Abstract
Although mitochondrial impairment has often been implicated in carcinogenesis, the mechanisms of its development in cancer remain unknown. We report here that autophagy triggered by oncogenic K-Ras mediates functional loss of mitochondria during cell transformation to overcome an energy deficit resulting from glucose deficiency. When Rat2 cells were infected with a retrovirus harboring constitutively active K-RasV12, mitochondrial respiration significantly declined in parallel with the acquisition of transformation characteristics. Decreased respiration was not related to mitochondrial biogenesis but was inversely associated with the increased formation of acidic vesicles enclosing mitochondria, during which autophagy-related proteins such as Beclin 1, Atg5, LC3-II and vacuolar ATPases were induced. Interestingly, blocking autophagy with conventional inhibitors (bafilomycin A, 3-methyladenin) and siRNA-mediated knockdown of autophagy-related genes recovered respiratory protein expression and respiratory activity; JNK was involved in these phenomena as an upstream regulator. The cells transformed by K-RasV12 maintained cellular ATP level mainly through glycolytic ATP production without induction of GLUT1, the low Km glucose transporter. Finally, K-RasV12-triggered LC3-II formation was modulated by extracellular glucose levels, and LC3-II formation increased only in hepatocellular carcinoma tissues exhibiting low glucose uptake and increased K-Ras expression. Taken together, our observations suggest that mitochondrial functional loss may be mediated by oncogenic K-Ras-induced mitophagy during early tumorigenesis even in the absence of hypoxia, and that this mitophagic process may be an important strategy to overcome the cellular energy deficit triggered by insufficient glucose.
Introduction
Mitochondria are organelles essential for aerobic ATP production in eukaryotes. Each eukaryotic cell contains hundreds to thousands of mitochondria, a number that is constantly maintained during normal cellular growth. However, the total cellular mitochondrial capacity can dramatically change under different energy demands and physiological conditions, implying that efficient and well-balanced mitochondrial biogenesis and degradation are required.Citation1,Citation2 Thus, prompt biosynthesis and assembly of mitochondrial components and appropriate elimination of defective or worn-out mitochondria by autophagy (mitophagy) are the key mechanisms for maintaining the optimal number of mitochondria within a cell. Through selective mitophagy, cells can avoid the abnormal accumulation of damaged and free-radical-generating mitochondrial remnants that may be related to cellular senescence and even cell death.Citation3–Citation5 While it is well known that mitophagy is triggered by mitochondrial defects including mitochondrial permeability transitionCitation5 and osmotic swellingCitation6 and mitochondrial stresses such as hypoxia, whether autophagy is directly involved in the degradation of intact mitochondria without pre-existing damage or stress remains unclear.
Most solid tumor cells are characterized by energy dependence on aerobic glycolysis accompanied by impairment of the mitochondrial respiratory capacity and these characteristics are consistent with the proposed Warburg effect.Citation7–Citation12 It is widely accepted that impaired mitochondria and aerobic glycolysis are merely cancer epiphenomena, as most solid tumors experience intermittent hypoxia due to the rapid increase in nodular mass during the early stages of cancer development.Citation13 On the other hand, the involvement of respiratory defects in neoplastic transformation has recently been re-evaluated as a marker for tumor progression or cancer prognosis,Citation14,Citation15 and respiration's importance and metabolic alteration in cancer development have further been emphasized.Citation16,Citation17 Although the contribution of mitochondrial gene mutation to cancer promotion has been confirmed,Citation18,Citation19 it is unclear how mitochondrial impairment or loss is induced and whether such defects are causative or correlative factors in cancer development. It is therefore critical to elucidate whether and how mitochondrial functional defects are initially involved in oncogenesis in the absence of hypoxic stimulus.
The Ras protein family members act as central transducers in growth factor-triggered signaling and exhibit potent transforming potential.Citation20,Citation21 The most frequently mutated genes in human cancers, these activating point mutations usually arise at codon 12 of the Ras proteins.Citation22 Interestingly, the vast majority of Ras mutations associated with human disease have been identified in K-Ras, with 32–57% prevalence in colorectal tumors and up to 90% prevalence in pancreatic cancers.Citation22,Citation23 K-Ras mutations also occur in early lesions, including hyperplasia, suggesting a role in driving the early stages of tumor development.Citation24,Citation25 On the other hand, Ras has been linked to upregulation of regulatory glycolytic proteins, including glucose transporters,Citation26,Citation27 and induction of hypoxia inducing factor (HIF)-1α,Citation28,Citation29 implying that glycolytic energy production may be activated by the Ras oncoprotein without hypoxia or pre-existing mitochondrial defects, enabling aerobic glycolysis. Antisense K-Ras treatment increased mitochondrial respiratory protein expression in pancreatic cancer cells,Citation30 demonstrating a potential link to mitochondrial biogenesis. These observations implicate K-Ras in both aerobic glycolysis and mitochondrial impairment, thus prompting us to develop a cell transformation system with oncogenic K-Ras to elucidate the causal relationship between mitochondrial impairment and transformation under nonhypoxic environmental conditions.
Here, we demonstrate that autophagy-mediated mitochondrial functional loss is prominent in cell transformation induced by oncogenic K-Ras even in the absence of pre-existing mitochondrial damage or hypoxic stress. We further identify JNK activation as a major mediator of K-Ras-induced autophagy. Finally, we emphasize that mitochondrial autophagy may serve as an important cellular strategy to overcome a cellular energy deficiency due to insufficient glucose import through expediting glycolysis, thus promoting cancer development.
Results
Mitochondrial respiration is impaired without altered biogenesis during oncogenic K-Ras-induced transformation.
To elucidate the causal relationship between mitochondrial impairment and transforming activity, we employed a cell transformation system lacking cell death. Transformation may accompany cell death, and mitochondrial defects commonly occur in dying cells, complicating the interpretation of the role of mitochondrial defects in transformation. Rat2 fibroblasts acquired transforming activity following infection with a retrovirus harboring activated K-Ras (K-RasV12), as evidenced by anchorage-independent growth on soft agar () and loss of contact inhibition on a plate (Fig. S1A). We clearly observed time-dependent declines in mitochondrial respiratory protein expression and respiratory activity (upper five parts of and C, and Fig. S1B) without additional hypoxic stress, suggesting a close link between the mitochondrial defects and oncogenic transformation. The K-RasV12-induced declines in respiratory subunit expression were also dose-dependent (Fig. S1C). In addition, expressions of non-respiratory mitochondrial proteins such as VDAC, TOM20 and TIM13 also decreased (, middle three parts), implying overall mitochondrial defects. To demonstrate that this mitochondrial impairment was not due to the presence of nearby dead cells, the K-RasV12-infected cells were continuously cultured up to 5 d and subjected to the LIVE/DEAD viability assay. We were unable to identify substantial numbers of dead cells (Fig. S1D); additionally, the time for one cell division was significantly shortened in K-RasV12-treated cells (11.6 ± 1.2 h) compared with control cells (16.8 ± 4.2 h), implying faster proliferation of the K-RasV12-treated cells (Fig. S2A and S2B).
Next, we examined whether the K-RasV12-induced mitochondrial defect was associated with mitochondrial biogenesis by monitoring mRNA levels of respiratory subunits and biogenesis-related transcription factors. We detected no changes in the mRNA levels of respiratory subunits despite a decrease in their protein levels (, upper two parts; , upper five parts). In addition, the mRNA levels of nuclear respiratory factor and mitochondrial transcription factor A, which control both mitochondrial DNA replication and transcription, were not altered (, 3rd and 4th parts). No clear decrease in protein expressions of other regulators of mitochondrial biogenesis, such as PGC1α, ERRα and GABP1α, was observed (Fig. S2C). These observations suggest that functional loss of mitochondria is closely associated with K-RasV12-induced cell transformation without affecting mitochondrial biogenesis, and that this system is suitable for investigating the relationship between mitochondrial impairment and transforming activity.
Functional loss of mitochondria is linked with autophagyassociated acidic vesicle (autolysosome) formation.
To identify the genes causing the K-RasV12-induced mitochondrial impairment, we used the GeneFishing DEG kits to screen genes that were differentially expressed following K-RasV12 overexpression. Out of 50 sequenced genes (either induced or repressed), two autophagy-related genes, V0 ATPase subunit D and becn1, were induced (Fig. S3). Induction of several autophagy-related genes (including these two genes) was further confirmed by RT-PCR or protein gel blot (lower two parts of and E). The inverse relationship between mitochondrial respiratory protein expression and autophagy-related protein expression persisted in the Rat2 clones that stably expressed K-RasV12 (Fig. S4A). We were interested to observe in phase-contrast images that the presence of intracellular vesicles progressively increased over the course of the transformation (Fig. S4B), implying that the vesicles may be autophagy-related.
We further evaluated whether mitochondrial impairment was linked with loss of mitochondrial mass and eventually with autolysosome (lysosome-linked acidic vesicle) formation, the functional step of autophagy-related organelle degradation, by monitoring changes in the masses of mitochondria and lysosome-associated particles or vesicles with membrane potential-independent MitoTracker Red (Figs. S5 and S6) and acidic lysosome-specific LysoTracker Green fluorescence dyes. During K-RasV12-induced transformation, the masses of the lysosomes (dense particles, open circles) and the lysosome-linked acidic vesicles (open squares) obviously increased (). It is noteworthy that most vesicles were integrated with a lysosome (, upper two parts), suggesting that the vesicles are activated autolysosomes. Although the increase in lysosome-associated acidic vesicles was inversely related with mitochondrial mass (), no mitochondria enclosed by the acidic vesicles were identifiable in images of cells co-stained with MitoTracker Red and LysoTracker Green (), possibly because the acidic environment may hinder staining efficiency. Rat2 cells expressing mitochondrially targeted RFP clearly contained RFP-tagged mitochondria entrapped by acidic vesicles (). Mitochondria entrapped by vesicles (autolysosomes) were further confirmed with electron microscopy (). We also tried to evaluate autophagic induction by monitoring punctate GFP-LC3. When Rat2 cells transfected with a GFP-LC3 plasmid were exposed to infection of K-RasV12 retrovirus, we could clearly observe colocalized mitochondria with punctate GFP-LC3, but most cells died (Fig. S7), implying that well-balanced control between anti-death (or survival) signal and autophagic activation by K-Ras is important for transformation.
If the autophagic flux is complete, autolysosome-related vesicles should rarely be seen in phase-contrast images due to their small size and efficient recycling. However, K-Ras induced a lot of large vesicles in size even in phase-contrast images. When we carefully examined electron microscopic images of 26 whole cells, cells possessed average 15 vesicles per cell and less cellular organelles, and about 91% of the vesicles were found to contain organelles or organellular debris (), implying inefficient degradation activity or/and defective recycling. Therefore, we further examined whether the autolysomes are active. When we added chloroquine, a lysosomal inhibitor, to K-Ras infected Rat2 cells, expression levels of mitochondrial proteins were recovered together with p62 and LC3 (). These results suggest that the autolysosomes induced by K-Ras are active in degradation of proteins, but may be insufficient to completely degrade all the entrapped organelles. In addition, existence of clear empty vesicles also implies inefficient amphisome clearance, the final step of autophagy flux. Further detailed mechanisms on the step of amphisome clearance have yet to be elucidated.
Next, we wished to determine whether autophagy was actively responsible for the functional loss of mitochondria, rather than the inevitable elimination of damaged mitochondria. We were surprised to observe that the mitochondrial respiratory function impaired by K-RasV12 was significantly restored by the pharmacological autophagy inhibitors bafilomycin A and 3-methyladenine, as evidenced by the recovery of mitochondrial mass ( and Fig. S8A), respiratory protein expression (), and cellular respiration rate (). This recovery of mitochondrial function was further confirmed by si-RNA-mediated knockdown of the autophagy-related genes ATG5, V0-ATPase and BECN1 ( and Fig. S8B and S8C). Accordingly, we propose that the autophagy activated by K-RasV12 is critically responsible for the functional loss of mitochondria without pre-existing functional disruption by environmental changes such as hypoxia.
JNK is an upstream regulator of autophagymediated mitochondrial loss and is essential for cell transforming activity.
Oncogenic K-Ras is known to promote tumor formation through the mitogenactivated protein kinase (MAPK) pathway.Citation28,Citation31 Indeed, we detected increased phosphorylation of all three MAPKs (Fig. S9A): extracellular signal-regulated kinase (ERK), p38 and Jun N-terminal kinase (JNK). Therefore, MAPK-specific inhibitors (PD98059 for ERK, SP600125 for JNK and PD169316 for p38) were used to evaluate whether a particular MAPK was directly involved in autophagy-associated mitochondrial functional loss. Only the JNK inhibitor SP600125 significantly restored the mitochondrial and lysosomal masses to control levels ( and Fig. S9B) and caused recovery of respiratory protein expression and function (Fig. S9C and , left part). Moreover, K-RasV12induced increased expression of autophagy-related proteins was effectively returned to control levels by treatment with the JNK inhibitor (), highlighting the importance of JNK activation in K-RasV12-induced mitochondrial loss. Recovery of mitochondrial respiratory protein expression and function was confirmed by siRNA-mediated JNK knockdown (, right part; Fig. S9D), and JNK-mediated mitochondrial loss was shown to be critical for K-RasV12-induced cell transformation via the soft-agar assay in the presence of the JNK inhibitor or JNK siRNA ( and Fig. S10).
Cells transformed by K-RasV12 overexpression overcome an energy deficit via mitophagy.
Next, we investigated intracellular energy status to address the underlying linkage between K-RasV12-induced transformation and autophagy-associated mitochondrial loss (mitophagy). During K-RasV12-induced transformation, faster cell growth was clearly observed despite defective mitochondrial function (); intracellular ATP levels were continuously maintained (, left part), whereas LDH activity significantly increased (, right part), implying that cellular ATP production was achieved by activated glycolysis.
We further evaluated dependency of cellular ATP generation on glycolysis of mitochondrial respiration by employing 2-deoxyglucose (DOG, glycolysis inhibitor) and oligomycin (Oli, mitochondrial Complex V inhibitor). When extracellular acidification rate (ECAR, an indication of glycolytic lactate production rate) and mitochondrial oxygen consumption rate (OCR) were monitored using Seahorse XF analyzer, K-RasV12-infected cells displayed higher ECAR (lactate production rate) and lower OCR than those of control cell ( and Fig. S11A). As expected, K-Ras transformed cell has higher glycolytic ATP level (DOG-sensitive ATP level, 49%) and lower mitochondrial ATP level (Oli-sensitive ATP level, 23%) than those of control cell (27% and 55%, respectively) ( and Fig. S11B). In addition, when autophagy of K-RasV12-infected cell was blocked by siRNA-mediated knockdown with siATG5 and si-Beclin 1, total cellular ATP level increased to 142% and 137%, and mitochondrial ATP production rate was recovered to 51% and 54%, respectively, compared with 17% of K-Ras-infected control cells ( and Fig. S11C). For these studies, oligomycin or DOG were added for 3 h in the presence of glucose and pyruvate to monitor cellular capacities for mitochondrial and glycolytic ATP production. These results indicate that autophagy by K-Ras inhibits mitochondrial ATP production by inducing functional loss of mitochondria and also imply that K-Ras may have independent pathways to activate glycolysis and autophagy.
Expression of the glucose transporter GLUT1 and its low Km (1∼2 mM) was previously reported to be critical for activated glycolysis, with Ras inducing GLUT1 expression via hypoxia-inducible factor 1.Citation32,Citation33 However, when we monitored the expression levels of the GLUTs, no significant changes in GLUT1 expression were observed, but GLUT2 (Km20∼25 mM) was induced (, left part). GLUT2 cannot function in the basal low glucose concentration (5 mM) of laboratory media; as expected, glucose uptake activity did not change in a low-glucose environment (, right part). These observations suggest that K-RasV12-induced transformation requires the maintenance of cellular ATP levels via activated glycolysis to support accelerated cell proliferation and transforming activity, a maintenance strategy without augmented uptake of extracellular glucose despite diminished mitochondrial respiration.
We examined whether K-RasV12-induced autophagy was modulated by extracellular glucose levels by monitoring LC3 expression. Interestingly, LC3-II formation clearly increased with lower glucose levels and decreased with higher glucose levels compared with normal media (5 mM glucose; ), implying that autophagy-mediated mitochondrial loss may overcome the energy deficit by tapping into an inefficient intracellular glucose supply. Recently, the enhanced glucose uptake by cancer cells enabled positron emission tomography (PET) imaging with 18F-fluoro-deoxyglucose (FDG) for earlier and simpler cancer diagnosis. However, not all tumor nodules can be detected by PET using 18F-FDG due to the existence of tumors with low glucose uptake.Citation34 When we compared the levels of LC3-II formation and K-Ras expression in human hepatoma tissues with high or low glucose uptake, surprisingly, increased LC3-II formation was observed only in tumor tissues with low glucose uptake and high K-Ras expression; no substantial changes were detected in tumors with high glucose uptake (). Taken together, these observations suggest that cancer cells may maintain cellular ATP levels with nutrients scavenged through autophagy-mediated organelle degradation when glucose is inefficiently supplied (regardless of cause). K-Ras may be in charge of the mitophagy induction required to meet cellular needs under these debris or were empty as shown in electron microscopic images stress conditions.
Discussion
Autophagy is an important degradative process mainly involved in the recycling and turnover of cellular constituents, including damaged proteins and organelles, for the maintenance of cellular homeostasis following environmental stimuli such as endoplasmic reticulum stress or hypoxic stress.Citation35–Citation37 In addition to the role of autophagy in cellular starvation and numerous pathogenic conditions, including bacterial and viral infections, and neurodegenerative and cardiovascular disorders, evidence is accumulating that autophagy contributes to carcinogenesis. However, whether autophagy functions as a guardian or killer in cancer is currently controversial.Citation35,Citation38,Citation39 An anticancer role is strongly supported by the observations that induction of autophagy by Beclin 1 inhibits tumorigenesis,Citation40,Citation41 that p53 and PTEN tumor suppressors induce autophagy,Citation42,Citation43 and that autophagy regulators function as tumor suppressors.Citation44,Citation45 However, it is hypothesized that autophagy may also contribute to tumorigenesis by keeping tumor cells alive when limited angiogenesis leads to nutrient deprivation and hypoxia.Citation46 This cancer-promoting role is partially supported by recent findings that autophagy plays a protective role against cell death, that growth factors regulate cell survival via autophagy,Citation46 and that autophagy promotes tumor cell survival by delaying apoptosis.Citation47,Citation48 However, direct evidence for a tumorigenic function of autophagy is still lacking.
In the current study we hypothesized that autophagy is activated for the maintenance of cellular homeostasis (removing damaged organelles and recycling degraded cellular components) if impaired mitochondria are critical in the initial stages of tumorigenesis, whether or not a hypoxic stimulus is present. From this point of view, we investigated the functional contribution of autophagy toward mitochondrial impairment and cell transformation by oncogenic K-Ras. Our most striking findings were the functional loss of mitochondrial respiration by K-RasV12 without pre-existing environmental damage such as hypoxia, and its reversal by blocking autophagy ( and ), implying the direct involvement of autophagy in mitochondrial loss without pre-existing damage. Furthermore, blocking autophagy effectively suppressed cell transformation. Thus, we provide direct evidence that activation of autophagy by oncogenic K-Ras is critical, not only for mitochondrial functional loss, but also for the transformation of Rat2 cells, highlighting the apparently tumorigenic role of autophagy. Although involvement of autophagy in mitochondrial loss by K-Ras is obvious, persistent appearance of large acidic vesicles (autolysosomes) prompted us to question whether the autolysosomes induced by K-Ras are active or not. The fact that most vesicles possessed organellar further increased our suspicion. However, the autophagosomes were proved to be active in protein degradation (), implying that degradation activity may not be sufficient to degrade all the organelles imported and/or the final recycling step may be impaired. Potential importance of p62, a link between LC3 and ubiquitinated substrates, in impairment of autophagy was well discussed.Citation49 Interestingly, dramatic increase and aggregation of p62 protein was induced by K-Ras infection although the level was further enhanced by lysosomal inhibitor, implying that the K-Ras-induced autophagic activity was insufficient. Induction of p62 by K-Ras was also reported.Citation50 So far, it is not clear whether p62 is also involved in amphisome recycling. Further studies will be necessary to elucidate the detailed mechanisms involved in this impaired autophagic process.
Why do transformed cells require active mitophagy? In order to maintain proliferative capacity, cells must continue to supply sufficient energy and cellular components such as amino acids, lipids and sugars, to constantly generate progeny. It is well known that many solid tumors depend on activated glycolysis to cope with the energy requirement for faster proliferation; this process requires effective uptake of glucose even in a stressful environment, such as hypoxic conditions, because tumor cells often experience intermittent hypoxia.Citation13 Therefore, it seems reasonable that many tumor cells possess increased levels of GLUT1 that can effectively uptake glucose present in low extracellular concentrations (1 mM). Since oncogenic Ras protein has been recognized as an inducer of GLUT1,Citation32,Citation33 it was therefore not surprising that we observed enhanced LDH activity and stable intracellular ATP levels in our transformation system (). Metabolic shift to glycolysis from mitochondrial ATP generation by K-Ras was clearly demonstrated (). Unexpectedly, GLUT2, rather than GLUT1, was induced by K-RasV12 (). This finding is well supported by the recent report that GLUT1 mRNA expression was not regulated by K-Ras in colon cancer.Citation51 GLUT2 can effectively take up glucose only when extracellular glucose concentrations are higher than 15 mM, such as during hyperglycemia and diabetes. To develop K-RasV12-induced transformation, we used low-glucose media (5 mM) and did not change the media for 3 d, expecting that media glucose levels would gradually drop over the course of the experiment; since the cells continued to grow, we conclude that the transforming cells are nourished from other sources. In addition, if activated glycolysis can meet the cellular energy requirement, then the maintenance of a high level of mitochondrial mass is not essential, at least for ATP production. Therefore, the degradation of unnecessary organelles including mitochondria via autophagy may serve as a useful mechanism to resupply nutrients, expediting glycolysis. These results are well supported by the recent report that autophagy facilitates glycolysis.Citation52 In addition, transformed cells may maintain small numbers of mitochondria during periods of rapid proliferation, minimizing the effort required to maintain subcellular organelles. Surprisingly, electron microscopic images show decreased mass of intracellular organelles within cytosol (), supporting our hypothesis. Further investigation will be necessary to fully develop this hypothesis.
To assess the possibility that similar phenomena may occur in human cancer, we investigated a tumor sample with low glucose uptake. A recent report has indicated the existence of peculiar tumor nodules that cannot be detected by PET imaging with 18F-FDG due to their low glucose uptake properties.Citation34 We therefore compared hepatocellular carcinoma tissues with high or low glucose uptake, precisely diagnosed using 18F-FDG PET imaging. Surprisingly, we observed increased levels of LC3-II formation only in tumor tissues with low glucose uptake and high K-Ras expression (), implying close links among glucose insufficiency, K-Ras and autophagy. Taken together, these observations suggest that cancer cells may activate autophagy-mediated organelle degradation to maintain cellular ATP levels, resupplying nutrients when glucose levels are insufficient; K-Ras may coordinate the activation of mitophagy necessary for the cell to meet this challenge. In addition, our results strongly support a tumorigenic role of autophagy, most likely via the provocation of mitochondrial loss in the absence of hypoxia.
Materials and Methods
Cell culture and small interfering RNA (siRNA) transfection.
Rat2 fibroblast cells were obtained from the American Type Culture Collection and were grown in RPMI 1640 medium (GIBCO, 31800-022) supplemented with 5% fetal bovine serum (GIBCO, 16000) and antibiotics at 37°C in a humidified incubator with 5% CO2. Oligonucleotides for Beclin1 siRNA (5′-CGA UCA AUA AUU UCA GAC Utt3′), JNK siRNA (5′-UCA GAC UCA UGC CAA GCG GTT-3′) and negative control siRNA (5′-UAG CGA CUA AAC ACA UCA A-3′) were produced by Ambion Inc., siRNA duplexes were introduced into cells using Lipofectamine 2000 (Invitrogen, 52887).
Human hepatocellular carcinoma samples from patients.
Surgical specimens (tumor samples and surrounding control tissues) were obtained from ten patients with HCC (age range 34–70 y) from August 2008 to January 2010 with informed consent through Ajou Institutional Review Board. No patient in the current study received chemotherapy or radiation therapy before the surgery. Prior to surgery, the patients were examined with a whole-body PET/CT camera (GE Advance). After fasting at least 4 h, patients received 370 MBq 18F-FDG intravenously. Whole-body PET/CT images were obtained on a Discovery ST scanner (GE Healthcare). Seven to eight frames (3 min/frame) of emission PET data were acquired in a two-dimensional mode after noncontrast CT scans from the base of the skull to the upper thigh (tube rotation time of 1 sec per revolution, 120 kV, 60 mA, 7.5 mm per rotation and acquisition time of 60.9 sec for a scan length of 867 mm). Emission PET images were reconstructed with noncontrast CT using iterative reconstruction. Attenuation-corrected PET/CT images were reviewed on a Xeleris workstation (GE Healthcare). Specialists in Nuclear Medicine evaluated the images qualitatively with four visual grades (VG 0, negative; VG 1, equal to surrounding; VG 2, slightly higher; VG 3, strongly higher uptake) to assess whether FDG uptake in the tumor was higher than that in the surrounding noncancerous tissue. The tumor samples of VG 0 (n = 5) and VG 3 (n = 5) were selected for our study and described as high uptake and low uptake groups.
Production of retrovirus containing activated K-ras (K-ras12V).
K-RasV12 retroviral vector was prepared from pSPORT-Ki-RasV12 provided by Dr. P. Kirshmeier (Schering-Plough Research Institute). K-RasV12 PCR fragment produced against pSPORT-Ki-RasV12 was cloned into MFG retroviral vector using NcoI and BamHI sites, generating MFG-K-RasV12. For retrovirus production, a modified 293T cell line were cultured in Dulbecco's modified Eagle's medium (DMEM; GIBCO, 31600-034) media supplemented with 10% FBS, 2 mM GlutaMAX (Invitrogen, 35050-06), 50 U/ml Penicillin-Streptomycin, 1 µg/ml Tetracyclin, 2 µg/ml Puromycin, 0.3 mg/ml G418 sulfate (Calbiochem, 345810) and transfected with plasmid containing K-RasV12 using the Lipofectamine Plus Reagent (Invitrogen, 10964-021). The virus-containing medium (supernatant) was harvested daily for 4 d. The harvested virus supernatant was filtered through 0.45 µm filter unit (Milipore corp., UFC 920008) and stored at 80°C.
Reverse transcription-polymerase chain reaction (RT-PCR).
Total RNA was isolated using Trizol (Invitrogen, 15596-02) and cDNA was prepared using avian myeloblastosis virus reverse transcriptase (Promega, Madison, M510A). PCR was performed with two primer sets, a set for target gene and a set of control gene (glyceraldehydes-3-phosphate dehydrogenase), in a single reaction tube. Oligonucleotide primers were produced by Bioneer (Seoul, Korea). The PCR primer sequences for RT-PCR were as follows: 5′-TTT GGA GAT GTT GGA GCA AA and 5′-GCA GCA TTG ATT TCA TTC CA for beclin-1; 5′-CTA ATC GCA GTA TTG GAG AC and 5′-GGA ACC ACT CAG TGA AGA GG for VATPase; 5′-CCA TGG AGA AGG CTG GGG and 5′-CAA AGT TGT CAT GGA TGA CC for glyceraldehyde 3-phosphate dehydrogenase; 5′-CCAAGAGTGGCACTGGCATCG and 5′-GAG GCT GAA ATC GTC ACT GG for ATPase; 5′-GAT TGC TAA CGC GAC TAA AA and 5′-ACC GAA TCC TCG GAT ATT TA for Comp I; 5′-TGT CTC CCA GGT GTA TGA AG and 5′-GGC ATT CCT AAG GAT GTT TT for Comp-III; 5′-ATT ATT CTG CTG TGG CTG AT and 5′-CAT AAT GAA TCC CTG TCC AA for NRF; 5′-GTG TGT AGG CAC AGT GTC and 5′-TTC TCA GAG ATG TCT CC for mtTFA.
Maximum cellular oxygen consumption.
Maximum cellular oxygen consumption rate was measured as described previously with slight modification.Citation53 Cells were transferred to Mitocell equipped with Clark oxygen electrode (782 Oxygen Meter, Strathkelvin Instrument) after adding 30 µM 2,4-dinitrophenol (DNP) to obtain maximum respiration rate, and its specificity for mitochondrial respiration was confirmed by adding 5 mM KCN. Maximum cellular respiration rates were determined by subtracting KCN-insensitive O2 consumption rate from DNP-uncoupled rate and expressed as percent of control.
Simultaneous measurement of extracellular acidification rate and cellular oxygen consumtion rate.
To determine cellular dependency on glycolysis or mitochondrial respiration, extracellular acidification rate (ECAR) and cellular endogenous oxygen consumption rate (OCR) were measured using Seahorse XF24 analyzer (Seahorse Bioscience Inc.) according to the manufacturer's protocol. Briefly, cells were plated at a density of 10,000 cells/well on XF24 tissue culture plate and preincubated with XF assay medium (Seahorse Bioscience) containing 1 mM pyruvate and 5 mM glucose in the presence or absence of 2 mM oligomycin (Oli) or 25 mM 2-deoxyglucose (DOG) for 1 h, and further equilibrated with the pre-calibrated probe inside the instrument for 2 h. Then, endogenous OCR and ECAR were measured under basal condition and normalized to cell number. ECAR represents the rate of released lactate into media. ECAR and OCR were expressed as percent of the values obtained from untreated control cells.
Estimation of lactate dehydrogenase (LDH) activity and cellular ATP levels.
Cell pellets were lysed in PES buffer (50 mM Na/K phosphate, pH 7.4, 1 mM EDTA, 0.5% sodium cholate) and a portion (25 µg) of lysate was used in total 150 µl of assay mixture. To determine LDH activities of the lysates, initial rates to oxidize NADH were monitored at 340 nm with freshly prepared 2 mg/ml NADH solution in 0.1 M potassium phosphate buffer, pH 7.4, after adding pyruvate to 4 mM final concentration using ThermoMax microplate reader (Molecular Devices Corp.). The activity was determined by comparing values with a calibration curve for known concentration of standard LDH performed at the same time. The LDH activities were calculated as unit of LDH per µg of cellular lysate protein and expressed as percent of control.
Intracellular ATP levels were measured by the bioluminescence assay as described previously with slight modification.Citation50
Glucose uptake assay.
Cellular glucose uptake activity was measured by exposing the cells to RPMI 1640 containing 5 mM glucose with 1 µCi/ml 2-deoxy-D-[1-3H] glucose (specific activity, 6 Ci/mmol) (Amersham, TRK 383). Briefly, cells were infected with the retrovirus harboring K-RasV12 for 3 d on 12-well plates and then further incubated with glucose free media for 1 h. Cellular uptake of 2-deoxy-D-[1-3H]glucose was measured after exposing cells to the radioisotope-containing media for 15 min or 1 h at 37°C. After cells were washed with ice-cold PBS, the radioactivity of cell lysates was measured by liquid scintillation counter (Tri-Carb2100TR, Perkin Elmer Life Science, Inc.).
Fluorescence and live microscopy and electron microscopy.
To visualize fluorescence images of live cells, cells cultured onto a Chamlide™ chamber (Live Cell Imaging) were imaged on Axiovert 200M equipped with HBO103 using a LD Plan-Neofluar x40 objective and x1.6 optovar (Carl Zeiss AM) at the interval of 1 min.
Electron microscopic images were obtained according to the method described previously in reference Citation54. After staining the sectioned cell slices with uranyl acetate and lead citrate, cells were observed and photographed under transmission electron microscope (Zeiss EM 902A, Leo).
Estimation of mitochondrial and lysosomal mass.
To assess concomitantly mitochondrial and lysosomal mass, the cells were co-stained with 200 nM MitoTracker™ Red (CMXRos; Molecular Probe, M-7512), a common mitochondria-specific dye and 50 nM LysoTracker™ Green DND-26 (L-7526, Molecular Probe) fluorescence dye for 10 min and visualized with fluorescence microscopy or quantitated using flow cytometry (FACS Vantage, Becton Dickinson corp.). Mitochondrial targeting of CMXRos (MitoR) is independent of mitochondrial membrane potential whereas that of CMH2XRos (Molecular Probe, M7513) is dependent (Fig. S5). Moreover, MitoR is more specific to mitochondria that MitoTracker™ Green FM (Molecular Probe, M7514) in Rat2 cell (Fig. S6).
Soft agar colony-formation assay.
Cellular anchorage-independent growth was analyzed using the soft agar colony-formation assay. Briefly, 5 × 104 trysinized cells were suspended in 0.45% agar and spread over a bottom layer of media with 0.9% agar. Colonies greater than 50 µm in diameter were counted 14 d after plating under a light microscope.
Western blot analysis and antibodies.
Cell lysates were applied to protein gel blot analysis as previously described in reference Citation54. Antibodies against subunits of OXPHOS complexes (complex II, A11142; complex I, A11140; complex III, A11143; complex IV-subunit 1, A6403; complex V, A11144; Molecular Probe), Beclin 1 (Cell Signaling Technology Inc., 3738), LC3 (MBL, PD014), V0-ATPase (Santa Cruz, SC21218), K-Ras (Calbiochem, OP24), VDAC (Merck, Darmstadt, Germany; PC548), TIM13 (Abnova Corp., Jhouzih, Taiwan; H00026517-A01), TOM20 (Santa Cruz, SC11415), LIMP-1 (Santa Cruz, SC25867), p62 (Abnova Corp., H00008878-M03) and α-tubulin (Calbiochem, CP06) were used for primary reaction.
Statistical analysis.
The graphs were expressed as mean ± standard deviation. They were analyzed by Student's t-test or by one-way ANOVA followed by post hoc comparisons (StudentNewman-Keuls) using the Statistical Package for Social Sciences 10.0 (SPSS Inc.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 Mitochondrial respiration was progressively impaired without changes in biogenesis, but with induction of autophagy-related proteins during oncogenic K-Ras-induced transformation. Rat2 cells were infected with retrovirus harboring K-RasV12 for the indicated periods. (A) soft-agar assay was performed as described in ‘Materials and Methods’. Representative images are shown (left part) and colonies greater than 50 µm in diameter were counted (right part). (B) cellular maximum respiration rates were determined as a KCN-sensitive DNP-uncoupled O2 consumption rate and expressed as percent of control. (C) expression levels of the respiratory and nonrespiratory mitochondrial proteins were analyzed by protein gel blot analysis. (D) mRNA levels with RT-PCR. (E) expression levels of autophagy-related proteins with protein gel blot analysis. *p < 0.05; **p < 0.01 vs. MFG control by student's t-test.
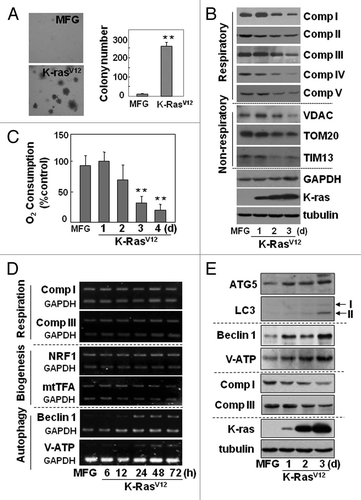
Figure 2 K-RasV12 activates autophagic vesicle formation, accompanied by mitochondrial loss. Rat2 cells were infected with retrovirus harboring K-RasV12 for the indicated periods. (A) Mitochondrial mass decrease and lysosomal mass increase were visualized after co-staining the cells with 50 nM LysoTracker Green (LysoG) and 200 nM MitoTracker Red (MitoR) without fixation using a Plan-Apochromat x100, 1.4 NA oil-immersion objective. Green indicates lysosomes and red indicates mitochondria. (B) Flow cytometric analysis was performed for quantification of mitochondrial mass (left part) or lysosomal mass (right part) of the co-stained cells with MitoR and LysoG. (C) To visualize live images, Rat2 cells were transiently transfected with mtRFP and infected with K-RasV12 retrovirus, and further cultured for 3 d on chamlide™ chamber as described in ‘Materials and Methods’. Targeting of mtRFP-labeled mitochondria into autophagic vesicles was visualized without fixation using a Plan-Apochromat x100, 1.4 NA oil-immersion objective. Representative images are presented. (D) Representative electron microscopic images are presented. The open arrows indicate representative mitochondria within autophagosome, thin arrows indicate autophagosome and arrowheads indicate endoplasmic reticulum entrapping mitochondria. (E) Numbers of total vesicles, clear empty vesicles (square), vesicles with debris (circle) and vesicles with organellar remnants (dotted square) were counted from 26 whole cell electron microscopic images (3,000x or 4,400x) of K-Ras infected Rat2 cells (right part). Representative images are shown in the left part. (F) Western blot analysis. After infected by K-RasV12 retrovirus for 24 h, cells were replenished with RPMi media containing 10 µM chloroquine (C6628, Sigma-Aldrich) for 2 d. *p < 0.05; **p < 0.01 vs. MFG control.
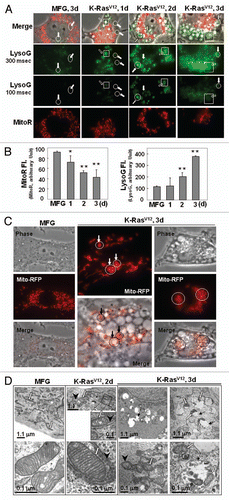

Figure 3 Recovery of mitochondriyal mass and function by blocking autophagy. (A–C) Rat2 cells were exposed to bafilomycin A (0.5 and 1 nM) or 3MA (1 to 3 mM) 1 h prior to K-RasV12 infection and further incubated for 3 d. (A) Mitochondrial (left part) and lysosomal (right part) mass were quantitated by flow cytometric analysis after co-staining cells with MitoR and LysoG. (B) western blot analyses for the recovery of respiratory proteins by pretreatment of bafilomycin A (left part) and 3MA (right part). (C) Maximum cellular oxygen consumption rates recovered by pretreatment of bafilomycin A (1 nM) and 3MA (3 mM). (D) Cellular oxygen consumption rates was estimated after Rat2 cells were transfected with si-ATG5, si-VATPaseE and si-Beclin 1 15 h prior to K-RasV12 infection and further incubated for 3 d. **p < 0.01 vs. MFG control and ##p < 0.01 vs. K-Ras-infected cells by one-way ANOVA.
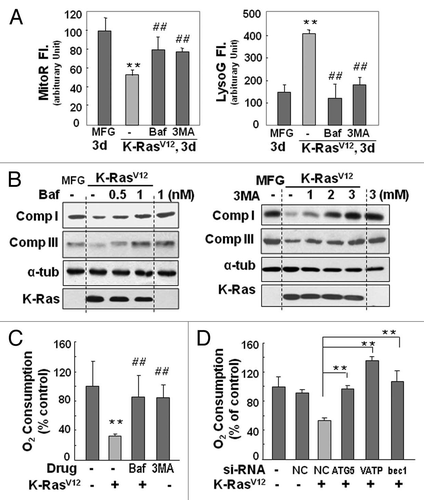
Figure 4 K-RasV12-induced autophagy is mediated through JNK. Rat2 cells were exposed to pharmacological inhibitors (15 µM PD98059, 15 µM SP600125 or 15 µM PD169316) or transfection of si-RNAs (si-NC, si-beclin-1 or si-JNK) prior to K-RasV12 infection and further incubated for 3 d as indicated. (A) Mitochondrial and lysosomal mass were estimated by flow cytometric analysis after co-staining cells with MitoR (left part) and LysoG (right part). (B) Cellular oxygen consumption rates. (C) Western blot analysis. (D) Soft-agar assay was performed as described in ‘Materials and Methods’. **p < 0.01 vs. MFG control and ##p < 0.01 vs. K-Ras-infected cells by one-way ANOVA.
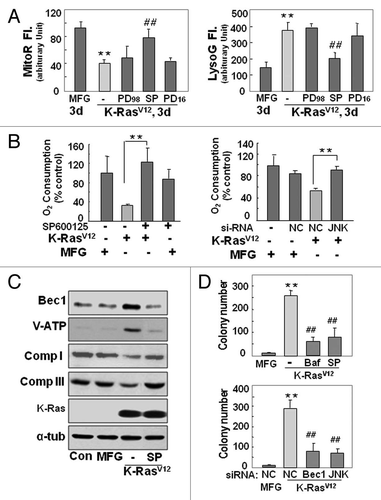
Figure 5 K-RasV12-induced autophagy is dependent on extracellular glucose levels. (A, B and F) Rat2 cells were infected with retrovirus harboring K-RasV12 for 3 d unless indicated. (A) Cell growth rates were measured by counting trypan blue-negative cells (left part) and phase contrast cell images are shown (right part). (B) Total cellular ATP levels were estimated by using luciferin/luciferase ATP assay kit (left part) and LDH activity to oxidize NADH was monitored at 340 nm using spectrophotometry (right part). (C and D) After infected by K-RasV12 retrovirus for 3 d, Rat2 cells were treated with 2 µM oligomycin (Oli) or 25 mM 2-deoxyglucose (DOG) for 3 h. (C) Extracellular acidification rate and cellular oxygen consumption rate were simultaneously measured by Seahorse XF analyzer as described in ‘Materials and Methods’. Dotted arrows indicate lactate increase (a) and respiratory defect (b) by K-Ras. (D) Intracellular ATP levels. DOG-sensitive (glycolysis dependent) and Oli-sensitive (mitochondrial-respiration dependent) ATP production were indicated as percentage of total cellular ATP levels. (E) Intracellular ATP levels. Rat2 cells were transfected with si-ATG5 or si-Beclin 1 (Bec-1) 15 h prior to K-RasV12 infection and further incubated for 3 d, then replenished with 2 µM oligomycin containing medium for 3 h. (F) Expression levels of GLUT proteins were monitored by protein gel blot analysis (left part) and cellular glucose uptake activity was measured as described in ‘Materials and Methods’ (right part). (G) LC3-II formation of Rat2 clone stably expressing K-RasV12 in response to different concentrations of glucose in media for 3 d was monitored by protein gel blot analysis (left part) and phase contrast cell images are shown (right part).
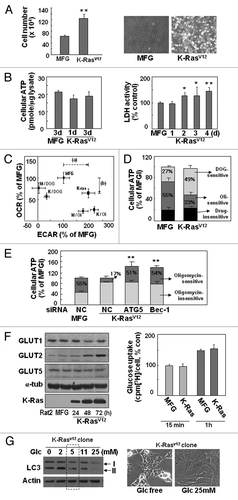
Figure 6 LC3-II formation increased only in the human hepatocellular carcinoma tissues with low glucose uptake. (A) Representative PET/CT images of human hepatocellular carcinoma patients. (B) LC3-II formation and K-Ras expression were monitored by protein gel blot analysis. The expression ratios for tumor/surrounding tissue (T/S) of the normalized values (LC3-II, total LC3, K-Ras) against actin are shown in the lower part.
Additional material
Download Zip (691.5 KB)Acknowledgements
This work was supported by the Korean Science and Engineering Foundation (KOSEF) funded by a grant from the Korean government (MEST) (R13-2003-019-01007-0), by the Mid-career Research Program through an NRF grant funded by the MEST (2009-0079076), and by the Korea Research Foundation Grant funded by the Korean Government (KRF-2008-314-C00286).
References
- Attardi G, Schatz G. Biogenesis of mitochondria. Annu Rev Cell Biol 1988; 4:289 - 333; PMID: 2461720; http://dx.doi.org/10.1146/annurev.cb.04.110188.001445
- Lee HC, Wei YH. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol 2005; 37:822 - 834; PMID: 15694841; http://dx.doi.org/10.1016/j.biocel.2004.09.010
- Brunk UT, Terman A, Cadenas E, Packer L. Understanding the process of aging 1999; New York Marcel Dekker 229 - 250
- Lemasters JJ. Selective mitochondrial autophagy or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction and aging. Rejuvenation Res 2005; 8:3 - 5; PMID: 15798367; http://dx.doi.org/10.1089/rej.2005.8.3
- Kim I, Rodriguez-Enriquez, Lemasters JJ. Selective degradation of mitochondrial by mitophagy. Arch Biochem Biophys 2007; 462:245 - 253; PMID: 17475204; http://dx.doi.org/10.1016/j.abb.2007.03.034
- Nowikovsky K, Reipert S, Devenish RJ, Schweyen RJ. Mdm38 protein depletion causes loss of mitochondrial K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ 2007; 14:1647 - 1656; PMID: 17541427; http://dx.doi.org/10.1038/sj.cdd.4402167
- Warburg O. On the origin of cancer cells. Science 1956; 123:309 - 314; PMID: 13298683; http://dx.doi.org/10.1126/science.123.3191.309
- Pedersen PL. Tumor mitochondria and the bioenergetics of cancer cells. Prog Exp Tumor Res 1978; 22:190 - 274; PMID: 149996
- Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene 2006; 25:4647 - 4662; PMID: 16892079; http://dx.doi.org/10.1038/sj.onc.1209607
- Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci 1999; 24:68 - 72; PMID: 10098401; http://dx.doi.org/10.1016/S09680004(98)01344-9
- Ziegler A, von Kienlin M, Decorps M, Remy C. High glycolytic activity in rat glioma demonstrated in vivo by correlation peak 1H magnetic resonance imaging. Cancer Res 2001; 61:5595 - 5600; PMID: 11454713
- Gatenby RA, Gawlinski ET. The glycolytic phenotype in carcinogenesis and tumor invasion: insights through mathematical models. Cancer Res 2003; 63:3847 - 3854; PMID: 12873971
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis?. Nat Rev Cancer 2004; 4:891 - 899; PMID: 15516961; http://dx.doi.org/10.1038/nrc1478
- Cuezva JM, Krajewska M, de Heredia ML, Krajewski S, Santamaría G, Kim H, et al. The bioenergetic signature of cancer: A marker of tumor progression. Cancer Res 2002; 62:6674 - 6681; PMID: 12438266
- Isidoro A, Casado E, Redondo A, Acebo P, Espinosa E, Alonso AM, et al. Breast carcinomas fulfill the Warburg hypothesis and provide metabolic markers of cancer prognosis. Carcinogenesis 2005; 26:2095 - 2104; PMID: 16033770; http://dx.doi.org/10.1093/carcin/bgi188
- Semenza GL. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J Bioenerg Biomembr 2007; 39:231 - 234; PMID: 17551816; http://dx.doi.org/10.1007/s10863-007-9081-2
- Kim JW, Dang CV. Cancer's molecular sweet tooth and the Warburg effect. Cancer Res 2006; 66:8927 - 8930; PMID: 16982728; http://dx.doi.org/10.1158/00085472.CAN-06-1501
- Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci USA 2005; 102:719 - 724; PMID: 15647368; http://dx.doi.org/10.1073/pnas.0408894102
- Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, et al. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res 2005; 65:1655 - 1663; PMID: 15753359; http://dx.doi.org/10.1158/0008-5472.CAN-04-2012
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature 1990; 348:125 - 132; PMID: 2122258; http://dx.doi.org/10.1038/348125a0
- Lowy DR, Willumsen BM. Function and regulation of ras. Annu Rev Biochem 1993; 62:851 - 891; PMID: 8352603; http://dx.doi.org/10.1146/annurev.bi.62.070193.004223
- Bos JL. Ras oncogenes in human cancer: a review. Cancer Res 1989; 49:4682 - 4689; PMID: 2547513
- Friday BB, Adjei AA. K-ras as a target for cancer therapy. Biochim Biophy Acta 2005; 1756:127 - 144
- Westra WH, Baas IO, Hruban RH, Askin FB, Wilson K, Offerhaus GJ, et al. K-ras oncogene activation in atypical alveolar hyperplasias of the human lung. Cancer Res 1996; 56:2224 - 2228; PMID: 8616876
- Kitamura H, Kameda Y, Ito T, Hayashi H. Atypical adenomatous hyperplasia of the lung. Implications for the pathogenesis of peripheral lung adenocarcinoma. Am J Clin Pathol 1999; 111:610 - 622; PMID: 10230351
- Flier JS, Mueckler MM, Usher P, Lodish HF. Elevated levels of glucose transport and transporter messenger-RNA are induced by ras or src oncogenes. Science 1987; 235:1492 - 1495; PMID: 3103217; http://dx.doi.org/10.1126/science.3103217
- Ramanathan A, Wang C, Schreiber SL. Perturbational profiling of a cell-line model of tumorigenesis by using metabolic measurements. Proc Natl Acad Sci USA 2005; 102:5992 - 5997; PMID: 15840712; http://dx.doi.org/10.1073/pnas.0502267102
- Lim JH, Lee ES, You HJ, Lee JW, Park JW, Chun YS. Ras-dependent induction of HIF-1alpha785 via the the Raf/MEK/ERK pathway: a novel mechanism of Ras-mediated tumor promotion. Oncogene 2004; 23:9427 - 9431; PMID: 15543236; http://dx.doi.org/10.1038/sj.onc.1208003
- Blum R, Jacob-Hirsch J, Amariglio N, Rechavi G, Kloog Y. Ras inhibition in glioblastoma downregulates hypoxia-inducible factor-1alpha, causing glycolysis shutdown and cell death. Cancer Res 2005; 65:999 - 1006; PMID: 15705901
- Ohnami S, Matsumoto N, Nakano M, Aoki K, Nagasaki K, Sugimura T, et al. Identification of genes showing differential expression in antisense K; rastransduced pancreatic cancer cells with suppressed tumorigeniecity. Cancer Res 1999; 59:5565 - 5571; PMID: 10554036
- Campbell PM, Groehler AL, Lee KM, Ouellette MM, Khazak V, Der CJ. K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phophatidylinositol-3-kinase signaling. Cancer Res 2007; 67:2098 - 2106; PMID: 17332339; http://dx.doi.org/10.1158/0008-5472.CAN-06-3752
- Amann T, Hellerbrand C. GLUT1 as atherapeutic target in hepatocellular carcinoma. Expert Opin Ther Targets 2009; 13:1411 - 1427; PMID: 19874261; http://dx.doi.org/10.1517/14728220903307509
- Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. J Biol Chem 2001; 276:9519 - 9525; PMID: 11120745; http://dx.doi.org/10.1074/jbc.M010144200
- Lee JD, Yun M, Lee JM, Choi Y, Choi YH, Kim JS, et al. Analysis of gene expression profiles of hepatocellular carcinoma with regard to 18F-fluorodeoxyglucose uptake pattern on positron emission tomography. Eur J Nucl Med Mol Imaging 2004; 31:1621 - 1630; PMID: 15278306; http://dx.doi.org/10.1007/s00259004-1602-1
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306:990 - 995; PMID: 15528435; http://dx.doi.org/10.1126/science.1099993
- Adhami F, Liao G, Morozov YM, Schloener A, Schmithorst VJ, Lorenz JN, et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol 2006; 169:566 - 583; PMID: 16877357; http://dx.doi.org/10.2353/ajpath.2006.051066
- Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14:230 - 239; PMID: 16794605; http://dx.doi.org/10.1038/sj.cdd.4401984
- Ogier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta 2003; 1603:113 - 128; PMID: 12618311
- Hippert MM, O'toole PS, Thorburn A. Autophagy in cancer: Good, bad or both?. Cancer Res 2006; 66:9349 - 9351; PMID: 17018585; http://dx.doi.org/10.1158/0008-5472.CAN-06-1597
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672 - 676; PMID: 10604474; http://dx.doi.org/10.1038/45257
- Liang XH, Yu J, Brown K, Levine B. Beclin 1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. Cancer Res 2001; 61:3443 - 3449; PMID: 11309306
- Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol-3-kinase/protein kinase B pathway. J Biol Chem 2001; 276:35243 - 35246; PMID: 11477064; http://dx.doi.org/10.1074/jbc.C100319200
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA 2005; 102:8204 - 8209; PMID: 15928081; http://dx.doi.org/10.1073/pnas.0502857102
- Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004; 23:2891 - 2906; PMID: 15077152; http://dx.doi.org/10.1038/sj.onc.1207521
- Levine B, Yuan J. Autophagy in cell death: an innocent convict?. J Clin Invest 2005; 115:2679 - 2688; PMID: 16200202; http://dx.doi.org/10.1172/JCI26390
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptisis. Cell 2005; 120:237 - 248; PMID: 15680329; http://dx.doi.org/10.1016/j.cell.2004.11.046
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation and tumorigenesis. Cancer Cell 2006; 10:51 - 64; PMID: 16843265; http://dx.doi.org/10.1016/j.ccr.2006.06.001
- Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 2007; 14:500 - 510; PMID: 16990848; http://dx.doi.org/10.1038/sj.cdd.4402039
- Klionsky DJ, Alveliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 175; PMID: 18188003
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NKκB mediator in tumorigenesis. Cancer Cell 2008; 13:343 - 354; PMID: 18394557; http://dx.doi.org/10.1016/j.ccr.2008.02.001
- Noguchi Y, Okamoto T, Marat D, Yoshikawa T, Saitoh A, Doi C, et al. Expression of facilitative glucose transpoter 1 mRNA in colon cancer was not regulated by k-ras. Cancer Lett 2000; 154:137 - 142; PMID: 10806301; http://dx.doi.org/10.1016/S03043835(00)00354-2
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Rasmediated oncogenic transformation. Mol Biol Cell 2011; 22:165 - 178; PMID: 21119005; http://dx.doi.org/10.1091/mbc.E10-06-0500
- Yoon YS, Byun HO, Cho H, Kim BK, Yoon G. Complex II defect via downregulation of iron-sulfur subunit induces mitochondrial dysfunction and cell cycle delay in iron chelation-induced senescence-associated growth arrest. J Biol Chem 2003; 278:51577 - 51586; PMID: 14512425; http://dx.doi.org/10.1074/jbc.M308489200
- Yoon YS, Yoon DS, Lim IK, Yoon SH, Chung HY, Rojo M, et al. Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J Cell Physiol 2006; 209:468 - 480; PMID: 16883569; http://dx.doi.org/10.1002/jcp.20753