Abstract
Autophagy is an evolutionarily conserved physiological process of self-digestion by a cell to adapt to various stresses, including starvation. Its molecular basis involves the concerted activation of proteins encoded by the family of autophagy-related (Atg) genes. The best characterized is the serine/threonine protein kinase Atg1 in yeast which appears to be essential at the early stage of autophagy. In mammals, five Atg1 homologues have been identified as uncoordinated (UNC) 51-like kinase 1 to 4 and STK36. ULK1 and ULK2 are the most closely related members of the family, sharing 78% homology within their protein kinase domains. However, the specific function of ULK1 and ULK2 in mammalian autophagy is not fully understood. Here, we demonstrate that ULK1 and ULK2 are functionally redundant protein kinases required to mediate autophagy under nutrient-deprived conditions in fibroblasts. In contrast, ULK1, but not ULK2, is critical to induce the autophagic response of cerebellar granule neurons (CGN) to low potassium concentration in serum-free conditions. Furthermore, we found that ULK1 has a cytoprotective function in neurons. Together, these results provide strong genetic evidence that ULK1 is an essential component of the autophagic signaling pathway. The ability of ULK2 to compensate for the loss of ULK1 function is cell-type specific.
Introduction
Autophagy is an evolutionarily conserved intracellular degradation system that contributes to maintaining energy homeostasis.Citation1 In particular, this process is crucial in promoting cell survival during embryogenesis and early neonatal development.Citation2,Citation3 In addition, autophagy contributes to the clearance of damaged organelles and aggregate-prone proteins.Citation4,Citation5 The physiological importance of this function is demonstrated by the accumulation of polyubiquitinated proteins with deleterious effects in the brain of mice in which autophagic genes have been specifically deleted in the central nervous system.Citation6,Citation7 Overall these genetic studies have established the first link between autophagy and neurodegeneration.Citation8 Interestingly, autophagy has also been implicated in promoting cell death under certain circumstances.Citation9
The molecular basis of autophagy involves the concerted activation of proteins encoded by the family of Atg genes, of which 31 to date have been described in reference Citation10 and Citation11. The core machinery is composed of three major functional groups that include the Atg1 complex which enables the localization of Atg proteins to the pre-autophagosomal structure (PAS) at the early stage of autophagosome formation.Citation11 Atg1 is a serine/threonine protein kinase and its activity requires Atg13 and Atg17.Citation12 Under nutrient-rich conditions, the target of rapamycin (TOR) phosphorylates Atg13, thereby preventing it from interacting with Atg1.Citation12,Citation13 Upon autophagy induction following nutrient starvation or rapamycin treatment, Atg13 becomes dephosphorylated and binds Atg1.Citation12,Citation13 The binding of Atg13 to Atg1 stabilizes the association of Atg17 to the complex.Citation12 Recent findings in mammals have identified a direct interaction between mammalian TOR (mTOR) and two Atg1 homologues, UNC51-like kinase (ULK) 1 and ULK2.Citation14–Citation16
ULK1 and ULK2 are ubiquitously expressed protein kinases localized to autophagosomal membranes in mammalian cells.Citation17 The ability of dominant negative mutants of ULK1 and ULK2 to block autophagy led to the hypothesis that ULK1 and ULK2 are redundant protein kinases.Citation18 Consistent with this, mice with a targeted deletion of either the ulk1 or the ulk2 gene displayed normal development.Citation19 The ability of ULK2 to compensate for the loss of ULK1 is further supported by evidence that ulk1-deficient mouse embryonic fibroblasts (MEFs) displayed autophagy induction following glucose withdrawal.Citation19 However, the study showed that ulk1, but not ulk2, mRNA was upregulated during erythroid differentiation, and that ULK1 deficiency resulted in delayed autophagic clearance of mitochondria in reticulocytes.Citation19 The selective requirement of ULK1 to promote autophagy in specific cell types and in response to distinct stimuli is consistent with evidence that ULK1, but not ULK2, is a critical regulator of autophagy in HEK293 cells and in MEFs following amino acid withdrawalCitation16,Citation20,Citation21 and rapamycin treatment.Citation15
To better understand the precise role of ULK1 and ULK2, we investigated the specific requirement of ULK1 and ULK2 in autophagy in response to starvation. Using ulk2+/− and ulk2−/− MEFs and cerebellar granule neurons (CGN) in which ulk1 was downregulated by siRNA, we revealed that the ULK1 and ULK2 have overlapping but also nonredundant function in regulating autophagy.
Results
The loss of ULK1 and ULK2 blocks nutrient deprivation-induced autophagy in MEFs.
To assess the specific function of ULKs in the regulation of autophagy, we tested the effect of the functional loss of ULK1 and ULK2 in MEFs following nutrient starvation. We obtained ulk2 null mice from the Mutant Mouse Regional Resource Centres (MMRRC). These mice are healthy and fertile while they carry a β-galactosidase and neomycin resistance (LZ-Neo) cassette containing a stop codon and a polyadenylation termination signal, in place of exons 1, 2 and 3 (). The ulk2−/− mice were bred with wild-type animals to generate heterozygous ulk2+/− mice. The genotyping of the offspring was performed by PCR on the genomic DNA extracted from tail snips using specific primers (). The loss of ulk2 mRNA in ulk2−/− MEFs did not affect the expression level of the ulk1 transcript (). Next, ulk2+/− and ulk2−/− MEFs were transfected with control siRNA or ulk1 siRNA for 48 h and then cultured in nutrient-deprived medium (Hanks' balanced salt solution, HBSS) for the indicated times. The suppression of ULK1 expression was confirmed by immunoblot analysis ().
Nutrient deprivation caused a decrease in LC3-I level concomitant with an increase in LC3-II level in MEFs after 1 h (). This was inhibited by incubating the cells with 3-methylamine (3-MA), a known pharmacological inhibitor of autophagy (data not shown), thereby confirming that starvation-induced LC3 conversion was a good indicator of autophagy induction in MEFs.Citation22 Time-course analysis demonstrated that the kinetics of LC3 conversion was significantly delayed in cells lacking both ULK1 and ULK2 ( and C). In contrast, no significant difference was observed between ulk2+/− MEFs and ULK1-depleted ulk2+/− or ulk2−/− MEFs ( and C). LC3-II produced in an autophagic-dependent manner is degraded after autophagosome-lysosome fusion. Consistently, nutrient-starved ULK1- or ULK2-deficient MEFs incubated with Bafilomycin A1 (Baf), an inhibitor of autophagosome-lysosome fusion, displayed higher levels of LC3-II (). Accumulation of LC3-II following Baf treatment was not observed in MEFs lacking both ULK isoforms ().
To confirm these results, autophagy flux was monitored by p62 degradation assay.Citation23 Consistent with the LC3 conversion assay, ulk2+/−, ULK1-depleted ulk2+/− and ulk2−/− MEFs displayed similar kinetics of p62 degradation, with almost 50% of the protein lost 1.5 h after starvation ( and C). The loss of p62 in nutrient-deprived ulk2−/− MEFs was abrogated by incubating the cells with Baf (), thereby confirming that decreased p62 level was due to autophagy-induced clearance. In contrast, the level of p62 did not significantly change in cells lacking both ULK1 and ULK2 incubated with or without Baf (, C and E). In addition, it is interesting to note that the basal level of p62 was slightly upregulated in ULK1- and ULK2-deficient MEFs. A similar observation has previously been reported in cells lacking essential autophagy genes such as atg5−/− MEFs compared to wild-type MEFs, irrespective of nutrient conditions.Citation17 This indicates that a low level of constitutive autophagy occurs in single gene-deficient cells, but not in MEFs lacking ULK1 and ULK2.
To further examine autophagy induction, we monitored the redistribution of ectopically expressed LC3 fused to the green fluorescent protein (GFP) from the cytosol to the membrane structures. GFP-LC3 localizes to the membranes of autophagosomes upon induction of autophagyCitation22 and the overexpression of GFP-LC3 does not affect endogenous autophagy.Citation24 Therefore, GFP-LC3 serves as a good autophagosomal marker. MEFs were co-transfected with a vector encoding GFP-LC3 and either control siRNA or ulk1 siRNA for 48 h prior to culturing the cells in HBSS. As expected, nutrient deprivation for 30 min increased the number of GFP-LC3 positive ulk2+/−, ULK1-depleted ulk2+/− and ulk2−/− MEFs (). This was suppressed by incubating the cells with 3-MA or following the loss of both ULK isoforms, consistent with the redundant function of ULK1 and ULK2 in mediating autophagy in MEFs.
ULK1 is required for low potassium-induced autophagy in CGN.
Autophagy has emerged as a pathway of critical importance in protecting neurons against starvation during development and in maintaining normal neuronal function by degrading aggregate-prone proteins.Citation3,Citation4 However, little is known about the molecular regulation of autophagy in neurons. To increase our knowledge, we tested the effect of the loss of ULK1 and ULK2 expression in neuronal autophagy using primary cultures of CGN. The survival of CGN in vitro is ensured by culturing the cells under depolarizing conditions with a high concentration of potassium (25 mM), a situation that is thought to mimic the physiological condition related to the development of excitatory synapses from mossy fibers onto differentiating CGN.Citation28 Mature CGN can be induced to undergo autophagy and apoptosis by reducing the potassium concentration to 5∼10 mM and removing serum.Citation25,Citation26
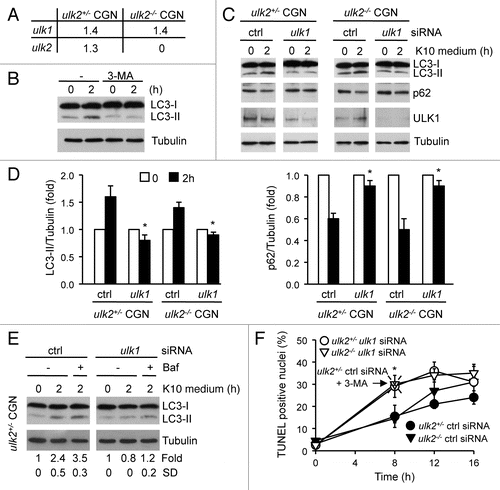
First, we confirmed that both ULK1 and ULK2 were expressed in CGN by real time PCR (). Like in MEFs, the loss of ulk2 mRNA in ulk2−/− CGN did not affect the expression level of the ulk1 transcript. Matured CGN were switched from a medium containing 10% serum and 25 mM potassium (K25 medium) to a serum-free low potassium concentration (10 mM) medium (K10 medium) for 2 h. The replacement of K25 medium with K10 medium increased the level of LC3-II (). However, in contrast to MEFs, this did not correlate with a decrease in the level of LC3-I. It is possible that the high level of LC3-I expressed in neurons prevents the detection of slight changes in protein amount by immunoblot analysis. Nonetheless, we confirmed that the detection of LC3-II was a good indicator of autophagy by demonstrating that increased LC3-II level following low potassium treatment in serum-free conditions was impaired in neurons incubated with 3-MA (). Together, these results showed that autophagy was induced in CGN incubated in K10 medium.
Next, a control experiment was carried out to establish the conditions for downregulating ULK1 in neurons. We found that 50 nM was the optimal concentration of ulk1 siRNA to specifically downregulate ULK1 expression in CGN without having any cytotoxic effect (data not shown). CGN prepared from ulk2+/− and ulk2−/− mice were transfected at two days in vitro (DIV2) with control siRNA or ulk1 siRNA prior to being cultured in K10 medium at DIV7. Consistent with our previous results (), LC3-II level significantly increased in ulk2+/− and ulk2−/− CGN incubated for 2 h in K10 medium ( and D). This correlated with a concomitant decrease in the level of p62 ( and D). In contrast, no significant changes in the levels of LC3-II and p62 were detected in ULK1-deficient CGN deprived of potassium and serum for 2 h, in the absence or in the presence of Baf (). Together these results demonstrated that ULK1, but not ULK2, was required for neuronal autophagy induced by low potassium concentration in serum-free conditions.
The loss of ULK1 sensitizes neurons to apoptosis.
The cytoprotective function of autophagy in neurons during starvation may, at least in part, be mediated by its ability to prevent apoptosis.Citation27 Apoptosis is a physiological process characterized by morphological changes that reflect the activation of a tightly regulated intrinsic cell signaling machinery that leads to the activation of caspases and DNA fragmentation.Citation28 However, under certain conditions, autophagy promotes cell death. For example, the downregulation of Atg5 suppresses the autophagic death of HeLa cells treated with interferon-γ.Citation29 Therefore, we investigated whether impaired autophagy in CGN associated with the functional loss of ULK1 affected the apoptotic response of neurons to starvation.
Consistent with previous data, incubation of CGN in K10 medium increased the number of neurons displaying apoptotic nuclei with fragmented DNA with a maximum after 16 h (). DNA fragmentation was detected by TUNEL (TdT-mediated dUTP nick end labeling), while neurons were visualized by immunostaining using a specific antibody to the microtubule-associated protein 2 (MAP2). DAPI was used as a nuclear marker. However, the loss of ULK1 significantly enhanced the percent of TUNEL positive cells after 8 h incubation in K10 medium, independently of the presence of ULK2 (). Inhibition of autophagy by incubating the cells with 3-MA caused a similar increase in the number of cells displaying fragmented DNA (). Together these results indicate that the requirement of ULK1 to induce autophagy in CGN transiently protects neurons from low potassium-induced apoptosis in serum-free conditions.
Discussion
Our results demonstrate that ULK1 and ULK2 are redundant protein kinases required for autophagy in primary fibroblasts under basal and deprived conditions. Consistently, we found that ULK1-depleted or ULK2-deficient MEFs displayed normal increase in LC3 conversion and p62 degradation in response to starvation, demonstrating that autophagy can be induced in the absence of ULK1 or of ULK2. Our conclusion is supported by evidence that ulk1−/− and ulk2−/− mice are viable and develop normally.Citation19 Furthermore, the targeted deletion of the ulk1 gene in MEFs did not prevent glucose withdrawal-induced autophagy.Citation19 However, previous studies have demonstrated that ulk1−/− MEFs were resistant to autophagy induced by rapamycin treatmentCitation15 or by amino acid withdrawal.Citation16 Consistently, ULK1, but not ULK2, was shown to be a critical regulator of autophagy in CGN (our results) and in HEK293 cells.Citation20,Citation21 The physiological significance of the selective requirement of ULK1 to promote autophagy is exemplified by the demonstration that ulk1, but not ulk2, mRNA was upregulated during erythroid differentiation, and that ULK1 deficiency resulted in delayed autophagic clearance of mitochondria in reticulocytes.Citation19 Although the ulk1−/− mice do not display any gross developmental defect of the brain, future studies will be required to determine the biological function of ULK1 in vivo using mouse models of brain injury and neurodegenerative disorders.
We have shown that CGN lacking ULK1 or incubated with 3-MA displayed higher sensitivity, under serum-free conditions, to low potassium-induced DNA fragmentation, one hallmark of apoptosis. Increased DNA fragmentation caused by the loss of ULK1 did not appear to correlate with elevated caspase 3 activity (data not shown). Mitochondrial release of the apoptosis inducing factor (AIF) has been shown to cause DNA fragmentation independently of caspases.Citation30 Together with evidence that potassium deprivation of CGN induces the nuclear translocation of AIF,Citation31 increased mitochondrial release of AIF may constitute one mechanism by which ULK1 inhibition could enhance DNA cleavage in neurons without affecting caspase 3 activity. Overall, the prosurvival function of ULK1-mediated autophagy in neurons is consistent with the demonstration that HeLa cells, in which Atg genes have been knocked down, are more sensitive to nutrient deprivation.Citation27 However, this observation contrasts with a previous study which demonstrated that the treatment of rat CGN with 3-MA decreases apoptosis.Citation32 Possible explanations for these controversial findings may lie in the difference in the species from which the neurons were prepared (i.e., rats compared to mice) and in the conditions of the cell cultures. This includes the concentration of potassium after deprivation (i.e., 5 mM compared to 10 mM), which can influence the signaling mechanisms in these postmitotic neurons.
However, the role of ULKs is not limited to autophagy and the regulation of cell survival. For example, we have found that ULK1- and ULK2-deficient CGN displayed shorter axons compared to control neurons (data not shown). The requirement of ULK1 and ULK2 for neurite development is supported by recent evidence that ULK1 and ULK2 are recruited to the TrkA-nerve growth factor (NGF) receptor complex and regulate non-clathrin-coated endocytosis in growth cones, filopodia extension and neurite branching of sensory axons.Citation33 Furthermore, expression of dominant negative mutants of ULKs (ULK1-K46R and ULK2-K39T) suppresses neurite extension in CGN.Citation34 However, these mutants displayed a residual kinase activity sufficient to autophosphorylate and did not affect the autophagic response of HEK293 cells.Citation20,Citation35 In contrast, ULK1-K46I or ULK2-K39I mutants defective in autophosphorylation inhibited starvation-induced autophagy.Citation35 Together, these studies suggest that different levels of protein kinase activity may determine the function of ULK1 and ULK2 as mediators of autophagy or neurite outgrowth.Citation36
Yeast Tor and its mammalian homolog mTOR function as negative regulators of autophagy.Citation5,Citation37,Citation38 This can be mediated, at least in part, by the ability of TOR to inhibit Atg1, the yeast homologue of ULK, by phosphorylating Atg13.Citation13 In mammals, a similar mechanism was described in which mTORC1, but not mTORC2, inhibited ULK activity by interacting with the ULK-mAtg13-FIP200 complex in a nutrient-dependent manner.Citation14–Citation16 mTORC1 phosphorylates ULK1, ULK2 and mAtg13 under nutrient-rich conditions, and ULK1 and ULK2 are dephosphorylated following rapamycin treatment or starvation.Citation14–Citation16 Conversely, a low level of phosphorylation of mTOR and its downstream substrates, AKT and p70S6K, was detected in cortical neurons deprived of nutrients.Citation39 However, the involvement of autophagy genes in inhibiting mTOR signaling was not addressed in this study. Therefore we tested the possibility that ULK1 and ULK2 induced autophagy by interfering with mTOR signaling. However, we found no significant difference in the level of AKT or p70S6K phosphorylation in nutrient-deprived ulk2+/− or ulk2−/− MEFs depleted or not of ULK1 (data not shown). Similarly, the loss of ULK1 did not affect decreased phosphorylation of AKT and p70S6K in ulk2+/− or ulk2−/− CGN following low potassium deprivation (data not shown). A key question in future studies will be to identify the mechanism by which ULKs induce autophagy to promote cell survival during development and in diseases. This will be addressed, at least in part, by finding novel physiologically relevant substrates of ULKs using proteomic approaches.
Materials and Methods
Materials.
Dulbecco's modified essential medium (DMEM 21969-035), Dulbecco's Phosphate Buffered Saline (D-PBS 14040), fetal bovine serum (FBS 10270), Lipofectamine 2000 (11668-019) and Opti-Minimum Essential Medium (Opti-MEM 31985) were purchased from Invitrogen. Eagle's Minimum Essential Medium (MEM, M2279), HBSS (H9394), poly-L-ornithine (P4957), 5-fluoro-2′-deoxyuridine (FDU, F0503) and 3-MA (M9281) were from Sigma. Baf (023-11641) was from Wako chemicals. In situ Cell Death Detection kit (TMR-red 12 156 792 910) was purchased from Roche. Amaxa electroporation transfection reagent was from Lonza (VPD-1005). Mouse ulk1 siRNA (SI01461999) and control siRNA (1027281) were obtained from Qiagen. pEGFP-LC3 was a kind gift from Dr. Noboru Mizushima (Tokyo Medical and Dental University, Japan).
ULK2 knockout mice.
The ulk2 null mice were obtained from the MMRRC (Mutant Mouse Regional Resource Centers, strain name: B6;129S5-Ulk2tm1Lex; stock number 011678-UNC). The ulk2+/− mice were generated by crossing ulk2−/− with wild-type mice. Offspring carrying the ulk2-null allele were identified by PCRs on tail DNA using forward for wild-type (P1; 5′-GCT TCA GCA TGA AAA CAT CG-3′), forward for knockout (P2; 5′-GCA GCG CAT CGC CTT CTA TC-3′) and reverse (P3; 5′-GCA GTC AGT GCT ACT AAC-3′) primers. 250 bp and 316 bp fragments were amplified from the wild type and ulk2-null allele, respectively. All mice were maintained in a pathogen-free facility at the University of Manchester. The animal studies were performed in accordance with the UK Home Office and institutional guidelines.
Tissue culture.
MEFs and CGN were prepared from ulk2+/− and ulk2−/− embryonic day 13 murine embryos and 7-day-old mice, respectively.Citation40,Citation41 MEFs were cultured in DMEM supplemented with 10% (v/v) FBS. For nutrient deprivation, MEFs were washed twice with D-PBS prior to incubation in HBSS for various times. CGN were cultured on poly-L-ornithine pre-coated six-well plates at a density of 0.25 × 106 cells/cm2 in MEM supplemented with 10% (v/v) FBS, 2 mM L-glutamine, 20 mM KCl and 30 mM glucose (K25 medium). At DIV2, the medium was replaced by fresh medium supplemented with 80 µM FDU to inhibit cell division of contaminant mitotic cells. To induce cell death in CGN, the K25 medium was removed at DIV7 and replaced by FBS-free medium containing 10 mM KCl (K10 medium). Where appropriate, the cells were incubated with inhibitors as follows: 3-MA (5 mM), Baf (100 nM).
siRNA transfection.
MEFs were transfected at 50% confluency with 10 nM siRNA using the Amaxa nucleofection system. Nucleofected cells were cultured for 48 h prior to being subjected to further treatments. CGNs at DIV2 were transfected with 50 nM siRNA by Lipofectamine 2000. Cells were cultured for additional 5 days prior to further treatments.
Preparation of cell lysates and immunoblot analysis.
Proteins were extracted from cells in triton lysis buffer.Citation42 20 to 50 µg proteins were subjected to immunoblot analysis with antibodies against ULK1 (Santa Cruz, N17), LC3 (MBL international, PM036), p62 (MBL international, PM045), and tubulin (Sigma, T6199). Immunecomplexes were detected by enhanced chemiluminescence with anti-goat, anti-rabbit or anti-mouse immunoglobulin G coupled to horseradish peroxidase as the secondary antibody.
DNA fragmentation assay.
Triple staining with DAPI (5 µg/ml), a mouse monoclonal anti-MAP2 antibody (Sigma, 4528) and TUNEL (In situ Cell Death Detection kit) was performed to detect apoptotic nuclei in CGN. Immunofluorescence was carried out in cells fixed in 4% paraformaldehyde (PFA). MAP2-immune complex was detected with secondary anti-mouse antibody conjugated to Alexa Fluor® 488 (Invitrogen, A11001). Fluorescence images were visualized using an Olympus Heinrich BX51 microscope attached to a CCD Coolsnap-ES camera and processed with the ImageJ software.
GFP-LC3 immunofluorescence.
MEFs were fixed in 4% PFA prior to being incubated with a specific antibody to GFP (Abcam, ab13970-100). Immune complexes were detected with secondary anti-chicken antibody conjugated to Alexa Fluor® 488 (Invitrogen, A11039). Fluorescence images were visualized using an Olympus Heinrich BX51 microscope attached to a CCD Coolsnap-ES camera and processed with the ImageJ software.
Quantitative real time PCR.
Total RNA was isolated from the cells using the Trizol™ reagent and cDNA synthesis was carried out as previously described in reference Citation42. Real-time quantitative PCRs were performed using the SYBR Green I Core Kit (Eurogentec, RT-SN10-05NR). Forward (5′-CGT CCT CCA AGA CGC TGT AT-3′) and reverse (5′-CCT GTT GCT TTC CTC CAA AG-3′) primers for ulk1, forward (5′-CAC AGA ACG ACC AAT GGA TG-3′) and reverse (5′-GGT GAA GAG GAC AGC TCT GG-3′) primers for ulk2, and forward (5′-CCC TAG GCA CCA GGG TGT GA-3′) and reverse (5′-GTC GTC CCA GTT GGT AAC AAT G-3′) primers for actin were used to generate PCR products that were detected in the ABI-PRISM 7700 sequence detection systems (Applied Biosystems). The CT values corresponding to ulk1 and ulk2 mRNA were normalized to that of actin mRNA.
Statistical analysis of the data.
Data were analyzed with Student's t test for comparison between groups. One-way ANOVA followed by post hoc Tukey's test was used for statistical analysis between CGN transfected with ulk1 siRNA versus ctrl.
Abbreviations
| ULK | = | uncoordinated 51-like kinase |
| ATG | = | autophagy-related |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| (m)TOR | = | mammalian target of rapamycin |
| mTORC | = | mTOR complex |
| GFP | = | green fluorescent protein |
Figures and Tables
Figure 1 The loss of ULK1 and ULK2 blocks the autophagic response of MEFs to nutrient deprivation. (A) (i) Schematic representation of the ulk2 wild-type and mutated allele. The LZ-Neo cassette is introduced in place of exons 1, 2 and 3 of the ulk2 gene; (ii) Genomic DNA isolated from the tails of heterozygous (ulk2+/−) and homozygous (ulk2−/−) null mice were amplified by PCR with primers specific for the wild type (P1 + P3) and for the mutated (P2 + P3) allele; (iii) Total RNA was extracted from ulk2+/− and ulk2−/− MEFs and the amount of ulk1 and ulk2 transcripts normalized to that of actin was measured by quantitative real-time PCR. The data are expressed as arbitrary units. (B–E) ulk2+/− and ulk2−/− MEFs were transfected with control (ctrl) siRNA or ulk1 siRNA for 48 h, and cultured in complete or starvation (HBSS) medium for the indicated times. Where indicated the cells were incubated with Baf (D and E) or with 3-MA (F). Immunoblot analysis was performed to detect LC3 conversion and p62 degradation and to confirm the suppression of ULK1 expression in cell transfected with siRNA against ULK1 (B, D and E). Tubulin was used as a loading control. The results are representative of three (B) or two (D and E) independent experiments. Immunoblot signals were quantified with the ImageQuantifier software (BioImage, Jackson MI) (C). LC3 ratio (LC3-II /LC3-I) and the level of p62 normalized to Tubulin (p62/Tubulin) are expressed as fold of nontreated cells for each condition. *p < 0.05, indicates a significant difference between ulk2+/− and ULK1-depleted ulk2−/− MEFs. (F) ulk2+/− and ulk2−/− MEFs were transiently transfected with pEGFP-LC3 (3 µg) together with either ctrl or ulk1 siRNA, and were incubated in complete or starvation medium for 30 min. GFP-LC3 positive cells were visualized by microscopy and counted by eye from a minimum of eight random fields of view, for each condition. The data correspond to the mean ± range of two independent experiments. Representative images are included. Scale bar, 20 µm.
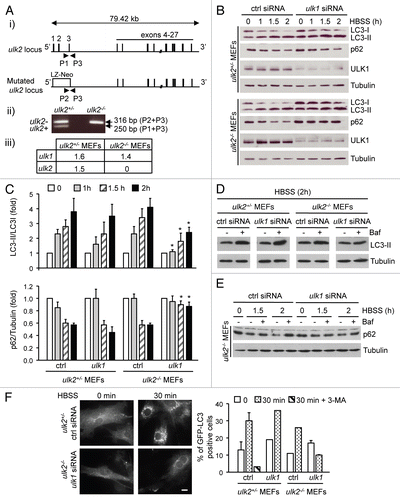
Figure 2 Impaired autophagy in CGN associated with the loss of ULK1 enhanced DNA fragmentation. (A) Total RNA was extracted from ulk2+/− and ulk2−/− CGN and the amount of ulk1 and ulk2 transcripts normalized to that of actin was measured by quantitative real-time PCR. (B) CGN at DIV7 were incubated in K10 medium for 2 h in the absence or in the presence of 3-MA. Autophagy induction by LC3 assay was detected by immunoblot analysis using a LC3 antibody. (C–F) ulk2+/− and ulk2−/− CGN were transfected with either ctrl siRNA or ulk1 siRNA at DIV2. At DIV7, the cells were incubated in K10 medium for the indicated times. Where indicated the cells were incubated with Baf (E) or with 3-MA (F). Immunoblot analysis was performed to detect LC3 conversion, p62 degradation and to confirm the suppression of ULK1 expression in CGN transfected with siRNA against ULK1 (C and E). Tubulin was used as a loading control. The results are representative of three (C) and two (E) independent experiments. Immunoblot signals were quantified with the ImageQuantifier software (BioImage, Jackson MI). The levels of LC3-II and of p62 normalized to Tubulin are expressed as fold of nontreated cells for each condition. The data correspond to the mean ± SE of three independent experiments (D) or to the mean ± SD of two independent experiments (E). *p < 0.05, indicates a significant difference between CGN transfected with ulk1 siRNA versus ctrl siRNA. Apoptotic nuclei were detected by TUNEL as purple (blue-DAPI + red-TUNEL). They were distinguished from DNA debris by immunostaining the cells with MAP2. A minimum of 100 cells were counted by eye from a minimum of eight random fields of view, for each condition. The percentage of TUNEL positive nuclei is plotted as a function of time (F). The data correspond to the mean ± SD of three independent experiments. *p < 0.05, indicates a significant difference between CGN transfected with ulk1 siRNA versus ctrl siRNA.
Acknowledgements
We are indebted to the Mutant Mouse Regional Resource Centers (MMRRC) for kindly providing us with the ulk2 null mice. We thank N. Mizushima for giving us the GFP-LC3 construct and A. Whitmarsh for critically reviewing the manuscript. This work was supported in part by a scholarship from the Korean National Research Foundation to E.L. and by a grant from the Wellcome Trust.
References
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463 - 477
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032 - 1036
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425 - 434
- Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 2002; 11:1107 - 1117
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36:585 - 595
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 889
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 884
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42
- Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differ 2005; 12:1528 - 1534
- Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett 1993; 333:169 - 174
- Suzuki K, Ohsumi Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett 2007; 581:2156 - 2161
- Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell 2005; 16:2544 - 2553
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 2000; 150:1507 - 1513
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981 - 1991
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992 - 2003
- Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297 - 12305
- Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008; 181:497 - 510
- Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol 2009; 29:157 - 171
- Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008; 112:1493 - 1502
- Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem 2007; 282:25464 - 25474
- Young AR, Chan EY, Hu XW, Köchl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 2006; 119:3888 - 3900
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 5728
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 614
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101 - 1111
- Contestabile A. Cerebellar granule cells as a model to study mechanisms of neuronal apoptosis or survival in vivo and in vitro. Cerebellum 2002; 1:41 - 55
- Canu N, Tufi R, Serafino AL, Amadoro G, Ciotti MT, Calissano P. Role of the autophagic-lysosomal system on low potassium-induced apoptosis in cultured cerebellar granule cells. J Neurochem 2005; 92:1228 - 1242
- Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25:1025 - 1040
- Rich T, Watson CJ, Wyllie A. Apoptosis: the germs of death. Nat Cell Biol 1999; 1:69 - 71
- Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 2005; 280:20722 - 20729
- Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001; 410:549 - 554
- Slagsvold HH, Rosseland CM, Jacobs C, Khuong E, Kristoffersen N, Gaarder M, et al. High molecular weight DNA fragments are processed by caspase sensitive or caspase independent pathways in cultures of cerebellar granule neurons. Brain Res 2003; 984:111 - 121
- Maycotte P, Guemez-Gamboa A, Moran J. Apoptosis and autophagy in rat cerebellar granule neuron death: Role of reactive oxygen species. J Neurosci Res 2010; 88:73 - 85
- Zhou X, Babu JR, da Silva S, Shu Q, Graef IA, Oliver T, et al. Unc-51-like kinase 1/2-mediated endocytic processes regulate filopodia extension and branching of sensory axons. Proc Natl Acad Sci USA 2007; 104:5842 - 5847
- Tomoda T, Bhatt RS, Kuroyanagi H, Shirasawa T, Hatten MEA. Mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron 1999; 24:833 - 846
- Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol 2009; 29:157 - 171
- Chan EY, Tooze SA. Evolution of Atg1 function and regulation. Autophagy 2009; 5:758 - 765
- Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell 2004; 7:167 - 178
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 1998; 273:3963 - 3966
- Young JE, Martinez RA, La Spada AR. Nutrient deprivation induces neuronal autophagy and implicates reduced insulin signaling in neuroprotective autophagy activation. J Biol Chem 2009; 284:2363 - 2373
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000; 288:870 - 874
- Courtney MJ, Nicholls DG. Interactions between phospholipase C-coupled and NMDA receptor in cultured cerebellar granule cells: protein kinase C-mediated inhibition of NMDA responses. J Neurochem 1992; 59:983 - 992
- Kayahara M, Wang X, Tournier C. Selective regulation of c-jun gene expression by the mitogen-activated protein kinases via the TPA-responsive element and the myocyte enhancer factor 2 binding sites. Mol Cell Biol 2005; 25:3784 - 3792