Abstract
Macro(autophagy) is a cellular mechanism which delivers cytoplasmic constituents to lysosomes for degradation. Due to its role in maintaining cellular integrity, autophagy protects against various diseases including cancer. p53 is a major tumor suppressor gene which can modulate autophagy both positively and negatively. p53 induces autophagy via transcriptional activation of Damage-Regulated Autophagy Modulator (DRAM-1). We report here that DRAM-1 encodes not just one mRNA, but a series of p53-inducible splice variants which are expressed at varying levels in multiple human and mouse cell lines. Two of these new splice variants, termed SV4 and SV5, result in mature mRNA species. Different to ‘full-length’ DRAM-1 (SV1), SV4 and SV5 do not localise to lysosomes or endosomes, but instead partially localise to peroxisomes and autophagosomes respectively. In addition, SV4 and SV5 can also be found co-localised with certain markers of the endoplasmic reticulum. Similar to SV1, SV4 and SV5 do not appear to be inducers of programmed cell death, but they do modulate autophagy. In summary, these findings identify new autophagy regulators that provide insight into the control of autophagy downstream of p53.
Introduction
Autophagy is a mechanism for degrading intracellular constituents in lysosomes.Citation1,Citation2 Depending on the mode of delivery to the lysosome, there are three types of autophagy: macroautophagy, microautophagy and chaperone-mediated autophagy.Citation1,Citation2 Macroautophagy, which for simplicity is referred to hereafter as autophagy, is the most extensively investigated and is active in all cells.Citation3 Upon induction, membranous structures termed phagophores or isolation membranes form from a variety of origins.Citation4–Citation7 As these membranes grow they accrue cytoplasmic constituents for digestion and form a double-membraned vesicle called an autophagosome which completely encapsulates the cargo.Citation1–Citation3 Autophagosomes can then fuse with additional organelles including endosomes and multivesicular bodies, but fusion ultimately occurs with a lysosome to form a new organelle termed an autolysosome. Acidic hydrolases provided by the lysosome promote the degradation of the contents of the autolysosome which are then released into the cytosol where they can be either further catabolized or utilized in biosynthetic pathways.Citation1–Citation3
Although active at basal levels in all cells as a homeostatic mechanism for the breakdown of misfolded proteins and damaged organelles, the levels and cargoes of autophagy can change in response to a variety of stimuli.Citation2,Citation3 For example, the rate of autophagy can be increased in response to starvation as a limited internal mechanism to fuel ATP synthesis until external nutrients are available.Citation8–Citation10 The selective removal of specific organelles can also occur such as the selective removal of mitochondria (mitophagy) which occurs during erythrocyte maturation.Citation11,Citation12 Due to these multiple roles in preserving cellular homeostasis, it is not a surprise that autophagy has key roles in protecting us against various forms of disease including Crohn disease, neurodegenerative diseases and cancer.Citation2,Citation13
The progression to cancer is a multistage process that involves the perturbation of genes which promote cancer (oncogenes) and genes which naturally repress cancer (tumor suppressor genes).Citation14 Many genes which are known to be altered during tumor development have been shown to be modulators of autophagy.Citation13 p53—the most frequently mutated gene in human cancer—is no exception in this regard and has been shown to have both positive and negative effects on autophagy.Citation15,Citation16 At basal levels, a proportion of p53 which is localized in the cytoplasm has been shown to inhibit autophagy by binding at the endoplasmic reticulum.Citation17 However, in response to various forms of cellular stress, for example DNA damage or ribosomal stress, the levels of p53 increase above basal levels and accumulate in the nucleus where p53 functions as a transcriptional activator of a series of genes involved in tumor suppression.Citation18 In this context, p53 has been shown to activate a number of genes which promote autophagy.Citation15,Citation16 Amongst these genes is the damage-regulated autophagy modulator (DRAM-1) which belongs to an evolutionarily conserved family of proteins.Citation19,Citation20 We have previously shown that DRAM-1 encodes for a lysosomal protein that is required for p53′s ability to induce autophagy.Citation21,Citation22 We report here that DRAM-1 also encodes for additional isoforms of DRAM-1 that are generated by alternative mRNA splicing in multiple cell types. We go on to show that these splice variants are induced by p53 and that two isoforms encoded by these new mRNA species are modulators of autophagy. These findings therefore not only identify new autophagy regulators, but also highlight additional complexity in p53′s control of autophagy.
Results
DRAM-1 encodes multiple splice variants that are induced by p53.
Many genes express a variety of alternatively spliced mRNA species which can encode for proteins with different functions.Citation23 We were interested therefore to know whether DRAM-1 also encodes for other splice variants beyond the one we had previously described.Citation21,Citation22,Citation24 To test this, we utilized a previously described p53 tetracycline-inducible (TetOn-p53) cell line to explore not only if DRAM-1 splice variants exist, but if so, whether they are also induced by p53.Citation25 RNA was isolated from this line and was subjected to semiquantitative RT-PCR using primers from exon 1 at the extreme 5′ of DRAM-1 and exon 7 at the extreme 3′. These primers were not only able to detect the mRNA species that we had previously reported as DRAM-1 (described here as SV1 for ‘splice variant 1’), but a number of smaller RNA species were also amplified (). Furthermore, activation of p53 in this cell line with the tetracycline analog, doxycyline (Dox) (), caused a marked upregulation of several RNA species which could be amplified with DRAM-1 primers (). We were able to clone eight of these new RNA species and sequencing confirmed that they were encoded by DRAM-1 and that they had the exon structure depicted in . Each of the splice variants contain exon 1 and exon 7, but lack different combinations of exons 2–6. Only the splicing of SV4 and SV5, however, results in mRNA species that continue in-frame to the same stop codon as SV1 at the end of exon 7 (Fig. S1). As a result, it is highly likely that these splice variants also have mature 3′ ends required for mRNA stability. We decided therefore to focus our analysis on SV4 and SV5.
To test if DRAM-1 splice variants were present in other cells and tissues, we isolated RNA from a panel of cell lines from various sites of origin. RT-PCR amplification of these RNAs with primers from exon 1 and exon 7 of DRAM-1, revealed that DRAM-1 splice variants were evident in all cell lines tested, but that comparative levels of splice variants were different in different cells (). We also considered whether DRAM-1 splice variants were a human-specific phenomenon or if they could be detected in cells from a different species. RNA was therefore isolated from mouse embryo fibroblasts (MEFs) and was amplified with primers from exons 1 and 7 of mouse DRAM-1. Similar to what was observed in human cells, a number of splice variants were detected in MEFs and the expression levels of a number of these mRNA species were increased in cells that had been treated with the p53 inducer Nutlin-3A ().Citation26
DRAM-1 splice variants do not induce programmed cell death.
Our previous work showed that knockdown of DRAM-1 by siRNAs which knocked down all of the splice variants reported here impeded p53-induced programmed cell death.Citation21 However, expression of DRAM-1 isoform 1 (the peptide encoded by SV1), did not cause cell death when ectopically expressed.Citation21 Since we now know that DRAM-1 encodes multiple isoforms, it remained possible that the isoforms encoded by SV4 and SV5 were cell-death inducers and were responsible for the role of DRAM-1 in cell death induced by p53. We therefore ectopically expressed isoforms 1, 4 and 5 alongside p53 to see if they were able to affect cell viability. After 48h of expression, p53 was able to cause a significant induction of apoptotic death as assessed by the appearance of cells with sub-G1 DNA content (p < 0.001), whereas DRAM-1 isoforms had no significant effect in this short-term cell death assay ( and B). To confirm these results, we also tested if DRAM-1 isoforms could cause translocation of phosphatidylserine to the external side of the cell membrane (a marker of apoptotic death)Citation27 and if they could cause cell permeability to propidium idodide (PI) (an indicator of cell membrane rupture which occurs late in apoptosis and also during necrosis).Citation27 In agreement with our findings using sub-G1 analysis, ectopic expression of p53 caused a statistically significant increase in both phosphatidylserine exposure (p < 0.001) and PI permeability (p < 0.001) indicating induction of apoptosis ( and D). No significant effects were observed, however, in cells expressing any of the DRAM-1 isoforms ( and D). Similarly, we also analyzed for changes in mitochondrial membrane potential (another characteristic change during apoptosis)Citation27 by staining with the dye tetramethylrhodamine (TMRE) which stains only active viable mitochondria.Citation27 In agreement with our other cell-death assays, ectopic expression of p53 was again able to cause a significant decrease in TMRE staining (p < 0.001) whereas none of the DRAM-1 isoforms had any significant effect in this assay ( and Fig. S2). Finally, we wanted to examine whether DRAM-1 isoforms had long-term effects on cell viability by monitoring their effects on cellular clonogenicity. In line with what was observed in short-term cell death assays, p53 caused a considerable decrease in the clonogenic potential of the cell population, but no effect was evident upon expression of DRAM-1 isoforms 1, 4 or 5 (). We can conclude from these data that DRAM-1 isoforms when expressed individually do not cause programmed cell death.
Although these assays show that DRAM-1 isoforms cannot induce death when expressed alone, since we know that knockdown of all isoforms of DRAM-1 impedes p53-induced death, it remains possible that any one of the isoforms may play an individual role in death that is induced in the context of p53 activation. To test this, we infected DRAM-1flox/flox mouse embryo fibroblasts (MEFs) with a retrovirus expressing the E1a oncogene fused to a portion of the estrogen receptor (E1aER). Under normal conditions, this construct is inactive due to binding by hsp90. Upon treatment with 4-hydoxytamoxifen, however, hsp90 binding is alleviated and E1a is active. In this state, MEFs become sensitive to the chemotherapeutic drug, doxorubicin, in a p53-dependent manner.Citation28,Citation29 These E1aER-expressing cells were then infected again with either DRAM-1 isoform 1, isoform 4, isoform 5 or empty vector as control (). Finally, each cell population was infected with a retrovirus expressing Cre recombinase (to inactivate all endogenous DRAM-1 expression) or empty retroviral vector as control (). This therefore resulted in cells that express either no DRAM-1 isoforms at all or express only either: isoform1, isoform 4 or isoform 5. These cells were then incubated with 4-hydoxytamoxifen to modulate E1a activity with or without the chemotherapeutic drug doxorubicin. As would be expected, activation of E1a (following treatment with 4-hydoxytamoxifen) caused sensitization to doxorubicin in control cells (-Cre) and in line with our previous studies,Citation21 this sensitization was significantly reduced following recombination and consequent loss of all endogenous DRAM-1 expression following infection with Cre (p < 0.001) (). Rescue of this effect was, however, not achieved by individual expression of either DRAM-1 isoform 1, isoform 4 or isoform 5 (). These data therefore show that neither isoform 1, isoform 4 or isoform 5 when expressed alone can reconstitute the role of DRAM-1 in p53-mediated programmed cell death. We can only assume therefore that DRAM-1's role in cell death downstream of p53 is either mediated by a combination of these isoforms or by an unidentified DRAM-1 protein species.
DRAM-1 isoforms 4 and 5 exhibit partial localization in peroxisomes and autophagosomes respectively as well as in the endoplasmic reticulum.
In addition to a role in cell death downstream of p53, we have also previously shown that DRAM-1 is required for p53-induced autophagy and that SV1 localizes to lysosomes.Citation21 We were keen therefore to know if isoform 4 and isoform 5 were also located in lysosomes. In agreement with our previous studies,Citation21 infection of cells with an adenovirus expressing isoform 1 revealed colocalization with the lysosomal enzyme cathepsin D and the lysosomal membrane protein LAMP-2 ( and Fig. S3A), however, no convincing lysosomal localization was observed in cells infected with adenoviruses expressing isoform 4 or isoform 5 ( and Fig. S3A). We therefore extended our immunofluorescent localization studies to see if isoforms 4 and 5 were colocalized with either: endosomes, autophagosomes, peroxisomes, mitochondria or the endoplasmic reticulum. Although colocalization was observed between isoform 1 and the late endosome markers, CD63 and Rab 7 ( and Fig. S3B), no colocalization of isoform 4 or isoform 5 was observed with either of these markers ( and Fig. S3B). Similarly, isoform 1 was also found to be colocalized with the early endosome markers EEA1 and Rab5 ( and Fig. S3C), but again no colocalization was observed with these markers and isoform 4 and isoform 5 ( and Fig. S3C).
Since isoform 4 and isoform 5 are not localized in lysosomes and late or early endosomes, we were interested to know where they reside in the cells. We first analyzed therefore whether isoform 4 and isoform 5 were located in autophagosomes. Cells were therefore infected with each isoform together with an adenovirus expressing the autophagy marker mCherry-LC3. This revealed that isoform 1 and also isoform 5, but not isoform 4, are partially located in autophagosomes/autolysosomes (). In light of this result, we also analyzed if DRAM-1 isoforms are colocalized with DFCP-1—a protein normally localized in the Golgi and endoplasmic reticulum, but which colocalizes with LC3 upon autophagy induction. This indeed revealed that all DRAM-1 isoform were found partially colocalized with DFCP-1 (). Since we only observed partial localization of DRAM-1 isoforms with LC3 and DFCP-1, further colocalization studies were carried out with markers of mitochondria, peroxisomes and the endoplasmic reticulum. Although no localization was observed for any of the DRAM-1 isoforms in mitochondria (Fig. S4), both isoforms 4 and 5, but not isoform 1 were found to be partially localized with the endoplasmic reticulum markers BIP and Calnexin ( and B). In addition, we also observed partial colocalization of isoform 4 with the peroxisome markers PMP70 and peroxisome-GFP (). However, no convincing peroxisomal localization was observed for isoform 1 and isoform 5 (). In light of these findings we consider that isoforms 4 and 5 are distributed across several cellular compartments explaining the partial colocalization that was observed in any individual compartment. It is also interesting to note that due to the different cellular localization of isoform 1 compared to isoform 4 and isoform 5, we observed that while isoform 1 is degraded in lysosomes as would be expected, isoform 4 and isoform 5 are not degraded in lysosomes (Fig. S5A), but instead are degraded in the proteasome (Fig. S5B).
DRAM-1 isoforms SV4 and SV5 are modulators of autophagy.
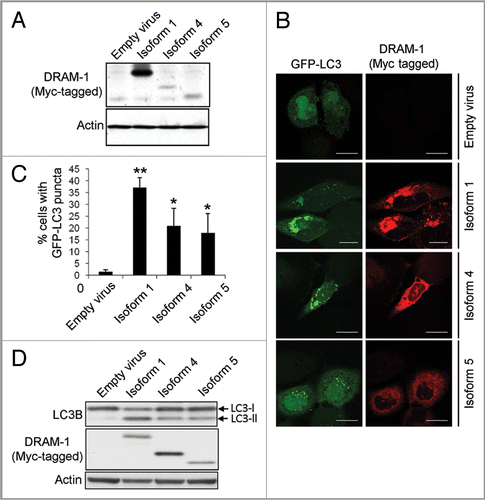
Although DRAM-1 isoforms 4 and 5 are not localized in lysosomes, their partial colocalization with the ER, autophagosomes and peroxisomes indicated that they may regulate autophagy. To test this, adenoviruses expressing isoforms 1, 4 and 5 were infected into cells together with an adenovirus expressing GFP-LC3.Citation30,Citation31 As expected, a marked accumulation of autophagosomes was observed following infection of cells with isoform 1 compared with cells infected with empty viral control (p , 0.001) (). Significant accumulation of autophagosomes was also observed in cells infected with isoform 4 and isoform 5 (p < 0.05) although the percentage of cells exhibiting an increase in GFP-LC3 puncta was lower than that observed with isoform 1 (). As a control for the effects of adenoviral infection, cells were also infected with equal numbers of infectious particles of an adenovirus that does not contain a transgene. Infection of cells with this virus did not result in an increase in GFP-LC3 puncta indicating that isoform 4 and isoform 5, like isoform 1, are modulators of autophagy (). To confirm that this was the case, we also assessed the change in ratio of LC3-I to LC3-II by western blot following infection with each of the DRAM-1 isoforms. In agreement with our analysis of GFP-LC3 puncta, this revealed that isoforms 1, 4 and 5 all increase the ratio of LC3-II to LC3-I (indicating formation of autophagosomes), but the change in ratio with isoform 1 was greater than that observed with isoform 4 or isoform 5 ().
Discussion
We describe here the identification and initial characterization of two new autophagy regulators in the form of isoforms of the previously described protein DRAM-1. Our previous studies had shown that knockdown of DRAM-1 with siRNAs that target all the DRAM-1 splice variants described in this study, caused impedance of the ability of p53 to induce autophagy and also programmed cell death.Citation21,Citation22 However, ectopic expression of what we at the time considered the only form of DRAM-1 (described here as isoform 1 or SV1 for splice variant 1) while able to modulate autophagy, had no impact on cell death.Citation21,Citation22 The discovery therefore of additional protein isoforms encoded by DRAM-1 opens up the possibility that these new isoforms may be responsible for phenotypic effects of DRAM-1 knockdown beyond the control of autophagy. As a result, our initial studies focused on whether these new isoforms could induce programmed cell death. This revealed, however, that the ectopic expression of either isoform 4 or isoform 5 had no impact on cell death even in long term clonogenic survival studies. Instead, we found that isoform 4 and isoform 5 were similar to isoform 1 in that they were modulators of autophagy.
Although isoform 4 and isoform 5 are factors which can regulate autophagy, it is interesting that they are different from isoform 1 with respect to their subcellular distribution. Following the extensive studies presented here, we found that DRAM-1 isoforms are partially localized in multiple cellular compartments and that isoform 4 and 5 differ in their cellular distribution when compared to isoform 1. It is intriguing therefore how these new isoforms modulate autophagy both individually, and perhaps also coordinately, in these different regions of the cell. Thus, additional studies in this area are certainly worthy of pursuit.
The accumulation of autophagosomes as a result of the ectopic expression of isoform 4 and isoform 5 in Saos-2 cells (an osteosarcoma cell line), was lower than that seen following expression of isoform 1. Although this may simply reflect lower levels of expression, it is important to consider the relative contribution of isoform 4 and isoform 5 to autophagy when compared with isoform 1. Our analysis of DRAM-1 splice variant expression in a variety of tissues revealed that they are widely expressed in both human and mouse. In many of the cells analyzed, it was also often the case that the expression level of these newly identified splice variants was in fact higher than that of SV1. This indicates therefore that while DRAM-1 splice variant expression is both widespread, and to a point at least, also evolutionarily conserved, it may also be that the balance of effects of different DRAM-1 isoforms and their relative importance is both cell type-and context-dependent.
Materials and Methods
Cell culture, infections and transfections.
TetOn-p53 and parental Saos-2 cells have been previously described.Citation21,Citation25 Wild-type and DRAM-1flox/flox MEFs (which contain a gene-targeting event in exon 2 of DRAM-1) were isolated from E13.5 wild-type or DRAM-1flox/flox embryos. All other cell lines are available from ATCC. All cell cultures were grown in D-MEM (Gibco, 21969) supplemented with 10% FCS and were, where indicated, either infected with adenoviral lysates or transfected using X-fect (Clontech, 631316) or calcium phosphate transfection as previously described.Citation32 TetOn-p53 cells were induced with 1 µg/ml doxycycline (Dox) (Sigma, D9891). Endogenous p53 was activated in wild-type MEFs with 10 µM Nutlin-3 (Sigma, N6287) for 48 h. E1aER-expressing cells were activated by incubation in 500 nM 4-hydoxytamoxifen (Sigma, H7904). Retroviral infections with pWZL-Blast-E1aER, pBabe-puro-Cre, pBabe-puro and viruses containing DRAM-1 isoforms as detailed below were carried out as previously described.Citation33
Identification and cloning of DRAM-1 splice variants.
PCR amplification of DRAM-1 mRNA species was performed with cDNA from TetOn-p53 cells ± dox (1 µg/ml) using ProofStart™ DNA polymerase (Qiagen, 202203). Cycling parameters used were 95°C 4 min 94°C 30 s, 52°C 30 s, 72°C 1 min 34 cycles, 72°C 10 min on a PTC-200 PCR machine. The primers used to amplify DRAM-1 splice variants were: Fwd, 5′-ATGCTGTGCT-TCCTGAGGGGAATG-3′ and Rev, 5′-CACAGAAATCAATG-GTGATATTTGA-3′. PCR products of DRAM-1 splice variants were ligated into pCR 2.1 vector using T4 DNA Ligase supplied in the TA cloning kit (Invitrogen, K2040-01) according to the manufacturer's instructions. Restriction digestion of pCR 2.1 constructs with HindIII and XbaI removed DRAM-1 coding sequences which were then inserted into pcDNA3.1 Myc-His A (Invitrogen, V80020). Retroviral constructs containing DRAM-1 splice variants were generated by digesting pcDNA3.1 Myc-His A expression constructs with HindIII and PmeI which removes the entire DRAM-1 coding region including Myc and His tags. Inserts were then blunt ended and cloned into the SnaB1 site of pWZL-Hygro.
Semi-Q and qPCR.
RNA was prepared using an RNeasy Mini kit (Qiagen, 74104) and cDNA was generated using reverse transcriptase. qPCR was carried out using the DyNAmo SYBR Green 2-step qRT-PCR kit (Finnzymes, F430L) and primers set as follows: Mouse DRAM-1, (Fwd, 5′-CAGCCTTCAT-CATCTCCTACG-3′; Rev, 5′-ATGCAGAGAAGTTTATCATG-3′. Expression was normalized relative to 18S rRNA levels (Fwd, 5′-GTAACCCGTTGAACCCCATT-3′; Rev, 5′-CCATCCAA-TCGGTAGTAGCG-3′). Data collection was carried out using a Chromo4 real-time PCR detector (CFB-3240) and MJ Opticon Monitor software.
For semiquantitative PCR of DRAM-1 mRNA species, cDNA was generated from cell lines as described. Amplifications were undertaken using ProofStart™ DNA polymerase (Qiagen, 202203). Cycling parameters used were 95°C 4 min 94°C 30 s, 52°C 30 s, 72°C 1 min 35 cycles, 72°C 10 min on a PTC-200 PCR machine. The primers used to amplify human DRAM-1 splice variants were: Fwd, 5′-ATGCTGTGCTTCCTGAGGGGAATG-3′ and Rev, 5′-CACAGAAATCAATGGTGATATTTGA-3′. For amplification of mouse DRAM-1 splice variants, the primers used were: Fwd, 5′-TTCCTCCTGGTGACTTGGTC3′ and Rev, 5′-AGTCGTCATTGATTTCTGTGG-3′. Expression was normalized relative to 18S rRNA levels (Fwd, 5′-GTAACCCGTTGAACCCCATT-3′; Rev, 5′-CCATCCAATCGGTAGTAGCG-3′)—amplification was under the same cycling parameters as described for DRAM-1, but for 20 cycles.
Construction of adenoviruses.
DRAM-1 splice variants SV1, SV4 and SV5 were excised from pcDNA3-Myc-His A with HindIII and PmeI and were cloned into the HindIII and EcoRV sites of pShuttle-CMV. pShuttle constructs were then recombined into pAdEasy and were subsequently amplified as previously described.Citation34,Citation35 Adenoviruses were titered using AdEasy™ Viral Titer Kit (Stratagene, 972500). Equal numbers of infective particles were used for each splice variant in comparative studies.
Cell death assays.
Cell death assays were undertaken in the following ways: (1) Flow cytometry analysis (BD FACSCalibur) for the percentage of cells with sub-G1 DNA content—a reliable measure of apoptotic death.Citation36 (2) Clonogenicity assays were performed on Saos-2 cells which had been transfected with either DRAM isoforms, p53 or empty vector as control. Cells were plated on 9 cm dishes at a density of 0.8 × 106 cells per dish. Twenty-four hours later, cells were transfected using calcium phosphate precipitation with 10 µg of either: DRAM-1 SV1, SV4 or SV5, p53 (positive control) or an empty plasmid pcDNA3.1 Myc-His A (negative control). After 48 h, transfected cell populations were replated with fresh media containing 600 µg/ml G418 (Gibco, 10131-019) for selection of transfected cells. Medium containing G418 was replaced every 48 h for 2 weeks. At this point, cells were washed in PBS and then stained for 10 min with Giemsa (Sigma, GS500). Cells were finally washed three times in 10%MeOH/H20 and allowed to dry prior to visualization. (3) Apoptosis was also assessed by double staining of live (un-fixed cells) with annexinV-Alexa Fluor 488 (Molecular Probes, A13201) and propidium iodide (Sigma, P4170). Apoptotic cells will exhibit positive staining for Annexin-V and at later time points also positive staining for propidium iodide due to cell membrane permeabilization. Necrotic cells stain positive for propidium iodide in the absence of positive Annexin-V staining. (4) Changes in mitochondrial membrane potential (another apoptotic marker) were also analyzed by measuring the accumulation of the mitochondrial probe MitoTracker Green FM (Molecular Probes; M7514) which stains mitochondria regardless of membrane potential (ΔΨm) and with TMRE (Molecular Probes, T-669) which is sequestered only by active mitochondria. Cells were incubated at 37 °C for 30 min in complete medium without phenol red (Gibco, 31053) containing 50 nM MitoTracker Green FM and 10 nM of TMRE and were then analyzed by fluorescent microscopy.
Immunoblotting.
Cells were lysed with RIPA buffer. Protein was quantitated using BCA assay (Sigma, B9643), separated on SDS-PAGE gels and transferred onto nitrocellulose membrane as previously described.Citation29 Membranes were probed with p53 (DO-1) (Pharmingen, 554293), myc (4A6) (Milipore, 05-724), LC3B (Abcam, ab48394) and actin (beta) (Abcam, ab8227). Proteins were detected by enhanced chemiluminescence (Thermo Scientific, 32106).
Immunofluorescence.
Saos-2 cells were seeded on coverslips and infected with appropriate adenoviral lysates as indicated. Cells were harvested and fixed in 4% paraformaldehyde (Electron Microscopy Sciences, 15710), and stained with the following antibodies: anti-myc tag (4A6) (Milipore, 05-724), Cathepsin D (Milipore, 06-467), LAMP-2 (BD Transduction; 555803), CD63 (BD Transduction; 556019), Calnexin (BD Transduction, 610523), GRP78 Bip (Abcam, ab21685), CoxIV (Abcam, ab16056), TOM20 (Santa Cruz, sc-11415), PMP70 (Abcam, ab3421), Peroxisome-GFP (Molecular Probes, c10604), EEA1 (BD Transduction, 610456) or Rab5 (BD Transduction, 610725) followed by Alexa Fluor 488 anti-mouse or rabbit IgG (Molecular Probes, A11001 or A11008), Alexa Fluor 647 antimouse or rabbit IgG (Molecular Probes, 21235 or 21244) or Texas Red X anti-mouse or rabbit IgG (Molecular Probes, T6390 or T6391). Cells were also transfected with mRFP-Rab7 (kindly provided by Jim Norman, Beatson Institute) and eGFP-DFCP1 (kindly provided by Nicholas Ktistakis, Babraham Institute, UK)4,37 or infected with adenoviral mCherry-LC3.Citation38 Following fixation, detection was via mRFP, GFP and mCherry fluorescence. In all cases, cells were imaged on Zeiss Axioplan 2 / ISIS for autophagosome formation and Leica Confocal 2 for DRAM-1 isoform colocalization assays.
Autophagy assays.
For assays to detect LC3 puncta as a marker of autophagosome formation, cells were infected with DRAM-1 isoforms or ‘empty virus’ as control and GFP-LC3 adenoviral lysates. After 48 h cells were fixed and imaged on Zeiss Axioplan 2 / ISIS. Images of cells were analyzed visually and cells with eight or more GFP-LC3 puncta were scored as positive for cells with accumulated autophagosomes. At least 200 cells were analyzed following infection of each isoform. Adenoviral GFP-LC3 was kindly provided by Aviva Tolkovsky (University of Cambridge) and has been previously described.Citation31
Analysis of LC3-I to LC3-II conversion was measured by western blotting following infection with either adenoviruses expressing DRAM-1 isoforms or an ‘empty’ adenovirus which contains no transgene as control. Cell lysates were separated on polyacrylamide gels and transferred to nitrocellulose membranes before being probed with antibodies against LC3B (Abcam, ab48394) and actin (beta) (Abcam, ab8227).
Statistical analysis.
Statistical analyses were undertaken via a univariate general linear model using SPSS software followed by correction using Tukey HSD. p values are described in the text at appropriate points. In all figures, *p < 0.05 and **p < 0.001.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| RNAi | = | RNA interference |
| siRNA | = | small interfering RNA |
| GFP | = | green fluorescent protein |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| SV | = | splice variant |
| DRAM-1 | = | damage-regulated autophagy modulator-1 |
| MEF | = | mouse embryo fibroblast |
| Dox | = | doxycycline |
Figures and Tables
Figure 1 DRAM-1 splice variants are induced by p53. (A) Tet-on p53 Saos-2 cells were treated with 1 µg/ml of doxycycline for 24 h. RNA was then harvested and analyzed on a 3% agarose gel. (B) p53 induction upon doxycycline treatment was verified by western blotting. (C) Schematic representation of splice variants from (A) which harbor different internal exon deletions. Images shown are representative of what was observed in at least three separate experiments.
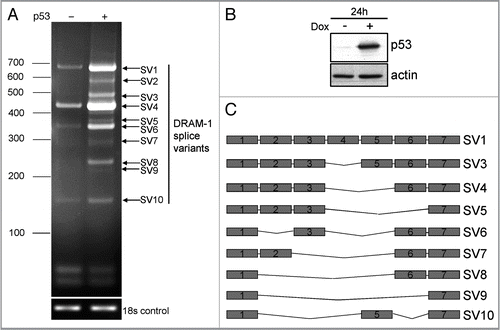
Figure 2 DRAM-1 splice variants are expressed in multiple human and mouse cells. (A) RNA from a panel of human cell lines was subjected to semiquantitative RT-PCR with primers from exon 1 and exon 7 of DRAM-1. Induction of p53 in p53-inducible Saos-2 cells was achieved with 1µg/ml of doxycycline for 24 h. (B) RNA from human p53-inducible Saos-2 cells and from MEFs that had been incubated in the absence or presence of Nutlin-3A (10 µM for 48 h) were subjected to semiquantitative RT-PCR with primers from exon 1 and exon 7 of human and mouse DRAM-1 respectively. Induction of p53 in p53-inducible Saos-2 cells was achieved with 1 µg/ml of doxycycline for 24 h. Images shown are representative of what was observed in at least three separate experiments.
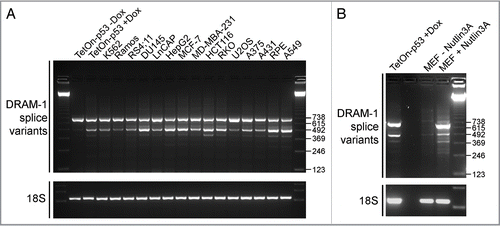
Figure 3 DRAM-1 isoforms 1, 4 and 5 do not induce cell death. (A and B) Saos-2 cells were infected with either an empty adenoviral vector, DRAM-1 isoforms 1, 4 or 5, or p53 (positive control). After 48 h, both adherent and floating cells were harvested and the extent of cell death measured by flow cytometry for the percentage of cells with sub-G1 DNA content. The data represented are from three independent experiments and error bars indicate standard deviation (s.d.) (B). Protein expression of p53 and DRAM-1 isoforms was validated by western blotting (A). Saos2 cells were infected with either an empty adenoviral vector, DRAM-1 isoforms 1, 4, 5 or p53. After 48 h, apoptosis was determined using annexinV-Alexa Fluor 488 and PI staining and analyzed by microscopy (C). Each histogram represents the mean percentage of either Annexin-V positive and PI negative or Annexin-V positive and PI positive cells. Results are the mean ± s.d. of seven determinations (D). Measurement of mitochondrial potential (ΔΨm) was assessed using MitoTracker Green FM which stains mitochondria regardless of ΔΨm and and TMRE which is sequestered only by active mitochondria. The staining was analyzed by microscopy. Each histogram represents the mean percentage of cells positive for both MitoTracker Green and TMRE. Results are the mean±s.d. of four determinations (E). Saos-2 cells were transfected with 10 µg of either: DRAM-1 SV1, SV4 or SV5, p53 (positive control) or an empty plasmid (pCMV; negative control). After 48 h, transfected cell populations were replated with fresh media containing G418 and assessed for clonogenic survival two weeks later (F). Images shown are representative of at least three independent experiments.
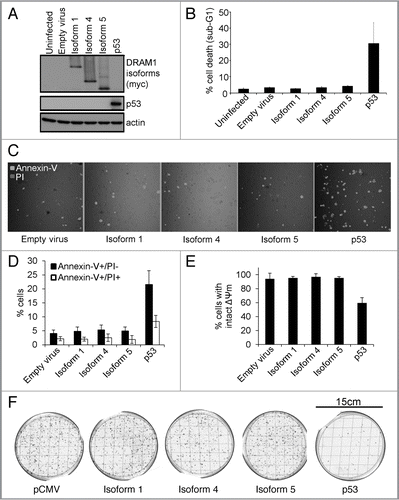
Figure 4 DRAM-1 isoforms 1, 4 or 5 cannot rescue the inhibition of oncogene-induced death caused by total loss of dram-1 expression. DRAM-1fl/fl MEFs expressing the tamoxifen-inducible adenovirus onco-protein E1A-ER were infected with DRAM-1 isoforms 1,4 and 5 or the empty retrovirus vector. Expression of DRAM-1 SV1, 4 and 5 were validated by western blot (A). These panels of cells were then infected with the Cre retrovirus to induce recombination, and an empty vector where indicated (-Cre). DRAM-1 excision was validated at the message level by RT-PCR (B). MEFs were treated with 500 nM of 4-hydroxy tamoxifen for 8 h to activate E1a in this system. The cells were then treated with 0.2 µg/ml doxorubicin. After 24 h, both adherent and floating cells were harvested and the extent of cell death measured by flow cytometry for the percentage of cells with sub-G1 DNA content. The data are representative of what was observed in three independent experiments and error bars indicate standard deviation (C). Tam, 4-hydroxytamoxifen.
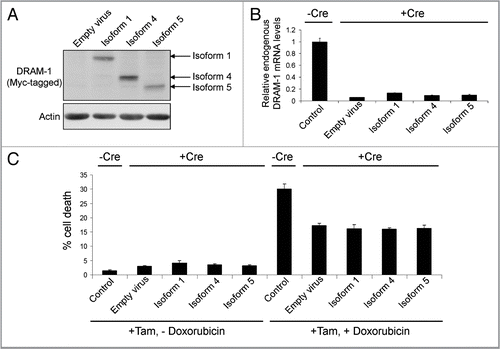
Figure 5 DRAM-1 isoform 1, but not isoforms 4 and 5, locates to lysosomes, as well as early and late endosomes. Saos-2 cells were infected with equivalent amounts of DRAM-1 adenoviral lysates for 48 h. Colocalization (yellow) of myc-tagged DRAM-1 (red) with the respective lysosome and endosome proteins, cathepsin D (A), CD63 (B) EEA1 (C) (green), was assayed by confocal microscopy. The scale bar shown represents 20 µm.
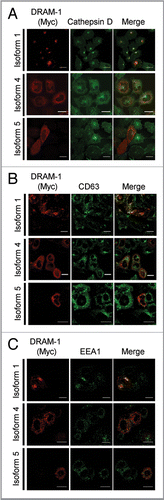
Figure 6 DRAM-1 isoforms 1 and 5 are partially localized with markers of autophagosomes/autolysosomes. Saos-2 cells were infected with an adenovirus carrying mCherry-LC3 transgene (A), and either DRAM-1 isoforms 1, 4 or 5 for 48 h. Colocalization (yellow) of myc-tagged DRAM-1 (green) with LC3 (red) was assayed by confocal microscopy. (B) Saos-2 cells were transfected with pEGFP-DFCP1 (green) using X-fect (Clontech) 24 h prior to infection with DRAM-1 isoforms (red) for 48 h, then harvested for immunofluorescent studies. The scale bar shown represents 20 µm.
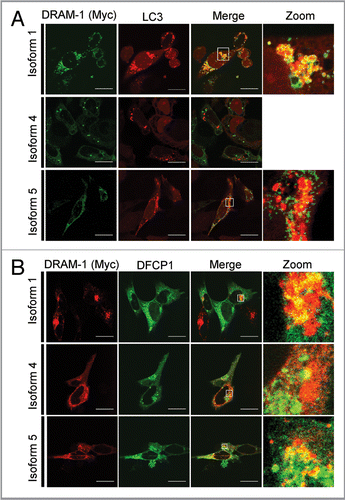
Figure 7 DRAM-1 isoforms 4 and 5 are partially localized in the endoplasmic reticulum. Saos-2 cells were infected with equivalent amounts of DRAM-1 adenoviral lysates for 48 h. Colocalization (yellow) of myc-tagged DRAM-1 (red) with the respective endoplasmic reticulum protein, Bip (A) and Calnexin (B) (green), was assayed by confocal microscopy. The scale bar shown represents 20 µm.
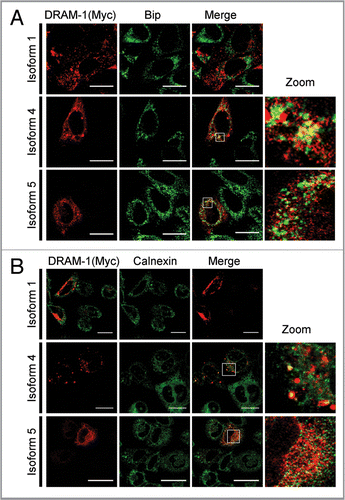
Figure 8 DRAM-1 isoform 4 is partially localized in peroxisomes. Saos-2 cells were infected with equivalent amounts of DRAM-1 adenoviral lysates for 48 h. Colocalization (yellow) of myc-tagged DRAM-1 (red) with the peroxisomal protein PMP70 (A) and peroxisome-GFP (B) (green), was assayed by confocal microscopy. The scale bar shown represents 20 µm.
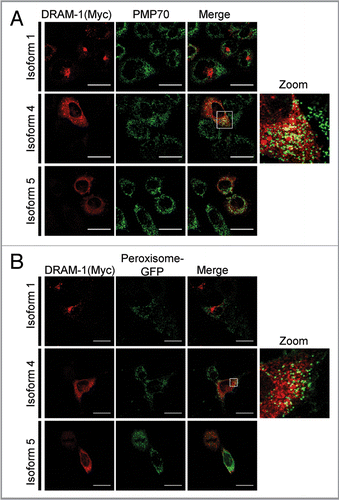
Figure 9 DRAM-1 isoform expression induces autophagosome formation. (A) Saos-2 cells were co-infected with GFP-LC3 and either DRAM-1 isoforms or empty vector for 48 h. Protein expression of DRAM-1 was validated by western blotting. (B) Representative images of cells overexpressing DRAM-1 isoforms displaying autophagosome formation. The scale bar shown represents 20 µm. (C) Quantitation of autophagosome formation. Cells with eight or more GFP-LC3 puncta were scored as positive for cells with accumulated autophagosomes. In each case at least 200 cells were counted and the error bar indicates standard deviation. (D) Saos-2 cells were infected with adenoviruses expressing either DRAM-1 isoforms 1, 4, 5 or ‘empty’ virus as control. Cell lysates were analyzed by western blotting for LC3, DRAM (myc) and actin. The data shown are representative of what was observed in at least three separate experiments.
Additional material
Download Zip (2 MB)Acknowledgements
We are grateful to Aviva Tolkovsky for kindly providing us with an adenovirus expressing GFP-LC3 and to Jim Norman, Beatson Institute and Nicholas Ktistakis, Babraham Institute for kindly provided mRFP-Rab7 and eGFP-DFCP1 respectively. We are also thankful to Tony McBryan for help with statistics and to members of the Tumour Cell Death Laboratory for critical reading of the manuscript. Work in the Tumour Cell Laboratory is supported by Cancer Research UK and the Association for International Cancer Research.
Related Research Data
References
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9:1102 - 1109; PMID: 17909521; http://dx.doi.org/10.1038/ncb1007-1102
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 1075; PMID: 18305538; http://dx.doi.org/10.1038/nature06639
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814 - 822; PMID: 20811353; http://dx.doi.org/10.1038/ncb0910-814
- Ax EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685 - 701; PMID: 18725538; http://dx.doi.org/10.1083/jcb.200803137
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010; 141:656 - 667; PMID: 20478256; http://dx.doi.org/10.1016/j.cell.2010.04.009
- Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol 2010; 12:831 - 835; PMID: 20811355; http://dx.doi.org/10.1038/ncb0910-831
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010; 12:747 - 757; PMID: 20639872; http://dx.doi.org/10.1038/ncb2078
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032 - 1036; PMID: 15525940; http://dx.doi.org/10.1038/nature03029
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005; 120:237 - 248; PMID: 15680329; http://dx.doi.org/10.1016/j.cell.2004.11.046
- Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol 2005; 6:439 - 448; PMID: 15928708; http://dx.doi.org/10.1038/nrm1660
- Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci USA 2010; 107:832 - 837; PMID: 20080761; http://dx.doi.org/10.1073/pnas.0913170107
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008; 454:232 - 235; PMID: 18454133; http://dx.doi.org/10.1038/nature07006
- Rosenfeldt MT, Ryan KM. The role of autophagy in tumour development and cancer therapy. Expert Rev Mol Med 2009; 11:e36; PMID: 19951459; http://dx.doi.org/10.1017/S1462399409001306
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 674; PMID: 21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer 2009; 9:691 - 700; PMID: 19759539; http://dx.doi.org/10.1038/nrc2715
- Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol 2010; 22:181 - 185; PMID: 20044243; http://dx.doi.org/10.1016/j.ceb.2009.12.001
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 2008; 10:676 - 687; PMID: 18454141; http://dx.doi.org/10.1038/ncb1730
- Crighton D, Ryan KM. Splicing DNA-damage responses to tumour cell death. Biochim Biophys Acta 2004; 1705:3 - 15; PMID: 15585169
- Kerley-Hamilton JS, Pike AM, Hutchinson JA, Freemantle SJ, Spinella MJ. The direct p53 target gene, FLJ11259/DRAM, is a member of a novel family of transmembrane proteins. Biochim Biophys Acta 2007; 1769:209 - 219; PMID: 17397945
- O'Prey J, Skommer J, Wilkinson S, Ryan KM. Analysis of DRAM-related proteins reveals evolutionarily conserved and divergent roles in the control of autophagy. Cell Cycle 2009; 8:2260 - 2265; PMID: 19556885; http://dx.doi.org/10.4161/cc.8.14.9050
- Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126:121 - 134; PMID: 16839881; http://dx.doi.org/10.1016/j.cell.2006.05.034
- Crighton D, Wilkinson S, Ryan KM. DRAM links autophagy to p53 and programmed cell death. Autophagy 2007; 3:72 - 74; PMID: 17102582
- Sharp PA. The discovery of split genes and RNA splicing. Trends Biochem Sci 2005; 30:279 - 281; PMID: 15950867; http://dx.doi.org/10.1016/j.tibs.2005.04.002
- Crighton D, O'Prey J, Bell HS, Ryan KM. p73 regulates DRAM-independent autophagy that does not contribute to programmed cell death. Cell Death Differ 2007; 14:1071 - 1079; PMID: 17304243; http://dx.doi.org/10.1038/sj.cdd.4402108
- Ryan KM, Ernst MK, Rice NR, Vousden KH. Role of NF-kappaB in p53-mediated programmed cell death. Nature 2000; 404:892 - 897; PMID: 10786798; http://dx.doi.org/10.1038/35009130
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844 - 848; PMID: 14704432; http://dx.doi.org/10.1126/science.1092472
- Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009; 16:1093 - 1107; PMID: 19373242; http://dx.doi.org/10.1038/cdd.2009.44
- Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 1993; 74:957 - 967; PMID: 8402885; http://dx.doi.org/10.1016/0092-8674(93)90719-7
- Helgason GV, O'Prey J, Ryan KM. Oncogene-induced sensitization to chemotherapy-induced death requires induction as well as deregulation of E2F1. Cancer Res 2010; 70:4074 - 4080; PMID: 20460519; http://dx.doi.org/10.1158/0008-5472.CAN-09-2876
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 5728; PMID: 11060023; http://dx.doi.org/10.1093/emboj/19.21.5720
- Bampton ETW, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells. Autophagy 2005; 1:23 - 36; PMID: 16874023;; http://dx.doi.org/10.4161/auto.1.1.1495
- Bell LA, O'Prey J, Ryan KM. DNA-binding independent cell death from a minimal proapoptotic region of E2F-1. Oncogene 2006; 25:5656 - 5663; PMID: 16652153; http://dx.doi.org/10.1038/sj.onc.1209580
- Ryan KM, O'Prey J, Vousden KH. Loss of nuclear factor-kappaB is tumor promoting but does not substitute for loss of p53. Cancer Res 2004; 64:4415 - 4418; PMID: 15231649; http://dx.doi.org/10.1158/0008-5472.CAN-04-1474
- Bell HS, Dufes C, O'Prey J, Crighton D, Bergamaschi D, Lu X, et al. A p53-derived apoptotic peptide derepresses p73 to cause tumor regression in vivo. J Clin Invest 2007; 117:1008 - 1018; PMID: 17347683; http://dx.doi.org/10.1172/JCI28920
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 1998; 95:2509 - 2514; PMID: 9482916; http://dx.doi.org/10.1073/pnas.95.5.2509
- Pellicciari C, Manfredi AA, Bottone MG, Schaack V, Barni S. A single-step staining procedure for the detection and sorting of unfixed apoptotic thymocytes. Eur J Histochem 1993; 37:381 - 390; PMID: 7510545
- Ridley SH, Ktistakis N, Davidson K, Anderson KE, Manifava M, Ellson CD, et al. FENS-1 and DFCP1 are FYVE domain-containing proteins with distinct functions in the endosomal and Golgi compartments. J Cell Sci 2001; 114:3991 - 4000; PMID: 11739631
- Wilkinson S, Croft DR, O'Prey J, Meedendorp A, O'Prey M, Dufes C, et al. The cyclin-dependent kinase PITSLRE/CDK11 is required for successful autophagy. Autophagy 2011; In press PMID: 21808150