed and ligated into pSIREN-RetroQ vector according to the manufacturer's instructions (Clontech, 631526). The target sequences for beclin 1 were shown as following: 5′-AAC TCA GGA GAG GAG CCA TTT-3′ for B-sh1, 5′-AAG ATT GAA GAC ACA GGA GGC-3′ for B-sh2, 5′-AAG GGT CTA AGA CGT CCA ACA-3′ for B-sh3. These target shRNAs and nontarget control shRNA (NC)-containing plasmids were transfected by retrovirus into NB4 cells. The viral supernatant was packaged in HEK293T cells by cotransfecting with pSIREN-RetroQ, pEQPAM (containing gag-pol, produced by Dr. Lishan Su in UNC Chapel Hill, USA) and VSVG (Clontech, PT-334350). After transfection for 48 h, the viral supernatant was collected, filter-sterilized and added onto NB4 cells (2 × 105 cells/well) in 6-well plates with the medium containing 4 µg/ml of polybrene (Millipore, TR-1003-G) and 0.6 µg/ml of puromycin (Calbiochem, 540411) to select for the stably transfected cells after another 48 h.
Statistical analysis.
Student's t-test was used to evaluate differences between two groups. A p value of less than 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Financial Support
This work is supported in part by grants from the National Basic Research Program of China (973 Program) (NO2009CB918404), National Natural Science Foundation of China (NSFC, 30630034, 30870523, 90813034, 81070431), Chinese Academy of Sciences (KSCX2-YW-R-097), Science and Technology Commission of Shanghai (08JC1413400) as well as leading academic discipline project of Shanghai Municipal Education Commission (J50201). The contribution of S.C.K. was supported by NIH grant CA95274. Dr. Guo-Qiang Chen is supported by Shanghai Ling-Jun Talent Program.
Abbreviations
| APL | = | acute promyelocytic leukemia |
| Atg | = | autophagy-related |
| hVps34 | = | human vacuolar protein sorting 34 |
| PtdIns3KC3 | = | class III phosphatidylinositol-3-kinase |
| LC3-I | = | microtubule-associated protein 1 light chain 3 type I |
| AML | = | acute myeloid leukemia |
| RARα | = | retinoic acid receptor alpha |
| PML | = | promyelocytic leukemia |
| ATRA | = | all-trans retinoic acid |
| As2O3 | = | arsenic trioxide |
| mTOR | = | mammalian target of rapamycin |
| EBSS | = | Earle's balanced salt solution |
| 3-MA | = | 3-methyladenine |
| TEM | = | transmission electron microscopy |
| AVs | = | autophagic vacuoles |
Figures and Tables
Figure 1 The effect of the ectopic expression of PML-RARα on autophagic activity in leukemic and nonleukemic cells. (A and B) U937/PR9 (A) and indicated cells (B) were respectively treated with or without 100 µM ZnsO4, or with EBSS for the indicated hours. Cell lysates were harvested for immunoblotting proteins as indicated. LC3-II protein expression was quantified according to the densitometric value and the relative folds against untreated cells were shown as means ± SD from three independent tests. (C) TEM micrographs of the indicated treatments in U937/PR9 cells. AV-like structures were indicated by arrowheads. “Nuc” stands for nucleus. The scale bars on control, EBSS- and Zn2+-treated cells were 1,000 nm. The high-magnification picture (right part of bottom row) was from the framed area in the Zn2+-treated cell (scale bar = 500 nm). (D) Quantification of data in part (C). The percentage of AV+ cells was shown in the left part and among these AV+ cells, the percentage of cells with indicated AV numbers per AV+ cell were indicated in the right part. The symbol *indicates a p value of less than 0.05 against U937/PR9 control cells. (E) U2Os cells were transfected with GFP-LC3 alone (top row) or co-transfected with GFP-LC3 and DsRed-PML-RARα, DsRed-PML or DsRed vector (bottom row). After 24 h, cells were examined by confocal microscopy. The representative images of the indicated transfected cells with the corresponding treatments were shown. The values (x ± SD) represent the percentage of GFP-LC3 puncta-positive cells from three independent experiments. The symbols * and # respectively indicated p values of less than 0.001 and 0.01 compared with the untreated cells with GFP-LC3 transfection alone. The symbol & indicated a p value of less than 0.05 compared with the cells co-transfected with DsRed and GFP-LC3 plasmids. All experiments were repeated at least three times and similar results were obtained.
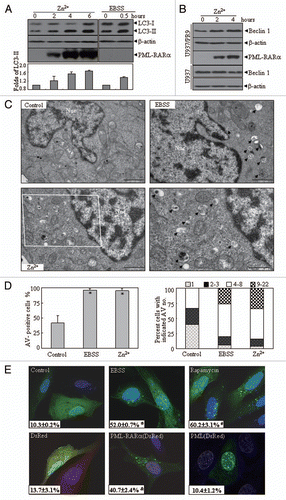
Figure 2 In vivo effect of PML-RARα expression on autophagy in leukemic mice. Leukemic cells (3 × 105) from BM and spleen of APL transgenic mice were injected into FVB/N mice via the tail vein. Animals were sacrificed about 29 d after leukemic cell transplantation. (A) Cytologic analysis by Wright's Giemsa staining of peripheral blood (PB) and BM. Histopathological sections of spleen from the indicated mice were stained with H&E. Images were observed by microscopy with a Nikon digital camera. (B) The proteins from BM cells and spleens of the normal and diseased mice were extracted and the indicated proteins were detected by protein gel blot. (C) Representative electron micrographs of myelocytes and promyelocytes in BM from normal and diseased mice were observed by TEM. The scale bars were 2,000 nm on the top images and the high-magnification picture (bottom part) was from the framed area of the middle part (scale bar = 1,000 nm). (D) The percentage of cells with the indicated AV numbers per immature granulocyte in BM from the indicated mice were calculated and summarized. All experiments were repeated three times and similar results were obtained.
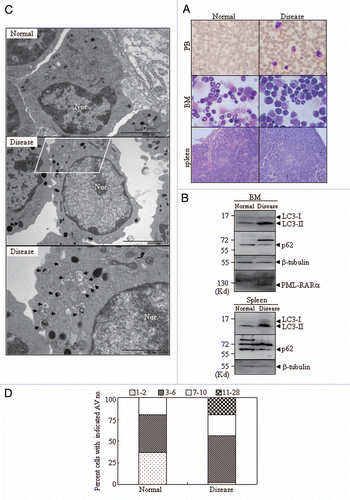
Figure 3 The effects of PLZF-RARα and NPM-RARα fusion proteins on autophagy. U2OS cells were co-transfected with GFP-LC3 and hcRed-PLZF-RARα, DsRed-PML-RARα, or the corresponding empty vectors (HcRed or DsRed), or with Myc-LC3 and CFP-NPM-RARα or the CFP vector. After transfection for 24 h, the cells were stained with anti-Myc antibody or directly analyzed by confocal microscopy. The Myc-LC3 signal was imaged on the red fluorescent protein (RFP) channel, and the CFP signal was obtained on the CFP channel. (A) Representative images of the cells transfected with the indicated constructs were shown. Arrowheads indicate cells with the expression of proteins as labeled. (B) The percentage of GFP-LC3 puncta-positive cells (left part) and the total number of GFP-LC3 dots per cell (right part) were calculated. The symbol *indicates a p value of less than 0.001 compared with the cells co-transfected with DsRed and GFP-LC3 plasmids. (C) After a transient transfection with the indicated plasmids, U2OS cells were extracted and detected by protein gel blot. The transfected expressions of APL-related fusion proteins were confirmed by a RARα antibody. (D) U937/PLZF-RARα cells were treated with 100 µM ZnSO4 for the indicated hours and the cell lysates were harvested for immunoblotting. Relative LC3-II in (C and D) was determined by the ratio of densitometric value of LC3-II relative to the corresponding empty-transfected or the untreated controls. All experiments were repeated three times with similar results, and all values were shown as means with bar as SD of three independent experiments.
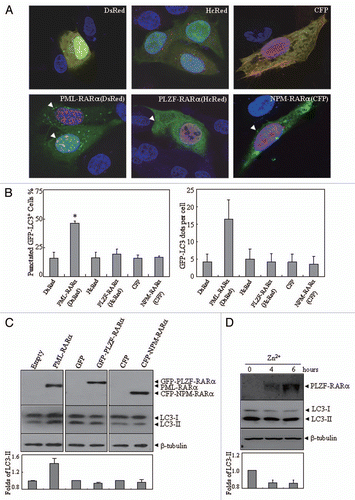
Figure 4 The effects of lysosomal enzyme inhibitors and 3-MA on altered localization and expression pattern of LC3 protein induced by PML-RARα. (A and B) U2OS cells were transiently co-transfected with GFP-LC3 and DsRed-PML-RARα (bottom parts) or DsRed (upper parts) for 24 h, followed by treatment with or without 3-MA (10 mM) and pepstatin A (10 µg/ml) plus E64d (10 µg/ml) for an additional 4 h. Then the cells were observed by confocal microscopy. The representative images for each treatment are shown (A). Quantification data of the percentage of GFP-LC3 puncta-positive cells and GFP-LC3 dots per cell are shown in the left and right parts, respectively (B). Symbol * stands for p < 0.05. (C and D) U2OS cells were transiently transfected with the indicated concentrations of Flag-PML-RARα (Flag-P-R) expression vector or the empty Flag vector (1.0 µg of each plasmid was transfected in C) for 24 h and then treated with or without pepstatin A (10 µg/ml) and E64d (10 µg/ml) (C), 3-MA (10 mM) (D) or an equal volume of the vehicle for an additional 4 h. Cell lysates were harvested and analyzed by protein gel blot with specific antibodies. Relative LC3-II expression in (C and D) was determined by the ratio of the densitometric value of LC3-II relative to the empty-transfected controls with vehicle treatment. All experiments were repeated three times with similar results, and values are shown as means with bar as SD of three independent experiments.
Figure 5 The effects of PML-RARα and PLZF-RARα overexpression on the Akt-mTOR pathway in leukemic cells. U937/PR9 and U937 cells (A) as well as U937/PLZF-RARα cells (B) were treated with or without 100 µM ZnSO4 for the indicated hours. Total proteins were harvested and the indicated proteins were analyzed by protein gel blot. The induced expression of PML-RARα and PLZF-RARα proteins in the respective cells were confirmed by the RARα antibody. All experiments were repeated at least three times and similar results were obtained.
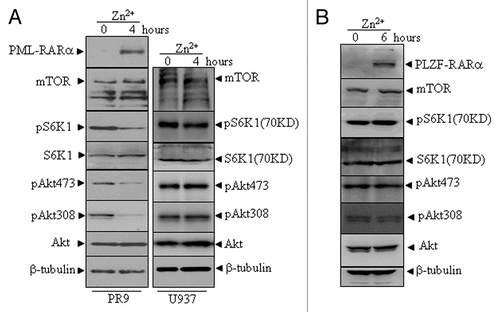
Figure 6 The effects of PML-RARα-induced autophagic activity on apoptosis in leukemic cells. (A) U937/PR9 cells were pre-treated with or without 100 µM ZnSO4 for 6 h, followed by treatment with or without etoposide. Then annexin V+ cells with or without PI staining were detected by flow cytometry. PML-RARα protein induction was also shown. (B) NB4 cells were stably transfected with B-sh1, B-sh2, B-sh3 or NC. The effect of Beclin 1 suppression by shRNAs was analyzed by protein gel blot (left part) and the percentage of growth inhibition against NC cells are shown as means ± SD from three independent experiments (right part). (C) NC- and B-sh3-expressing cells were respectively treated with or without etoposide (1.5 µM), or with Ara-C (2 µM) for 24 h. Then annexin V+ cells with or without PI staining were detected by flow cytometry. In (A and C), the values represent the percentages of the corresponding cells in means ± SD of triplicates from an independent experiment. The symbol * indicates p values of less than 0.01 compared with etoposide-treated U937/PR9 cells without Zn2+ induction (A) or NC cells with corresponding treatments (C). The symbol **points to a nonspecific band. All the experiments were repeated three times and similar results were obtained.
Additional material
Download Zip (733.2 KB)Acknowledgements
We thank Dr. Shuo Dong and Dr. Qing Zhong for generously providing the plasmids pSG5-PML-RARα, GFP-PLZF-RARα and CFP-NPM-RARα, as well as Myc-LC3. U937/PLZF-RARα cells were kindly provided by Dr. Jian-Hua Tong from the Shanghai Institute of Hematology (SIH). We also very much appreciated the help from Dr. Han-Yi Zhuang in our laboratory for her careful English editing of the manuscript.
References
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931 - 937; PMID: 17712358; http://dx.doi.org/10.1038/nrm2245
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463 - 477; PMID: 15068787; http://dx.doi.org/10.1016/S1534-5807(04)00099-1
- Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J 2001; 20:5971 - 5981; PMID: 11689437; http://dx.doi.org/10.1093/emboj/20.21.5971
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001; 2:211 - 216; PMID: 11265251; http://dx.doi.org/10.1038/35056522
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; PMID: 19653858; http://dx.doi.org/10.1146/annurev-genet-102808-114910
- Meijer AJ, Codogno P. Autophagy: regulation and role in disease. Crit Rev Clin Lab Sci 2009; 46:210 - 240; PMID: 19552522; http://dx.doi.org/10.1080/10408360903044068
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861 - 2873; PMID: 18006683; http://dx.doi.org/10.1101/gad.1599207
- Mann SS, Hammarback JA. Molecular characterization of light chain 3. A microtubule binding subunit of MAP1A and MAP1B. J Biol Chem 1994; 269:114927; PMID: 7908909
- Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007; 130:165 - 178; PMID: 17632063; http://dx.doi.org/10.1016/j.cell.2007.05.021
- Tanida I, Sou YS, Ezaki J, Minematsu-Ikeguchi N, Ueno T, Kominami E. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J Biol Chem 2004; 279:36268 - 36276; PMID: 15187094; http://dx.doi.org/10.1074/jbc.M401461200
- Levine B, Yuan J. Autophagy in cell death: an innocent convict?. J Clin Invest 2005; 115:2679 - 2688; PMID: 16200202; http://dx.doi.org/10.1172/JCI26390
- Hippert MM, O'Toole PS, Thorburn A. Autophagy in cancer: good, bad or both?. Cancer Res 2006; 66:9349 - 9351; PMID: 17018585; http://dx.doi.org/10.1158/0008-5472.CAN-06-1597
- Lefranc F, Facchini V, Kiss R. Proautophagic drugs: a novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist 2007; 12:1395 - 1403; PMID: 18165616; http://dx.doi.org/10.1634/theoncologist.12-12-1395
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961 - 967; PMID: 17972889; http://dx.doi.org/10.1038/nrc2254
- Grimwade D, Lo Coco F. Acute promyelocytic leukemia: a model for the role of molecular diagnosis and residual disease monitoring in directing treatment approach in acute myeloid leukemia. Leukemia 2002; 16:1959 - 1973; PMID: 12357347; http://dx.doi.org/10.1038/sj.leu.2402721
- Licht JD. Reconstructing a disease: What essential features of the retinoic acid receptor fusion oncoproteins generate acute promyelocytic leukemia?. Cancer Cell 2006; 9:73 - 74; PMID: 16473273; http://dx.doi.org/10.1016/j.ccr.2006.01.024
- Zelent A, Guidez F, Melnick A, Waxman S, Licht JD. Translocations of the RARα gene in acute promyelocytic leukemia. Oncogene 2001; 20:7186 - 7203; PMID: 11704847; http://dx.doi.org/10.1038/sj.onc.1204766
- Nasr R, Guillemin MC, Ferhi O, Soilihi H, Peres L, Berthier C, et al. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat Med 2008; 14:1333 - 1342; PMID: 19029980; http://dx.doi.org/10.1038/nm.1891
- Lallemand-Breitenbach V, Jeanne M, Benhenda S, Nasr R, Lei M, Peres L, et al. Arsenic degrades PML or PML-RARα through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat Cell Biol 2008; 10:547 - 555; PMID: 18408733; http://dx.doi.org/10.1038/ncb1717
- Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, et al. Arsenic trioxide controls the fate of the PML-RARα oncoprotein by directly binding PML. Science 2010; 328:240 - 243; PMID: 20378816; http://dx.doi.org/10.1126/science.1183424
- Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J, Cai X, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood 1997; 89:3345 - 3353; PMID: 9129041
- Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 1997; 89:3354 - 3360; PMID: 9129042
- Melnick A, Licht JD. Deconstructing a disease: RARα, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 1999; 93:3167 - 3215; PMID: 10233871
- Qiu JJ, Lu X, Zeisig BB, Ma Z, Cai X, Chen S, et al. Leukemic transformation by the APL fusion protein PRKAR1A-RARα critically depends on recruitment of RXRα. Blood 2010; 115:643 - 652; PMID: 19965660; http://dx.doi.org/10.1182/blood-2009-07-232652
- Isakson P, Bjoras M, Boe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010; 116:2324 - 2331; PMID: 20574048; http://dx.doi.org/10.1182/blood-2010-01-261040
- Qian W, Liu J, Jin J, Ni W, Xu W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via upregulation of Beclin-1. Leuk Res 2007; 31:329 - 339; PMID: 16882451; http://dx.doi.org/10.1016/j.leukres.2006.06.021
- Grignani F, Ferrucci PF, Testa U, Talamo G, Fagioli M, Alcalay M, et al. The acute promyelocytic leukemia-specific PML-RARα fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell 1993; 74:423 - 431; PMID: 8394219; http://dx.doi.org/10.1016/0092-8674(93)80044-F
- Grignani F, Testa U, Rogaia D, Ferrucci PF, Samoggia P, Pinto A, et al. Effects on differentiation by the promyelocytic leukemia PML/RARα protein depend on the fusion of the PML protein dimerization and RARα DNA binding domains. EMBO J 1996; 15:4949 - 4958; PMID: 8890168
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 326; PMID: 20144757; http://dx.doi.org/10.1016/j.cell.2010.01.028
- Munafó DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci 2001; 114:3619 - 3629; PMID: 11707514
- Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2001; 2:330 - 335; PMID: 11306555; http://dx.doi.org/10.1093/emboreports/kve061
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 24145; PMID: 17580304; http://dx.doi.org/10.1074/jbc.M702824200
- Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, Hangen E, et al. Methods for assessing autophagy and autophagic cell death. Methods Mol Biol 2008; 445:29 - 76; PMID: 18425442; http://dx.doi.org/10.1007/978-1-59745-157-4_3
- Brown D, Kogan S, Lagasse E, Weissman I, Alcalay M, Pelicci PG, et al. A PMLRARα transgene initiates murine acute promyelocytic leukemia. Proc Natl Acad Sci USA 1997; 94:2551 - 2556; PMID: 9122233; http://dx.doi.org/10.1073/pnas.94.6.2551
- Liu W, Guo M, Xu YB, Li D, Zhou ZN, Wu YL, et al. Induction of tumor arrest and differentiation with prolonged survival by intermittent hypoxia in a mouse model of acute myeloid leukemia. Blood 2006; 107:698 - 707; PMID: 16166593; http://dx.doi.org/10.1182/blood-2005-03-1278
- Le Beau MM, Davis EM, Patel B, Phan VT, Sohal J, Kogan SC. Recurring chromosomal abnormalities in leukemia in PML-RARA transgenic mice identify cooperating events and genetic pathways to acute promyelocytic leukemia. Blood 2003; 102:1072 - 1074; PMID: 12689927; http://dx.doi.org/10.1182/blood-2003-01-0155
- Catalano A, Dawson MA, Somana K, Opat S, Schwarer A, Campbell LJ, et al. The PRKAR1A gene is fused to RARA in a new variant acute promyelocytic leukemia. Blood 2007; 110:4073 - 4076; PMID: 17712046; http://dx.doi.org/10.1182/blood-2007-06-095554
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol-3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 2000; 275:992 - 998; PMID: 10625637; http://dx.doi.org/10.1074/jbc.275.2.992
- Araki N, Hamasaki M, Egami Y, Hatae T. Effect of 3-methyladenine on the fusion process of macropinosomes in EGF-stimulated A431 cells. Cell Struct Funct 2006; 31:145 - 157; PMID: 17146146; http://dx.doi.org/10.1247/csf.06029
- Díaz-Troya S, Perez-Perez ME, Florencio FJ, Crespo JL. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 2008; 4:851 - 865; PMID: 18670193
- Nason-Burchenal K, Takle G, Pace U, Flynn S, Allopenna J, Martin P, et al. Targeting the PML/RARα translocation product triggers apoptosis in promyelocytic leukemia cells. Oncogene 1998; 17:1759 - 1768; PMID: 9778041; http://dx.doi.org/10.1038/sj.onc.1202075
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; PMID: 18191218; http://dx.doi.org/10.1016/j.cell.2007.12.018
- Guibal FC, Alberich-Jorda M, Hirai H, Ebralidze A, Levantini E, Di Ruscio A, et al. Identification of a myeloid committed progenitor as the cancer-initiating cell in acute promyelocytic leukemia. Blood 2009; 114:5415 - 5425; PMID: 19797526; http://dx.doi.org/10.1182/blood-2008-10-182071
- Schardt JA, Weber D, Eyholzer M, Mueller BU, Pabst T. Activation of the unfolded protein response is associated with favorable prognosis in acute myeloid leukemia. Clin Cancer Res 2009; 15:3834 - 3841; PMID: 19470730; http://dx.doi.org/10.1158/1078-0432.CCR-08-2870
- Ni M, Zhou H, Wey S, Baumeister P, Lee AS. Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP. PLoS ONE 2009; 4:6868; PMID: 19718440; http://dx.doi.org/10.1371/journal.pone.0006868
- Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, et al. PML inhibits HIF-1α translation and neoangiogenesis through repression of mTOR. Nature 2006; 442:779 - 785; PMID: 16915281; http://dx.doi.org/10.1038/nature05029
- Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature 2006; 441:523 - 527; PMID: 16680151; http://dx.doi.org/10.1038/nature04809
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett 2010; 584:1287 - 1295; PMID: 20083114; http://dx.doi.org/10.1016/j.febslet.2010.01.017
- Rego EM, Ruggero D, Tribioli C, Cattoretti G, Kogan S, Redner RL, et al. Leukemia with distinct phenotypes in transgenic mice expressing PML/RARα, PLZF/RARα or NPM/RARα. Oncogene 2006; 25:1974 - 1979; PMID: 16331271; http://dx.doi.org/10.1038/sj.onc.1209216
- Goussetis DJ, Altman JK, Glaser H, McNeer JL, Tallman MS, Platanias LC. Autophagy is a critical mechanism for the induction of the antileukemic effects of arsenic trioxide. J Biol Chem 2010; 285:29989 - 29997; PMID: 20656687; http://dx.doi.org/10.1074/jbc.M109.090530
- Wang Z, Cao L, Kang R, Yang M, Liu L, Zhao Y, et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARα oncoprotein. Autophagy 2011; 7; [Epub ahead of print] 14397 PMID: 21187718
- Alcalay M, Meani N, Gelmetti V, Fantozzi A, Fagioli M, Orleth A, et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J Clin Invest 2003; 112:1751 - 1761; PMID: 14660751
- Zhao KW, Li X, Zhao Q, Huang Y, Li D, Peng ZG, et al. Protein kinase Cdelta mediates retinoic acid and phorbol myristate acetate-induced phospholipid scramblase 1 gene expression: its role in leukemic cell differentiation. Blood 2004; 104:3731 - 3738; PMID: 15308560; http://dx.doi.org/10.1182/blood-2004-04-1630