Abstract
Cationic lipid DNA complex (CLDC) is an immunostimulatory preparation that has significant anti-leukemic effects in multiple murine models of leukemia: BCR-ABL+ myelogenous leukemia in C3H/HeJ animals, and myelomonocytic leukemia in BALB/c mice. Following leukemic challenge, CLDC treatment inhibits tumor cell growth in vivo and extends survival, sometimes resulting in apparent eradication of tumor cells. CLDC induces multiple cytokines including interferon-gamma (IFN-γ), and intravenous treatment results in a more rapid and robust response than subcutaneous treatment. IFN-γ is induced in a dose-dependent manner, and tachyphylaxis results from repeated doses of CLDC. Tachyphylaxis of therapeutic effects is exacerbated at higher doses, thus the optimal survival benefits are seen at intermediate doses. Animals whose leukemia has been successfully treated with CLDC exhibit a survival advantage when faced with a secondary leukemic challenge, suggesting the existence of an adaptive anti-leukemic response. This work demonstrates the effectiveness of CLDC in multiple experimental leukemias and is consistent with a stimulation of a lasting TH1 anti-leukemic immune response.
Introduction
CLDC has been shown to be a potent activator of innate immunity and to induce natural killer (NK) cells, IFNγ, IL-12 and other cytokines. This immune activation is dependent upon CpG motifs present in the non-coding plasmid DNA, as non-CpG plasmids have been shown in other models to fail to elicit a protective immune response.Citation1 The CpG plasmid stimulates immune activation via the Toll-like Receptor 9 (TLR-9), but evidence suggests the involvement of unknown pathway(s) independent of TLR-9.Citation2–Citation4 TLR-9 cues an immune response via myeloid differentiation factor 88 (MyD88),Citation5 mimicking the body's response to bacterial DNA. Either component alone (cationic lipid or CpG plasmid DNA) is incapable of invoking this immune response, as the lipids likely provide protection from degradation as well as a delivery vehicle enabling cellular entry of the plasmid DNA, but in themselves have no therapeutic effects.Citation1,Citation6 The cationic liposomal components may also direct the plasmid DNA subcellularly, favoring endosomal retention over lysosomal degradation.Citation7 When combined with soluble protein or peptide antigens, CLDC has been shown to induce cross-priming and a potent T-cell response, potentially contributing to the control of a neoplastic condition.Citation5
Acute leukemias have long been recognized as suitable targets for immunotherapy.Citation8 Allogeneic blood and marrow transplantation is an active form of such treatment frequently used in younger patients.Citation9,Citation10 Graft versus leukemia (GVL) effects have been documented in animal and human settings to be mediated by both antigen specific cytotoxic lymphocytesCitation11,Citation12 and by NK cells.Citation13 As might be expected, these activities operate most efficiently when the burden of residual leukemia in patients is small, as in patients who are in complete remission but are expected to experience disease relapse. More recently, non-myeloablative conditioning regimens have enabled investigators to extend GVL effects to older patients with positive results.Citation14,Citation15 Autologous immunotherapy has also been studied by a number of investigators and a significant body of experimental data suggests that in multiple animal models of acute myelogenous leukemia, the autologous immune system can control the leukemia effectively. Several investigators have documented the ability of HLA restricted and antigen specific cytotoxic lymphocytes to contribute to this control.Citation16,Citation17 Chaudhury et al. clearly showed cytokine induced differentiation of chronic myelogenous and acute myelogenous leukemia cells into dendritic antigen presenting cells (APCs) that could induce leukemia-specific cytotoxicity in autologous lymphocytes.Citation18–Citation20 Other investigators have demonstrated the potential of antigen-specific immunotherapy in leukemia. Molldrem et al. have shown that chronic myelogenous leukemia specific cytotoxicity may be mediated by cytotoxic lymphocytes specific for the PR-1 antigen.Citation21–Citation24 His group has also shown anti PR-1 cytotoxicity induced by vaccination may yield anti-leukemic responses in a group of myeloid leukemia patients.Citation25 There is also evidence that the WT1 protein, known to contribute to leukemogenesis, may be immunogenic both in mice and humans.Citation26,Citation27
Previous studies have demonstrated that CLDC is effective in the treatment of mouse solid tumorsCitation6 and the treatment of canine patients with spontaneous malignancies.Citation28 The current studies were designed to establish the efficacy of CLDC immunotherapy in multiple murine models of leukemia and to gauge the contribution of the adaptive response in a situation where a specific antigen is not identified or administered therapeutically. A further goal of these studies is to evaluate the dose-response relationship of CLDC, so as to investigate appropriate dosing schedules to avoid tachyphylaxis and maximize benefit in clinical trials.
Results
Leukemogenic cell lines result in predictable leukemic death.
Leukemic challenge of C3H/HeJ mice with BCR-ABL transfected 32D cells (32D/BCR-ABL) regularly produced death of animals between days 26 and 34 and the 32D/BA-GFP cells (an independently derived BCR-ABL clone carrying a fluorescent marker) yielded deaths somewhat earlier (day 18–26), as has been reported previously.Citation30 In both of these clones, animals either ill with leukemia or succumbing to leukemia showed considerable splenomegaly and histological infiltration with blastic immature progenitor cells (data not shown). The 32D/BA-GFP cells yielded circulating GFP-expressing cells detectable by flow cytometry (described below). WEHI-3B cells were similarly lethal, yielding splenic enlargement and leukemic deaths in challenged syngenic BALB/c mice between days 21 and 41, consistent with previous reports.Citation31
CLDC causes innate immune activation.
CLDC was administered subcutaneously (SC) or intravenously (IV) with no evidence of toxicity. Histologically, IV administration of CLDC was associated with accumulation of numerous pale-staining macrophages within the splenic marginal zones, consistent with innate immune activation () as identified by previous authors.Citation6
In an effort to maximize the immune response following administration of CLDC, we compared innate immune activation using CLDC given by SC and IV administration. An IV dose of only 5 µg of CLDC results in rapid and robust response, with IFNγ levels peaking at 2,600 pg/ml at 6 hours following administration ( and ). The systemic IFNγ response following SC administration of 20 µg CLDC peaks at 18 hours at a level of 70 pg/ml (). Substantially elevated levels of other cytokines such as IL-12, IFNα and CCL-2 were also detected following IV administration of CLDC, but not with SC administration of CLDC (). IV administration of CLDC at lower doses results in lower levels of systemic cytokines as expected, with at least a 100-fold range of response (0.05 µg–5 µg CLDC, data not shown). The cytokine release profile of IV administration as well as previous GLP toxicology in non-human primatesCitation32 and prior human experience with CLDC given by IV administration (unpublished data) suggested further evaluation of IV dosing of CLDC.
To examine the effect of multiple CLDC doses on cytokine response, the time between repeated doses was altered. IFNγ levels were measured following a second CLDC dose that came 1, 3, 7, 10 or 14 days after the initial dose in unchallenged mice (). When a second dose of CLDC is administered only one day after the first, no IFNγ induction is apparent indicating that CLDC induces a refractory period. The IFNγ response returns as the time between multiple doses is lengthened, and by 14 days, the response has returned to the levels induced in a naïve mouse. These data indicate that timing of multiple CLDC doses is critical to achieving optimal IFNγ response.
Doses greater than 5 µg were also tested by IV injection. Because IFNγ levels peak at 6 hours following IV CLDC, a 6-hour time point was used for all subsequent measurements. C3H/HeJ mice were challenged with 32D/BA-GFP and weekly CLDC treatments were started on day 3. Following the initial dose of 10, 20 or 30 µg IV CLDC, serum IFNγ levels increased in a dose-dependent manner (). However, upon repeated weekly treatments, a trend of tachyphylaxis became apparent, with higher doses exhibiting a more dramatic decrease in IFNγ ().
IV treatment with CLD C is associated with a dose-dependent survival benefit.
To test the efficacy of CLDC via IV administration and to determine a dose response relationship with efficacy, 32D/BA-GFP was tested in a CLDC dose titration experiment. Mice were treated with CLDC on days 2 and 9 following 32D/BA-GFP challenge and then monitored for survival. This treatment conferred a significant survival benefit in a dose-dependent manner ().
Larger IV doses (10, 20 and 30 µg) were also tested in the 32D/BA-GFP model, and GFP tumor burden was followed closely to assess the kinetics of tumor cell growth in each treatment group. In the absence of treatment, circulating GFP-positive cells were measurable by flow cytometry as early as 11 days after challenge ( and B). This population expanded aggressively, resulting in death in 24–26 days (). Weekly IV treatments with CLDC beginning on day 3 significantly inhibited growth of the GFP-positive population at all doses tested ( and B). The 10 µg dose resulted in the best control of tumor burden and the greatest number of long-term surviving (apparently cured) animals (). Treatment with 20 µg CLDC significantly improved survival over control mice, but was not as beneficial as 10 µg. The 30 µg dose yielded mixed results, with some animals dying before or concurrently with the untreated controls, and others surviving until >80 days. This dose-response relationship indicates a threshold effect, where an intermediate dose is more beneficial than higher or lower doses.
The IV dose-response relationship was also examined in the myelomonocytic leukemia induced by WEHI-3B in BALB/c mice. In this model, weekly administration of 20 µg IV CLDC is more effective than 10 µg (), indicating that the survival benefit of CLDC is not specific to the C3H/HeJ leukemia model, and that dose must be determined empirically for individual cancer types and animal strains.
Successful treatment with CLD C extends survival following secondary challenge.
We then addressed the question of immunological memory after CLDC therapy of established leukemia. This work exploited two versions of BCR-ABL-transformed 32D cells: non-fluorescent 32D/BCR-ABL (challenge 1) and fluorescent 32D/BA-GFP (challenge 2). Animals that received challenge 1 without treatment died within 34 days (). Animals in Group 1 receiving challenge 2 without treatment died on day 16–19 (). CLDC treatment did not affect survival when given well in advance of challenge 2, as is seen by the death of Group 2 animals on days 14–19, similar to Group 1. Group 3 animals received challenge 1 on day-60 with 32D/BCR-ABL and were successfully treated with CLDC given on day 46 and 32. These animals then received challenge 2 on day 0 and survived significantly longer than groups 1 and 2 (p < 0.01).
Discussion
CLDC has been shown to be a systemic immunostimulant, activating effector cells of the innate and adaptive immune systems.Citation6 This formulation has shown therapeutic activity for experimental solid tumors.Citation6 Available data suggests that this activity stems in part, but likely not fully, from stimulation through TLR-9. Our results presented here clearly show significant in vivo activity for this material in therapy of three murine models of acute leukemia. Both subcutaneous and intravenous routes of administration are active, and available evidence suggests that some animals have been cured of this leukemia via CLDC therapy. Given the effectiveness, reproducibility and therapeutic range of the IV route, it may prove optimal for future study.
In this report we show the dose dependency of CLDC treatment of leukemic neoplasms. Among CLDC doses tested, higher doses yielded higher IFNγ levels but showed tachyphylaxis of both IFNγ levels and anti-leukemic effect. The optimal dose also seems to vary from one leukemia model to another ( and ). In the C3H/HeJ model, the 10 µg dose provided the best tumor burden control and survival benefit while in the WEHI-3b model, 20 µg was best. This difference may result from differences in genetic background in the two mouse strains. Alternatively, the type of leukemia induced by the myeloid 32D/BCR-ABL cells and the myelomonocytic WEHI-3B cells may respond differently to CLDC dosing. It is clear that clinical application of this therapeutic preparation will require careful attention to dosing, as genetic and leukemic differences will also be present in human patient populations.
Timing of treatment administration also appears to be critical for optimization of therapeutic benefit. IFNγ measurements on mice given repeated CLDC treatments suggest the induction of a refractory period following treatment. Together, these data suggest that optimization of therapeutic effects may be expected to require some care and attention to the amount and timing of CLDC doses. It is notable however that there is no identified correlation between optimal IFNγ increase and therapeutic effect.
Previous data in other experimental systems shows involvement of CTLs and NK cells in biological responses to CLDC,Citation5 but important questions remain regarding the mechanism(s) of the CLDC anti-leukemic therapeutic effect. In particular, the possible role of adaptive, antigen-specific immune response merits investigation.
Work involving multiple subsequent leukemic challenges is useful in probing the possibility of immunological memory. In , mice that were treated successfully following an initial challenge with 32D/BCR-ABL fared better when faced with an additional challenge with 32D/BA-GFP than mice being challenged for the first time with 32D/BA-GFP. This suggests a lasting anti-leukemic benefit results from successful treatment with CLDC. Immunological memory provides a logical possible explanation for this lasting anti-leukemic effect. In this experiment, successful therapy of 32D/BCR-ABL cells contributed to in vivo inhibition of growth of 32D/BA-GFP. This is consistent with persistence of cellular effectors with cross reactivity for both clones. These results do not however exclude the possibility of participation of humoral immunity or macrophage mediated cytotoxicity in the therapy. Furthermore, the relative contributions of NK cells including NKT cells and antigen specific CTLs remain unclear. This latter issue is of particular importance because of the usefulness of long term immunity associated with the development of antigen specific responses.
Anti-leukemic immunotherapy utilizing CLDC is demonstrated here in multiple transplantable murine models. CLDC induces a robust IFNγ response and results in control of tumor burden and extension of survival. The treatment is highly effective, reproducible and provides a lasting survival benefit in the face of secondary leukemic challenge. The application of these observations to the therapy of human neoplasms is clearly of interest.
Materials and Methods
Leukemic clones.
Three mouse models were studied. 32D cells transformed with BCR-ABL (32D/BCR-ABL) and congenic with C3H/HeJ mice were obtained from Dr. S.S. Clark, University of Wisconsin as a kind gift.Citation29 A similar clone of 32D derived cells independently transformed with BCR-ABL but also transfected to stably express the green fluorescent protein molecule (32D/BA-GFP) was obtained as a kind gift from Dr. Ling.Citation30 WEHI-3B cells (congenic with BALB/c mice) were obtained from ATCC. Animals received leukemia infusions via IV tail vein injection. Injection of 1 × 106 cells yielded reliable leukemic death and thus was administered for all subsequent experiments.
Mice.
Male C3H/HeJ and BALB/c mice were obtained from Charles River Laboratories at 7–8 weeks of age. Animals were kept in conventional animal facilities using minimum conventional feed and water and were monitored for illness and survival. Using the models described here, lethargy and inability to feed preceded death by less than 24 hours. Thus, the primary experimental endpoint was death or near moribund status requiring sacrifice. Some animals were subjected to necropsy after sacrifice or spontaneous death. Tissues were processed by routine technique after formalin fixation and hematoxylin and eosin staining of sections. All procedures were reviewed and approved by the Penn State Institutional Animal Care and Use Committee.
Analysis of peripheral blood.
To analyze circulating tumor burden in some experiments, blood was subjected to flow cytometric analysis for quantification of 32D/BA-GFP cells. Blood (15 µL) was drawn via tail snip and red blood cells were lysed in 200 µL RBC lysis solution (5 Prime, Gaithersburg, MD). White blood cells were pelleted by centrifugation for 2 min in an Eppendorf 5415 C centrifuge at 300 × g, washed in 1 ml phosphate-buffered saline (PBS) and resuspended in 400 µL PBS. Analysis was performed on a FACSCalibur instrument. List-mode multiparameter data files (each file with forward scatter, side scatter and 4 fluorescent parameters) were analyzed using CellQuest Pro Software. Files were gated to exclude red cell debris. A minimum of 20,000 gated events was acquired.
For IFNγ analysis, mice were treated as indicated with CLDC and serum was collected after the specified timepoint. Cytokine levels were assayed using ELISA reagents per manufacturer's instructions (eBioscience, San Diego, CA).
CLDC treatment.
CLDC was prepared from liposomes of the cationic lipid DOTIM, the neutral lipid cholesterol and non-coding bacterial plasmid DNA. The pMB75.6 plasmid was produced from non-pathogenic E. coli DH5α cells and is 4,242 base-pairs in length. Concentrations of CLDC components were verified by HPLC analysis and spectrophotometry. The DNA was found to be 100% circular plasmid DNA and the product was manufactured and tested under Good Manufacturing Practice regulations and found to be sterile and free from endotoxin and particulate matter. Treated animals received CLDC either SC (subcutaneous base of tail injection) or IV (intravenous tail vein injection). CLDC dosage was defined by the content of DNA in the delivered dose. SC administration volume was adjusted to 200 µL while IV administration volume was 100 µL.
Statistical analysis.
Animal survival was documented by the method of Kaplan and Meier and statistical significance of survival data was analyzed by the Mantel-Cox log-rank test using GraphPad Software version 5.00 for Windows, San Diego California USA, www.graphpad.com. This software was also used to plot and analyze data using the Student's t-test.
Financial Disclosure
Supported by NCI SBIR R41 CA114803-01 A1, The Penn State University Kiesendahl Family Endowed Leukemia Research Fund and The Kenneth Noel Memorial Fund.
Abbreviations
| APC | = | antigen presenting cell |
| CLDC | = | cationic lipid DNA complex |
| GVL | = | graft versus leukemia |
| GFP | = | green fluorescent protein |
| IFNγ | = | ingerferon-γ |
| IV | = | intravenous |
| NK | = | natural killer |
| PBS | = | phosphate-buffered saline |
| SC | = | subcutaneous |
| TLR-9 | = | toll-like receptor 9 |
Figures and Tables
Figure 1 Single IV doses of CLDC were associated with accumulation within the splenic marginal zones of numerous pale-staining macrophages, consistent with innate immune activation (7 days post IV administration).
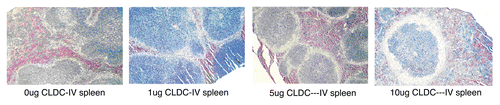
Figure 2 IFNγ is induced following IV CLDC treatment. Unchallenged BALB/c mice (n = 5 per group) were treated with 5 µg CLDC by IV injection (A) and serum was collected at indicated time points for ELISA analysis. Unchallenged BALB/c mice (n = 5 per group) were given 5 µg CLDC by IV injection (B). The IV dose was repeated 1, 3, 7, 10 or 14 days later and IFNγ levels were measured from serum samples taken 6 hours later.
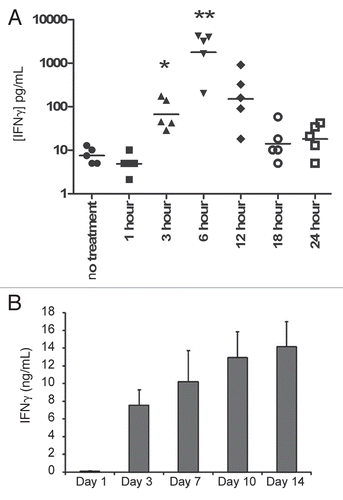
Figure 3 CLDC administration results in a dose-dependent induction of IFNγ that exhibits tachyphylaxis. (A) IFNγ levels of 32D/BA-GFP-challenged C3H/HeJ mice display a dose-dependent induction. Serum samples were drawn six hours after the indicated IV dose of CLDC. Individual measurements are displayed for each mouse following the day 3 treatment. p < 0.01 for 0 µg vs. 10 µg, 0 µg vs. 30 µg and 10 µg vs. 30 µg. p < 0.05 for 0 µg vs. 20 µg and 10 µg vs. 20 µg. 20 µg vs. 30 µg not significantly different. (B) Average and standard deviation values from treatments on days 3, 10, 18 and 24 are graphed, revealing a decrease in IFNγ levels following repeated CLDC treatment. Significant differences are noted: 10 µg Day 3 vs. 18: p < 0.01. 20 µg Day 3 vs. 18, 10 vs. 18, 3 vs. 24 and 10 vs. 24, p < 0.01. 30 µg Day 3 vs. 10 and 3 vs. 24: p < 0.05, Day 3 vs. 18 p < 0.001.
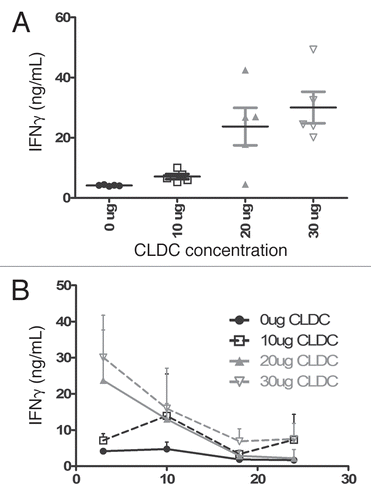
Figure 4 CLDC therapy shows dose dependent anti-leukemic effects. Animals received IV CLDC at doses of 0.1 µg, 1 µg or 5 µg on days 2 and 9 following 32D/BA-GFPchallenge. p < 0.05 for control vs. 0.1 µg and 1 µg vs. 5 µg. p < 0.01 for all other pair-wise comparisons.
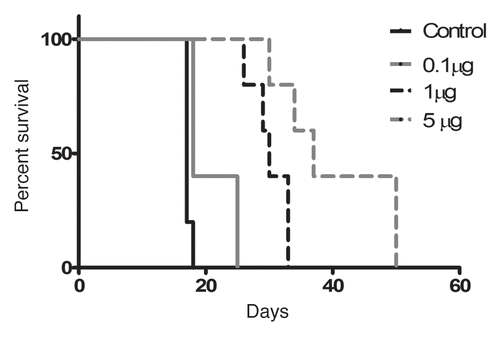
Figure 5 CLDC treatment inhibits tumor burden growth and extends survival. Mice were challenged with 32D/BA-GFP and treated with weekly IV injections of 0, 10, 20 or 30 µg CLDC from days 3 through 24 after challenge. Mice were periodically monitored for circulating tumor burden by flow cytometry. (A) Flow cytometric analysis of a representative mouse is shown from each treatment group. GFP-positive cells are displayed in green, with % GFP-positive cells noted for each plot. (B) Percent GFP-positive cells in individual mouse is graphed versus time (n = 5 per group). (C) Survival of CLDC-treated mice depends on IV dose administered; p < 0.01 for control vs. 10 µg, control vs. 20 µg and 10 µg vs. 20 µg.
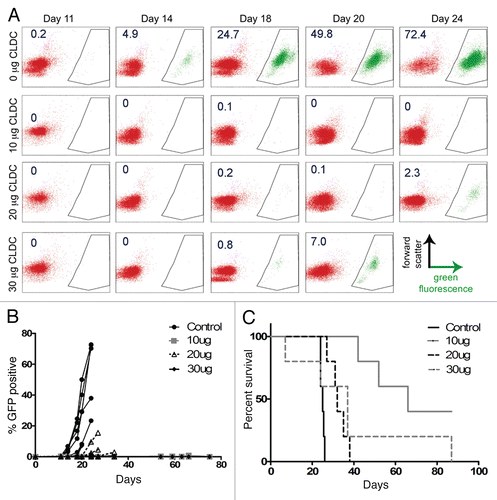
Figure 6 CLDC extends survival in BALB/c mice in a dose-dependent manner. WEHI-3B-challenged BALB/c mice were treated on days 3, 10, 17 and 24 with IV injections of 0, 10 or 20 µg CLDC and followed for survival. Control vs. 20 µg and 10 µg vs. 20 µg, p < 0.01.
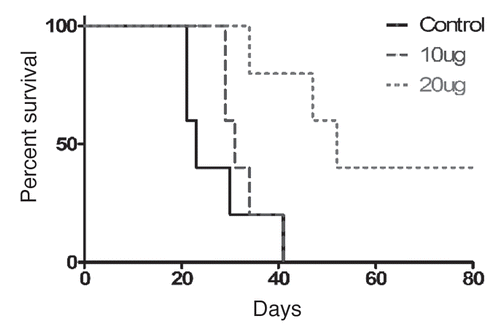
Figure 7 Successful treatment with CLDC provides benefit during secondary challenge. Mice challenged with 32D/BCR-ABL die within 34 days if left untreated (upper right corner insert, n = 10). Group 1 mice received 32D/BA-GFP challenge on Day 0 and died by day 19 (n = 5 per group). Group 2 mice were treated with 10 µg IV CLDC on Day-46 and Day-32 relative to 32D/BA-GFP and also died by day 19. Group 3 mice received an initial challenge with 32D/BCR-ABL on day-60, were treated with CLDC as in Group 2 and survived significantly longer in response to 32D/BA-GFP challenge (10–17 day increase in survival, p < 0.01).
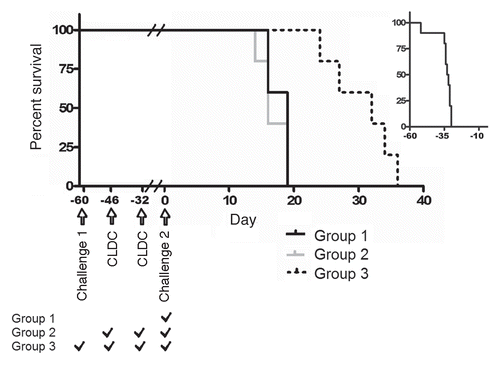
Table 1 Cytokine levels following IV or SC CLDC treatment
Acknowledgements
We thank Nate Sheaffer and David Stanford of the Flow Cytometry Core Facility at Penn State Hershey Cancer Institute/Milton S. Hershey Medical Center for their help with acquisition and analysis of flow cytometry data. We thank the faculty and staff of the Penn State Comparative Medicine Department at Penn State College of Medicine for their care of animals used in this work. This work was funded in part by The Penn State University Kiesendahl Family Endowed Leukemia Research Fund and The Kenneth Noel Memorial Fund.
References
- Gowen BB, Fairman J, Dow S, Troyer R, Wong MH, Jung KH, et al. Prophylaxis with cationic liposome-DNA complexes protects hamsters from phleboviral disease: importance of liposomal delivery and CpG motifs. Antiviral Res 2009; 81:37 - 46
- Yasuda K, Ogawa Y, Kishimoto M, Takagi T, Hashida M, Takakura Y. Plasmid DNA activates murine macrophages to induce inflammatory cytokines in a CpG motif-independent manner by complex formation with cationic liposomes, Biochem. Biochem Biophys Res Commun 2002; 293:344 - 348
- Yasuda K, Ogawa Y, Yamane I, Nishikawa M, Takakura Y. Macrophage activation by a DNA/cationic liposome complex requires endosomal acidification and TLR9-dependent and -independent pathways. J Leukoc Biol 2005; 77:71 - 79
- Yasuda K, Yu P, Kirschning CJ, Schlatter B, Schmitz F, Heit A, et al. Endosomal translocation of vertebrate DNA activates dendritic cells via TLR9-dependent and -independent pathways. J Immunol 2005; 174:6129 - 6136
- Zaks K, Jordan M, Guth A, Sellins K, Kedl R, Izzo A, et al. Efficient immunization and cross-priming by vaccine adjuvants containing TLR3 or TLR9 agonists complexed to cationic liposomes. J Immunol 2006; 176:7335 - 7345
- Dow SW, Fradkin LG, Liggitt DH, Willson AP, Heath TD, Potter TA. Lipid-DNA complexes induce potent activation of innate immune responses and antitumor activity when administered intravenously. J Immunol 1999; 163:1552 - 1561
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005; 434:772 - 777
- Charak BS, Brynes RK, Chagyoli M, Kortes V, Tefft M, Mazumder A. Graft versus leukemia effect after transplantation with IL-2 activated bone marrow. Correleation with erradication of residual disease. Transplantation 1993; 56:31
- Butturini A, Gale RP. Graft versus leukemia in humans. Cancer Treat Res 1995; 76:299 - 314
- Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002; 295:2097 - 2100
- Mutis T, Verdijk R, Schrama E, Esendam B, Brand A, Goulmy E. Feasibility of immunotherapy of relapsed leukemia with ex vivo-generated cytotoxic T lymphocytes specific for hematopoietic system-restricted minor histocompatibility antigens. Blood 1999; 93:2336 - 2341
- Rufer N, Wolpert E, Helg C, Tiercy JM, Gratwohl A, Chapuis B, et al. HA-1 and the SMCY-derived peptide FIDSYICQV (H-Y) are immunodominant minor histocompatibility antigens after bone marrow transplantation. Transplantation 1998; 66:910 - 916
- Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002; 295:2097 - 2100
- Storb RF, Champlin R, Riddell SR, Murata M, Bryant S, Warren EH. Non-myeloablative transplants for malignant disease. Hematology 2001; 375 - 391
- Claxton DF, Ehmann C, Rybka W, Claxton DF, Ehmann C, Rybka W. Control of advanced and refractory acute myelogenous leukaemia with sirolimus-based non-myeloablative allogeneic stem cell transplantation. Br J Haematol 2005; 130:256 - 264
- Mutis T, Verdijk R, Schrama E, Esendam B, Brand A, Goulmy E. Feasibility of immunotherapy of relapsed leukemia with ex vivo-generated cytotoxic T lymphocytes specific for hematopoietic system-restricted minor histocompatibility antigens. Blood 1999; 93:2336 - 2341
- Rufer N, Wolpert E, Helg C, Tiercy JM, Gratwohl A, Chapuis B, et al. HA-1 and the SMCY-derived peptide FIDSYICQV (H-Y) are immunodominant minor histocompatibility antigens after bone marrow transplantation. Transplantation 1998; 66:910 - 916
- Choudhury A, Toubert A, Sutaria S, Charron D, Champlin RE, Claxton DF. Human leukemia-derived dendritic cells- ex vivo development of specific antileukemic cytotoxicity. Crit Rev Immunol 1998; 18:121 - 131
- Choudhury A, Gajewski JL, Liang JC, Popat U, Claxton DF, Kliche KO, et al. Use of leukemic dendritic cells for the generation of antileukemic cellular cytotoxicity against Philadelphia chromosome-positive chronic myelogenous leukemia. Blood 1997; 89:1133 - 1142
- Choudhury A, Liang JC, Thomas EK, Flores-Romo L, Xie QS, Agusala K, et al. Dendritic cells derived in vitro from acute myelogenous leukemia cells stimulate autologous, antileukemic T-cell responses. Blood 1999; 93:780 - 786
- Molldrem J, Dermime S, Parker K, Jiang YZ, Mavroudis D, Hensel N, et al. Targeted T-cell therapy for human leukemia: cytotoxic T lymphocytes specific for a peptide derived from proteinase 3 preferentially lyse human myeloid leukemia cells. Blood 1996; 88:2450 - 2457
- Molldrem JJ, Clave E, Jiang YZ, Mavroudis D, Raptis A, Hensel N, et al. Cytotoxic T lymphocytes specific for a nonpolymorphic proteinase 3 peptide preferentially inhibit chronic myeloid leukemia colony-forming units. Blood 1997; 90:2529 - 2534
- Molldrem JJ, Lee PP, Wang C, Felio K, Kantarjian HM, Champlin RE, Davis MM. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat Med 2000; 6:1018 - 1023
- Molldrem JJ, Lee PP, Wang C, Champlin RE, Davis MM. A PR1-human leukocyte antigen-A2 tetramer can be used to isolate low-frequency cytotoxic T lymphocytes from healthy donors that selectively lyse chronic myelogenous leukemia. Can Res 1999; 59:2675 - 2681
- Qazilbash MH, Wieder ED, Thall PF, Wang X, Rios RL, Lu S, et al. PR1 Peptide Vaccine-Induced Immune Response Is Associated with Better Event-Free Survival in Patients with Myeloid Leukemia. ASH Annual Meeting Abstracts 2007; 110:283
- Gaiger A, Reese V, Disis ML, Cheever MA, Gaiger A, Reese V, et al. Immunity to WT1 in the animal model and in patients with acute myeloid leukemia. Blood 2000; 96:1480 - 1489
- Asemissen AM, Keilholz U, Tenzer S, Muller M, Walter S, Stevanovic S, et al. Identification of a highly immunogenic HLA-A*01-binding T cell epitope of WT1. Clin Cancer Res 2006; 12:7476 - 7482
- Kamstock D, Guth A, Elmslie R, Kurzman I, Liggitt D, Coro L, et al. Liposome-DNA complexes infused intravenously inhibit tumor angiogenesis and elicit antitumor activity in dogs with soft tissue sarcoma. Cancer Gene Ther 2006; 13:306 - 317
- Sahin MB, Perman SM, Jenkins G, Clark SS. Perillyl alcohol selectively induces G0/G1 arrest and apoptosis in Bcr/Abl-transformed myeloid cell lines. Leukemia 1999; 13:1581 - 1591
- Ling X, Wang Y, Dietrich MF, reeff M, Arlinghaus RB. Vaccination with leukemia cells expressing cell-surface-associated GM-CSF blocks leukemia induction in immunocompetent mice. Oncogene 2006; 25:4483 - 4490
- Murphy WH, Urnovitz HB, Maryanski JL, Abrams GD. Characterization of transplantable myelomonocytic leukemia WEHI-3B in syngeneic BALB/c mice. Proc Soc Exp Biol Med 1978; 157:556 - 564
- Fairman J, Moore J, Lemieux M, Van RK, Geng Y, Warner J, Abel K. Enhanced in vivo immunogenicity of SIV vaccine candidates with cationic liposome-DNA complexes in a rhesus macaque pilot study. Hum Vaccin 2008; 5