Abstract
Head and neck squamous cell carcinoma (HNSCC) is a biologically aggressive disease that has been modestly impacted by improvements in therapeutic strategies. Several lines of evidence support the role of TrkB for invasion and metastasis in various solid tumor models, and we have shown an important function of this receptor in HNSCC tumor biology. Therapeutic modulation of TrkB function has been supported in the literature by the development of small molecule inhibitors (SMI) with minimal success. To assess the validity of targeting TrkB in HNSCC, we tested a novel agent, AZ64, and show significant dose and time-dependent inhibition of cellular proliferation in cell lines. Genetic studies revealed the specificity of this compound for the TrkB receptor, as exposure of cells that had genetic suppression of TrkB did not demonstrate abrogated oncogenic signaling. We next assessed the impact of AZ64 as a chemotherapy-sensitizer, and identified an enhancement of cisplatin-mediated anti-proliferation across all cell lines. We then demonstrated that AZ64 can overcome chemotherapy resistance in a novel model of cisplatin resistance in HNSCC. Modulation of the pro-oncogenic STAT3 and Src pathways was identified, suggesting molecular mechanisms of action for AZ64. In this study, we demonstrate the feasibility of targeting TrkB, and suggest a novel approach for the treatment of some chemotherapy-resistant HNSCC.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is a biologically aggressive disease, with an annual incidence of approximately 42,000 cases in the United States and over 650,000 worldwide.Citation1 While strides have been made in surgical techniques, refinement of radiation delivery and intensification of local-regional treatment with chemotherapeutic strategies, a significant number of patients succumb to distant metastasis and local-regional failure. The rationale that molecularly-targeted therapy in the treatment of HNSCC is based upon the observation that specific molecular growth and survival signaling pathways are amplified in HNSCC and EGFR-targeting, either with antibody- or small molecule inhibitor-based approaches, has been the most well studied in this disease.Citation2,Citation3 However, the incremental improvements in patient outcomes with EGFR-directed therapy have been marginal.Citation4 While there are abundant positive pre-clinical data supporting the use of EGFR as a therapeutic target, it is clear that other molecular mediators of HNSCC tumor progression should also be evaluated as targets for this disease.
The tropomyosin-related kinase B receptor (TrkB) is a 145 kD member of the neurotrophin (NT) receptor family that is activated by brain-derived neurotrophic factor (BDNF) and, to a lesser degree, neurotrophic factor 4 (NT4). Several lines of evidence support the role of TrkB for invasion and metastasis in various solid tumor models.Citation5–Citation7 Our laboratory has established the critical role of this receptor in human HNSCC tumors and as a mediator of epithelial-mesenchymal transition (EMT) in this disease.Citation8 As a receptor tyrosine kinase (RTK), TrkB activates diverse downstream signaling cascades that ultimately induce cellular proliferation and pro-survival mechanisms, through AKT, STAT3 and MAPK pathways.Citation9 Thus, similar to other RTKs, TrkB is a potentially important target for novel treatment approaches for HNSCC.
Therapeutic modulation of TrkB function has been supported in the literature by the development of agents that have putative activity against TrkB.Citation10 The growth-inhibitory and apoptosis-inducing effects of compounds that target TrkB have not been well-described for HNSCC, raising the issue as to whether targeting TrkB is a viable approach for this disease. We have recently demonstrated the importance of a BDNF-TrkB axis as a mediator of aggressive biological behavior in HNSCC.Citation8 While standard systemic therapy regimens for the treatment of HNSCC are based upon high-dose cisplatin (CDDP), many tumors demonstrate de novo or acquired resistance cisplatin,Citation11 which raises the question as to whether TrkB-targeted therapy might sensitize tumors to CDDP. To address these questions, a novel inhibitor against TrkB was used to determine its efficacy alone and in combination with CDDP in pre-clinical models of HNSCC. In this study, we demonstrate the feasibility of targeting TrkB and suggest a novel approach for the treatment of some chemotherapyresistant HNSCC.
Results
Trk receptor specificity of AZ64.
A small molecule inhibitor of the Trk-receptor family was designed and assayed for its ATP-competitive effects.Citation14 The Ki for TrkA was determined using an enzyme-based kinase assay and was found to be 2.0 ± 0.25 nM. Based upon these initial findings, AZ-64 selectivity was further evaluated against other known tyrosine kinases and showed a high degree of specificity to Trk at a 500 nm concentration across a large part of 177 diverse kinases, including those related to the Trk family. There were 11 enzymes that showed some level of inhibition at 500 nm, which were subsequently evaluated to more definitively assess selectivity versus TrkA and TrkB (). AZ-64 showed an IC50 value of 0.2 nM against TrkA and 2 nM against TrkB. These results suggested that AZ-64 is a potent and selective Trk kinase inhibitor, with enzyme IC50 values in the range of 0.2–2 nm.
We next characterized the phospho-specificity of AZ64 against TrkA and TrkB kinases in cell-based assays to determine whether AZ-64 had similar activity against different Trk isoforms. Stably transfected NIH-3T3 cells overexpressing full-length forms of human TrkA and TrkB were evaluated for pTrk inhibition following stimulation with NGF or BDNF, respectively (). Significant and potent suppression of TrkA and TrkB receptor phosphorylation between 5–50 nM AZ64 (p < 0.05) confirmed the in vitro kinase results and further suggested the potential impact of this compound in tumor models of HNSCC (). To assess whether the TrkB signaling axis was altered in response to AZ64, HEK-293 cells harboring a stable TrkB construct were exposed to AZ64 and downstream mediators of TrkB activity were monitored (Suppl. Fig. 1). In a dose-dependent fashion, AZ64 was found to suppress TrkB and STAT3 activation, suggesting that cell-based Trk receptor activity was specifically altered by the inhibitor.
TrkB is frequently expressed in HNSCC tumors.
The expression of TrkB was determined in a part of HNSCC tumor specimens with SDS-PAGE analysis of whole-tumor lysates and by immunohistochemistry (). TrkB receptor was identified in approximately 30% of tumor samples (n = 20), but was absent in normal mucosa (n = 3), suggesting its importance in tumorigenesis in HNSCC. Independent evaluation of TrkB expression in orthotopically-implanted HNSCC tumors also confirmed these results (n = 10, ). Whole-cell lysates of normal murine tongues had undetectable levels of TrkB, whereas orthotopic tumors had elevated levels of receptor expression. These data confirmed the relevance of TrkB in the biology of HNSCC and suggested a rationale for targeting TrkB in this disease.
In vitro anti-proliferative response of HNSCC to AZ64.
To determine whether AZ64 was effective in HNSCC, we assayed HNSCC cell lines for the anti-proliferative response to AZ64. In vitro studies demonstrated that the inhibitor had significant activity against a part of cancer cell lines, with IC50 ranging from 0.6 to 5.3 µM (). The relatively consistent growth-suppressive characteristics of AZ64 within a narrow dose range suggested that HNSCC cell lines were uniformly responsive to AZ64 and prompted further analysis. Five HNSCC cell lines (FADU, UMSCC22A, HN5, HN30, UMSCC10B) were then selected for further analysis to determine the temporal effects of AZ64. As shown in and C, AZ64 significantly suppressed cellular proliferation among all cell lines in a time-dependent and dose-dependent manner. These effects were seen at nearly all doses within 48 or 72 hours, consistent with the IC50 for each cell line.
AZ64 abrogates tumor migration.
Prior studies suggested that TrkB mediates migration in numerous tumor models including HNSCC.Citation8 We therefore hypothesized that suppressing Trk activity would downregulate cellular motility in vitro. Wound scratch assays revealed that AZ64 inhibited tumor cell migration in both a time- and dose-dependent manner across all tested cell lines (). These data, coupled with the growth suppressive efficacy, suggested that AZ64 may be a potent antagonist to local progression in the tumor microenvironment and prompted subsequent in vivo analysis. These data also raised the possibility that Trk receptors may play distinct roles in mediating cell migration or EMT, rather than driving proliferation in HNSCC, consistent with our previous findings.
Genetic inhibition of TrkB sensitizes HNSCC to CDDP inhibition.
Single-agent strategies are uncommon in the therapeutic paradigm for HNSCC, as CDDP-based programs are standard for the majority of both recurrent and primary tumors treated with systemic therapy. To assess whether TrkB suppression, in combination with a standard cytotoxic agent, might be a biologically rational approach, we first evaluated the impact of genetic inhibition of TrkB in sensitizing HNSCC cells to CDDP. After stable retroviral infection with a shRNA construct targeting human TrkB, the levels of TrkB were determined using western blotting (WB). We previously demonstrated that TrkB knockdown suppressed cellular response to BDNF exposure, mediated in part through AKT-mediated effects.Citation8 After confirming downregulation of TrkB, cells harboring either a non-targeting construct (shRNA-NT) or the TrkB-targeting construct (shRNA-TrkB) were exposed to escalating doses of CDDP. Genetic downregulation of TrkB lowered the effective IC50 of HNSCC cells by 60%, suggesting that sensitization to CDDP in HNSCC cells can be enhanced by downregulation of TrkB (Suppl. Fig. 2A). We next assessed the signaling specificity of AZ64 on known downstream mediators of TrkB activation. Both phosphorylation of STAT3 and Src were suppressed by AZ64 in the shRNA-NT cells (Suppl. Fig. 2B), but this effect was moderately abrogated in the shRNA-TrkB cells, suggesting that AZ64 is a specific inhibitor of TrkB signaling in HNSCC and confirming the concept of CDDP-sensitization with TrkB targeting.
AZ64 enhances the anti-proliferative activity of CDDP.
To further determine the potential role of AZ64 in combinational therapies for HNSCC, we studied its impact on cell viability as an adjunct to CDDP, an established systemic agent for this disease. We first determined the sensitivity of each cell line to CDDP and identified a broad range of responsiveness in HNSCC cells (). In all tested cell lines, AZ64 interacted in an additive manner to enhance the cytotoxic properties of cisplatin (). Interestingly, the profound anti-proliferative effects of AZ64 were noted even with concentrations of CDDP below its IC50 for each cell line (). Particularly, HN5, with the highest IC50 for CDDP, was remarkably sensitized to CDDP with TrkB inhibition.
AZ64 sensitizes CDDP-resistant cells to cytotoxic therapy.
To further elaborate the potential role of TrkB inhibition in a relevant pre-clinical model of HNSCC, we developed a cell line characterized by acquired resistance to CDDP (OSC19-CR) and assessed the impact of Trk inhibition on cellular viability. We first tested the isogenic lineage of the OSC19-CR cells by short tandem repeat genotyping (STR) and confirmed that, by DNA genetic fingerprinting, the OSC19-CR cells were identical to the parental cell lines ().Citation17 After confirming the relative resistance of the OSC19-CR cell line to CDDP, levels of TrkB were assessed via WB and the CDDP-resistant cells demonstrated significant overexpression of TrkB compared to the parental cells, while levels of EGFR were unchanged. Additionally, STAT3 and Src were upregulated in the CDDP-resistant cells () and consistent with prior studies, STAT3 upregulation was associated with TrkB overexpression. However, although BDNF stimulation induced cellular migration of both the resistant and the sensitive cell lines, the degree of migration is not significantly different between the two groups (Suppl. Fig. 3). Exposure of the OSC19-CR cells to AZ64 profoundly abrogated cellular growth, but these cells were unaffected by treatment with CDDP (). Further, dose-response curves and IC50 levels to AZ64 were identical between the OSC19-CR and the parental OSC19 cell lines (). Exposure of both the parental OSC19 and OSC19-CR cells resulted in downstream modulation of STAT3 and Src signaling in a dose-dependent fashion, suggesting that AZ64 can overcome deregulated signaling in HNSCC by targeting TrkB (). Taken together, these findings supported the hypothesis that AZ64 has potential additive properties with CDDP and can overcome chemotherapy resistance in selected models of HNSCC by suppressing oncogenic signaling.
Discussion
The concept of targeting TrkB as a therapeutic modality for HNSCC has received little attention, primarily due to the lack of clinical relevance of this RTK in squamous cell carcinomas. In this report, we demonstrate the potential application of a novel inhibitor with putative specificity for TrkB utilizing in vitro models of HNSCC. We first demonstrated the nanomolar specificity of AZ64 for Trk receptors, as well as differential expression of TrkB in both human HNSCC tumors and orthotopic mouse models of HNSCC. In HNSCC cell lines, we identified a narrow therapeutic window for AZ64 at 1–5 µM, with a defined temporal relationship between drug exposure and anti-proliferative activity. The specificity of AZ64 in this system was confirmed in cells that had been genetically engineered to suppress TrkB expression and we demonstrated that AZ64 may have potential applications as a chemosensitizer for CDDP-based therapy. Finally, our data support the feasibility of targeting TrkB to overcome cisplatin resistance and associated oncogenic signaling in HNSCC.
Prior studies have demonstrated the potential feasibility of Trk inhibition for the treatment of neuroblastoma and other solid tumors.Citation15,Citation16 While initially described as a Trk inhibitor for neurogenic tumors, CEP-701 has been resurrected as a FLT3 inhibitor and has potent activity in hematologic malignancies.Citation17 Other inhibitors with selectivity against Trk receptors have been described but have not been transitioned to clinical evaluation.Citation18,Citation19 To date, there have been no studies targeting Trk receptors for novel approaches in HNSCC.
Specifically in the realm of HNSCC, targeting Trk receptors is an attractive target, due to the high levels of receptor expression, activation and function in both in vivo and human HNSCC. Conceptually, AZ64 is unique in its Trk receptor selectivity in the nanomolar range. Although its cellular effects appear to be demonstrable only in the low micromolar range, these are within therapeutically-achievable doses in humans. Conceivably, at these concentrations, the anti-proliferative effects may involve inhibition of Src or other molecular targets, enhancing the therapeutic profile of the inhibitor. It is possible that other targets are inducing some of the observed biological effects and further studies are warranted to explore this. In our model of HNSCC, cell-based systems revealed a relatively narrow window of activity, which was uniform across all tested cell lines. Therefore, a Trk-based therapeutic strategy may not be limited to tumors with high TrkB expression.
The addition of cisplatin to standard HNSCC therapy, either as a radio-sensitizing agent or in the neoadjuvant setting, has had a modest impact on disease progression, suggesting that HNSCC tumors have an innate degree of CDDP resistance.Citation20,Citation21 Support for combination therapy has been proffered by several lines of evidence in HNSCC, utilizing both cytotoxic agents and distinct molecular targeted approaches.Citation4,Citation22 To further define the potential of TrkB targeting in HNSCC with AZ64, we approached this by addressing three fundamental issues: (1) the relevance of TrkB as a viable target for therapy in HNSCC; (2) the specificity of AZ64 for TrkB and its efficacy as a single agent for cytotoxic-based therapy in HNSCC; and (3) enhancing the effects of CDDP-based therapy in HNSCC through TrkB targeting.
In line with previously published data, TrkB appears to play an important role in cellular migration and AZ64 antagonizes this effect, likely through Src or STAT3 pathways. While Src is known to mediate cellular migration in HNSCC, a link to TrkB has not been elucidated to date.Citation23 The downregulation of Src phosphorylation with exposure to AZ64 coincides temporally with the abrogation of cellular migration in multiple cell lines, revealing a novel signaling cascade which links TrkB to migration, possibly through Src pathway activation. These findings are also in line with our data that support a critical role for TrkB in epithelial-mesenchymal transition, suggesting that targeting TrkB may impact not only proliferation, but also cellular migration. In response to AZ64, altered Src and STAT3 signaling were particularly notable in the CDDP-resistant cells. It is thus conceivable that a dual-targeting approach with small molecules targeting Src, such as dasatinib, may be effective in HNSCC. Due to the importance of TrkB in neuronal maintenance and neurite sprouting, one limitation of this approach is the potential enhancement of CDDP-associated neurotoxicity that is noted in 5–10% of patients.Citation24
Resistance to CDDP has been postulated to be due to various mechanisms, including altered p53 expression, an anti-apoptotic transcriptional shift, as well as upregulation of EGFR.Citation25 Previous approaches to “re-sensitizing” CDDP-resistant HNSCC include targeting BCL-xl and BCL2,Citation26 as well as the proteosomal complex,Citation27 to induce apoptosis. In this study, we demonstrate the feasibility of TrkB inactivation to enhance CDDP-based therapy using both pharmacological and genetic approaches. What was particularly interesting was the relatively modest doses of CDDP necessary to induce an anti-proliferative effect with AZ64 in all tested cell lines. The variable response of the combinational therapy could be explained by the differential of CDDP-sensitivity of the tested cell lines. Despite this variability, the uniform responses suggested that AZ64, as an adjunct to a CDDP-based regimen, could have therapeutic potential in the treatment of HNSCC.
Further elucidation of the precise mechanism of a TrkB-directed re-sensitization of HNSCC to CDDP therapy is warranted.
The unique biology of HNSCC and the central role of CDDP in its treatment highlight novel mechanisms of chemoresistance and drug targeting through TrkB. In this study, we demonstrate the feasibility of a novel small molecule inhibitor that has significant anti-proliferative properties against HNSCC by targeting Trk receptors. Using both genetic and pharmacological approaches, we established the specificity of this compound for Trk receptors and demonstrate its efficacy against chemotherapy-resistant HNSCC. Inhibiting the downstream oncogenic STAT3 and Src pathways revealed a novel link between TrkB and these signaling cascades, suggesting an important avenue for drug development in HNSCC.
Methods
Cell lines and reagents.
The head and neck squamous cell carcinoma cell lines OSC19, MDA1986, Tu138, MDA686LN, HN5, UMSCC-22A, UMSCC10A, UMSCC1, DM14, Tu167, JMAR, Tu159, UMSCC-10B, FADU, HN30 and the lung cancer cell lines HCC827 and H1975, were maintained as described.Citation12,Citation13 NIH3T3 cells and HEK-293 were purchased from ATCC (Manassas, VA). The following antibodies were utilized: TrkB (sc-8316, Santa Cruz), BDNF (sc-546), GAPDH, STAT3 (9132, Cell Signaling), phospho-STAT3 (9145), Src (2108) and phospho-Src (2113). Cisplatin (CDDP) was purchased from Calbiochem (San Diego, CA) and dissolved in DMSO. AZ64 (MW = 356) was synthesized as previously described and dissolved in DMSO.Citation14
Establishment of CDDP-resistant cell line and DNA finger-printing.
The OSC19 cell line was grown in escalating doses of CDDP, starting at 0.1 µM, over a 6-month period, to a final concentration of 40 µM. Genomic DNA was extracted from both the parental OSC19 cells and from the CDDP-resistant cell lines (Qiagen) and analyzed as previously described.
Plasmid transfections.
Short-hairpin RNA (shRNA) constructs targeting TrkB (Cat. TR320436, Origene, Rockville, MD) were introduced into cells via retroviral infection according to the manufacturer's protocol, as previously described.Citation8 For TrkA and TrkB overexpression, pcDNA3.1 plasmids containing the full length cDNA sequences for human TrkA and TrkB were stably transfected into NIH-3T3 cells and selected with G418.
Western blotting (WB).
Cells were grown to 80% confluency, washed with PBS and lysed for 30 minutes on ice (Tris-HCL 50 mM, NaCl 100 mM, Triton-X 1%, deoxycholute 0.5%, MgCl2 10 mM, NaVO3 1 mM, NaF 50 mM, PMSF 1 mM, protease inhibitor in PBS). SDS-PAGE analysis was performed and membranes were incubated overnight at 4° with antibodies directed against the indicated proteins. Membranes were washed, incubated with the appropriate secondary antibodies and exposed with the ECL chemiluminescent substrate kit (Pierce, Rockford, IL). Images were analyzed with ImagePro (MediaCybernetics, Bethesda, MD) and Prism (GraphPad Software, La Jolla, CA).
Measurement of cell proliferation.
The anti-proliferative effects of CDDP and AZ64 were determined by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, as previously described.Citation3 Statistical significance was determined using Student's t-test (SPSS, Chicago, IL). A two-way ANOVA was performed to test the interaction between Cisplatin and AZ64 on cell proliferation rate. If the interaction was significant, we further compared mean differences between any of two treatment groups using Tukey's multiple comparison adjustment to control the overall type I error rate at a 0.05 significance level. A p value of <0.05 was considered significant. SAS version 9.1 and S-Plus version 7.0 were used to carry out the computations for all analyses. IC50 levels were determined using Prism.
Human tumor analysis and immunohistochemistry.
Fresh-frozen head and neck squamous cell carcinoma tumors were obtained from the M.D. Anderson Cancer Center Head and Neck Tumor Tissue Repository under an Institutional Review Board-approved protocol. Specimens were homogenized, lysed and analyzed by WB.
Wound scratch assay.
Wound scratch assays were performed as previously described.Citation8 Measurements were taken at indicated time-points and were quantified with ImagePro. Statistical significance was determined using Student's t-test. A p value of <0.05 was considered significant.
Financial Support
Dan Duncan Cancer Center Award at Baylor College of Medicine (GD); RNR Cross Foundation (EYH); Strategic Alliance Agreement between Astra Zeneca and UT M.D. Anderson Cancer Center (J.N.M.); Physician-Scientist Award (M.E.K.), Head and Neck SPORE Program (M.E.K.), NCI Short Term Research Scientist Exchange Program (T.Y., M.E.K.).
Figures and Tables
Figure 1 (A) Stably-transected cells harboring either TrkA or TrkB were exposed to increasing concentrations of AZ64. Cell lysates were separated by SDS-PA GE and membranes were exposed to the indicated antibodies. Total MAP K levels were assayed as a loading control. (B) Quantification of phosphorylation intensity signal change in response to AZ64 for TrkA and TrkB, with MAP K used for loading control. Data were normalized to the MAP K signal and results were analyzed by Student's t-test. (C) TrkB is overexpressed in human HNSCC tumors. Left—Protein lysates from representative human squamous cell carcinoma tumors (n = 20) and normal mucosa (n = 3) were separated by 7% SDS-PA GE and assessed with the indicated antibodies. Mouse brain (MB) was used as a positive control for TrkB expression. Right—Immunohistochemistry (IHC) was performed on paraffin-embedded HNSCC tumor sections with TrkB antibody for confirmation. (Magnification 200x; inset 40x). (D) Orthotopically-implanted HNSCC tumors from nude mice (n = 10) were assessed by both 7% SDS-Page (left) and IHC (right, magnification 200x) for TrkB expression.
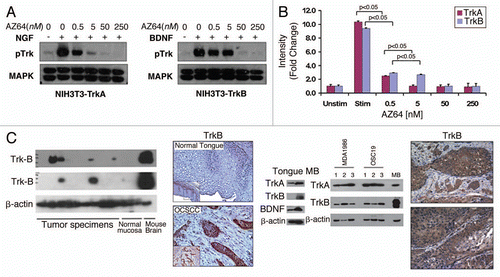
Figure 2 AZ64 profoundly inhibits cellular proliferation in HNSCC. (A) IC50 results for depicted HNSCC cell lines after exposure to AZ64. (B and C) Dose-dependent effects of AZ64 on cellular viability in representative HNSCC cell lines by the MTT assay at 48 hours (C) and 72 hours (C). Data were normalized to control (untreated group) and analyzed with ANOVA and Tukey's Multiple Comparison Tests. p values represent cell viability across each concentration in comparison to results at 24 hours. Data is representative of three independent experiments, with eight replicated for each concentration.
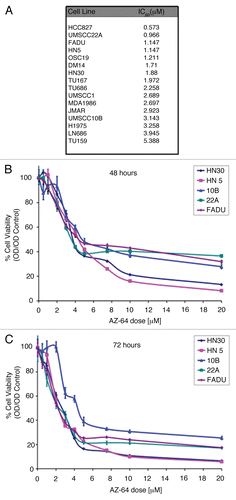
Figure 3 Suppression of cellular migration through TrkB inhibition in HNSCC. (A-E) Indicated cells were grown to 90% confluence in 6-well plates and a standard sized wound was made in five locations on each plate. Cells were then exposed to AZ64 at variable doses and cell migration was measured at 5 spots for each wound at the indicated time points. Data represent the percent wound closure (from control) for 5 spots from each wound. Results were analyzed by the Student's t-test. Data is representative of three independent experiments.
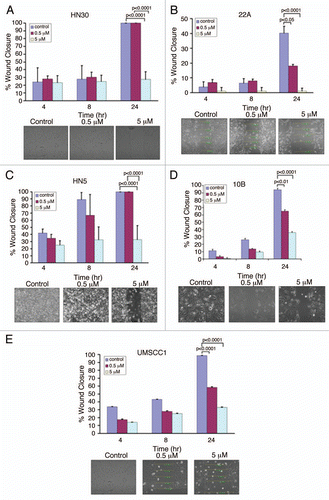
Figure 4 AZ64 synergizes with CDDP to suppress HNSCC growth. (A) IC50 results for CDDP in depicted HNSCC cell lines. (B-F) Indicated HNSCC cell lines were exposed to increasing concentrations of AZ64 in the presence of CDDP. Cell viability was measured at the indicated time points. Data were normalized to control (untreated group) and analyzed with ANOVA and Tukey's Multiple Comparison Tests. p values represent cell viability across each concentration in comparison to results at 24 hours. Data is representative of three independent experiments, with eight replicated for each concentration.
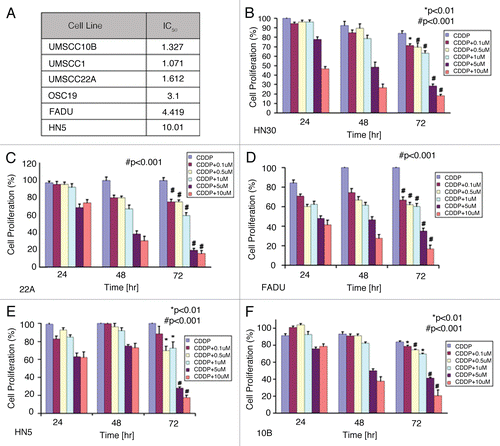
Figure 5 TrkB inhibition re-sensitizes chemotherapy-resistant cells to CDDP-based therapy. (A) OSC19 cells were exposed to increasing concentrations of CDDP over a 6-month period of time to develop the OSC19-CR cell line. Parental OSC19 and OSC19 cells were then exposed to increasing concentrations of CDDP and analyzed by the MTT protocol. Inset, lysates from the indicated cell lines were separated by 10% SDS-PA GE and membranes were exposed to the indicated antibodies. (B) OSC19-CR cells were exposed to either AZ64 or CDDP and analyzed by the MTT protocol to assess response to AZ64. (C) Parental OSC19 and OSC19-CR cells were exposed to increasing doses of AZ64, demonstrating equivalent anti-proliferative effects through TrkB inhibition. (D) After exposing cells to increasing concentrations of AZ64 for 4 hours, OSC19 and OSC19-CR cells were lysed and proteins were separated by 10% SDS-PA GE. Membranes were incubated with the indicated antibodies.
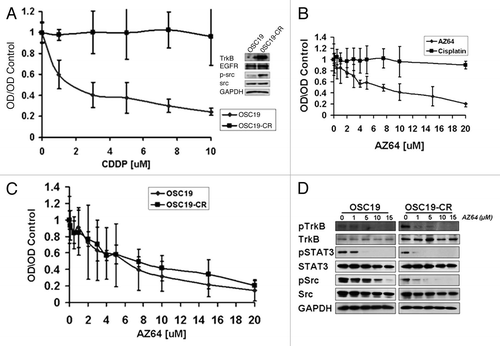
Table 1 Selectivity profile of AZ64
Table 2 Effects of combined cisplatinTable Footnote† and AC64 on cell proliferation
Table 3 STR Locus
Additional material
Download Zip (62.6 KB)Acknowledgements
We wish to thank Ms. Terri Astin for preparation of the manuscript, Ms. Jun Ju for assistance with the research and Dr. Katherine Hale of the Department of Systems Biology for the STR Analysis.
References
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer Statistics 2007. CA Cancer J Clin 2007; 57:43 - 66
- Karamouzis MV, Grandis JR, Argiris A. Therapies directed against epidermal growth factor receptor in aerodigestive carcinomas. Jama 2007; 298:70 - 82
- Yigitbasi OG, Younes MN, Doan D, et al. Tumor cell and endothelial cell therapy of oral cancer by dual tyrosine kinase receptor blockade. Cancer Res 2004; 64:7977 - 7984
- Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus Cetuximab for Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med 2006; 354:567 - 578
- Pearse RN, Swendeman SL, Li Y, Rafii D, Hempstead BL. A neurotrophin axis in myeloma: TrkB and BDNF promote tumor-cell survival. Blood 2005; 105:4429 - 4436
- Eggert A, Grotzer MA, Ikegaki N, et al. Expression of the neurotrophin receptor TrkB is associated with unfavorable outcome in Wilms' tumor. J Clin Oncol 2001; 19:689 - 696
- Jaboin J, Kim CJ, Kaplan DR, Thiele CJ. Brain-derived neurotrophic factor activation of TrkB protects neuroblastoma cells from chemotherapy-induced apoptosis via phosphatidylinositol 3′-kinase pathway. Cancer Res 2002; 62:6756 - 6763
- Kupferman ME, Jiffar T, El-Naggar AK, et al. TrkB induces EMT and plays a key role in invasion of head and neck squamous cell carcinoma. Oncogene 2010;
- Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature 2004; 430:1034 - 1039
- Camoratto AM, Jani JP, Angeles TS, et al. CEP-751 inhibits trk receptor tyrosine kinase activity in vitro and exhibits anti-tumor activity. International Journal of Cancer 1997; 72:673 - 679
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 2007; 7:573 - 584
- Zhou G, Xie TX, Zhao M, et al. Reciprocal negative regulation between S100A7/psoriasin and beta-catenin signaling plays an important role in tumor progression of squamous cell carcinoma of oral cavity. Oncogene 2008; 27:3527 - 3538
- Nakashima T, Pak SC, Silverman GA, Spring PM, Frederick MJ, Clayman GL. Genomic cloning, mapping, structure and promoter analysis of HEADPIN, a serpin which is downregulated in head and neck cancer cells. Biochimica et Biophysica Acta (BBA)—Gene Structure and Expression 2000; 1492:441 - 446
- Wang T, Lamb ML, Scott DA, et al. Identification of 4-Aminopyrazolylpyrimidines as Potent Inhibitors of Trk Kinases. Journal of Medicinal Chemistry 2008; 51:4672 - 4684
- George DJ, Dionne CA, Jani J, et al. Sustained in Vivo Regression of Dunning H Rat Prostate Cancers Treated with Combinations of Androgen Ablation and Trk Tyrosine Kinase Inhibitors, CEP-751 (KT-6587) or CEP-701 (KT-5555). Cancer Res 1999; 59:2395 - 2401
- Marshall JL, Kindler H, Deeken J, et al. Phase I trial of orally administered CEP-701, a novel neurotrophin receptor-linked tyrosine kinase inhibitor. Investigational New Drugs 2005; 23:31 - 37
- Hexner EO, Serdikoff C, Jan M, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood 2008; 111:5663 - 5671
- Wood ER, Kuyper L, Petrov KG, Hunter Iii RN, Harris PA, Lackey K. Discovery and in vitro evaluation of potent TrkA kinase inhibitors: oxindole and azaoxindoles. Bioorganic & Medicinal Chemistry Letters 2004; 14:953 - 957
- Lippa B, Morris J, Corbett M, et al. Discovery of novel isothiazole inhibitors of the TrkA kinase: Structure-activity relationship, computer modeling, optimization and identification of highly potent antagonists. Bioorganic & Medicinal Chemistry Letters 2006; 16:3444 - 3448
- Induction chemotherapy plus radiation compared with surgery plus radiation in patients with advanced laryngeal cancer. The Department of Veterans Affairs Laryngeal Cancer Study Group. N Engl J Med 1991; 324:1685 - 1690
- Brizel DM, Albers ME, Fisher SR, et al. Hyperfractionated irradiation with or without concurrent chemotherapy for locally advanced head and neck cancer. N Engl J Med 1998; 338:1798 - 1804
- Forastiere AA, Goepfert H, Maor M, et al. Concurrent Chemotherapy and Radiotherapy for Organ Preservation in Advanced Laryngeal Cancer. N Engl J Med 2003; 349:2091 - 2098
- Johnson FM, Saigal B, Talpaz M, Donato NJ. Dasatinib (BMS-354825) Tyrosine Kinase Inhibitor Suppresses Invasion and Induces Cell Cycle Arrest and Apoptosis of Head and Neck Squamous Cell Carcinoma and Non-Small Cell Lung Cancer Cells. Clin Cancer Res 2005; 11:6924 - 6932
- Gibson MK, Li Y, Murphy B, et al. Randomized Phase III Evaluation of Cisplatin Plus Fluorouracil Versus Cisplatin Plus Paclitaxel in Advanced Head and Neck Cancer (E1395): An Intergroup Trial of the eastern Cooperative Oncology Group. J Clin Oncol 2005; 23:3562 - 3567
- Michaelis M, Bliss J, Arnold SC, et al. Cisplatin-Resistant Neuroblastoma Cells Express Enhanced Levels of Epidermal Growth Factor Receptor (EGFR) and Are Sensitive to Treatment with EGFR-Specific Toxins. Clin Cancer Res 2008; 14:6531 - 6537
- Bauer JA, Trask DK, Kumar B, et al. Reversal of cisplatin resistance with a BH3 mimetic, (â‘’)-gossypol, in head and neck cancer cells: role of wild-type p53 and Bcl-xL. Molecular Cancer Therapeutics 2005; 4:1096 - 1104
- Li C, Li R, Grandis JR, Johnson DE. Bortezomib induces apoptosis via Bim and Bik upregulation and synergizes with cisplatin in the killing of head and neck squamous cell carcinoma cells. Molecular Cancer Therapeutics 2008; 7:1647 - 1655