Abstract
Commentary to:
Prioritization of driver mutations in pancreatic cancer using cancer-specific high-throughput annotation of somatic mutations (CHASM)
Hannah Carter, Josue Samayoa, Ralph H. Hruban and Rachel Karchin
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer death in the western world, with a 5 year survival rate that is under 5%. The diagnosis of pancreatic cancer is often established at an advanced stage, precluding at least 80% of these patients from undergoing attempts at curative resection. Moreover, following surgery many patients develop recurrence of their cancer. Sadly, non-surgical treatment modalities for this malignancy have all too often yielded very limited results. Therapeutic failure is caused by several factors, including the presence of non-specific symptoms, which delays the diagnosis; the absence of serum markers for reliably screening for early stage disease, the tumor's propensity to metastasize, the high frequency of local lymphatic and peri-neural invasion, the intense desmoplasia which interferes with drug delivery to the cancer cells, and the intrinsic resistance of these cells to chemotherapeutic agents.
The dismal response to current therapies in PDAC is also due to the biological aggressiveness of the cancer, which is caused by the presence of multiple molecular alterations in the cancer cells within the tumor mass. These include a high frequency of activating KRAS mutations, and loss of function mutations in the P16/CDKN2A, TP53 and SMAD4/DPC4 genes.Citation1 Mutations in all four genes are recognized as “driver mutations” in PDAC because they drive neoplastic transformation (KRAS) and tumor progression (P16/CDKN2A, TP53 and SMAD4/DPC4). The concept that these are indeed driver mutations is underscored by studies with mouse models of PDAC in which it has been demonstrated that targeted pancreatic activation of KrasG12D from its own endogenous locus results in pancreatic intraepithelial neoplasia (PanIN) lesions that advance to PDAC after a long latency,Citation2 and that concomitant deletion and/or mutation of p16, p53 or Smad4 greatly accelerates PanIN progression and PDAC formation.Citation3–Citation5
Carter, et al. applied the analytical technique of Cancer-specific High-throughput Annotation of Somatic Mutations (CHASM) to study 963 somatic missense mutations discovered in 24 PDACs.Citation6 They correctly pointed out that many mutations are not readily characterized as driver mutations, and they provided evidence that CHASM has allowed them to identify putative driver mutations in 15 genes, including those encoding kinases (PIK3CG, DGKA, STK33, TTK and PRKCG), cell cycle related proteins (NEK8) and cell adhesion proteins (CMAS, PCDHB2). By identifying putative driver genes that are mutated at relatively low frequency, their findings will ultimately allow for the delineation of subgroups of patients with PDAC, identify specific components within a particular pathway that are altered in a given cancer, and potentially provide a roadmap for the design of individualized therapeutic strategies.
In order for the findings from this study to yield an improved understanding of the role of these driver genes in the pathobiology of PDAC, it is now important to assess their function in several biological systems, including pancreatic cancer cell lines and genetic mouse models of PDAC. Such studies could also ultimately guide the development of novel drug therapies for this deadly malignancy. It should be appreciated that PDACs overexpress multiple tyrosine kinase receptors and their ligands,Citation7 all of which signal, in part, through Kras, and that Kras activates crucial signaling pathways, including the Raf/ERK, Ral, Tiam1 and Rac pathways, phospholipase Cε-mediated activation of protein kinase C and phosphatidylinositol 3-kinase (PI3K).Citation8 It is noteworthy, therefore, that 6 of the new driver genes were kinases, and that at least 4 of these genes (PIK3CG, DGKA, STK33, PRKCG) encode proteins that have the potential to cross-talk with Kras-driven pathways (), thereby potentially leading to the synergistic and aberrant activation of multiple signaling networks that promote cancer cell growth, survival, migration, invasion and metastasis.
Perhaps the most intriguing aspect of the findings reported by Carter et al. is the observation that mutations in PIK3CG and NEK8 genes are driver mutations,Citation6 given that they encode proteins that participate in the modulation of important signaling pathways. Thus, PIK3CG encodes the p110 gamma catalytic subunit of PI3K (PI3Kγ) which forms a heterodimer by associating with either a p101 or a p84/p87PIKAP regulatory subunit, and which interacts with G-protein-coupled receptors (GPCRs). In contrast, NEK8 encodes a serine-threonine kinase that is a member of the “never-in-mitosis” or NIMA-related kinase (NRK) family, and generally localizes to primary cilia. A better understanding of the role of these two genes, neither of which has been previously implicated in PDAC, would constitute a significant advance in our understanding of the pathobiology of this malignancy.
PI3Ks are grouped into three distinct classes (I, II and III), and PI3Kγ represents the 1B subclass.Citation9 By contrast to PI3Kγ, members of the 1A subclass of PI3Ks are activated by tyrosine kinase receptors and consist of a 110 Kda catalytic subunit (α, β or δ isoforms) that associates with an SH2-containing p85 regulatory subunit. The catalytic domain of class I PI3Ks has a Ras binding site, and activation of either the IA or IB subclass leads to the generation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) from phosphatidylinositol (4,5)-bisphosphate (PIP2). PIP3 can recruit pleckstrin homologydomain containing proteins such as the serine-threonine kinase AKT to the cell membrane. There are three genes encoding three distinct but related AKTs (1, 2 and 3), which become activated following phosphorylation on threonine 308 by the actions of 3′-phophoinositide-dependent kinase 1 (PDK1) and on serine 473 by mammalian target of rapamycin (mTOR) complex-2 (TORC2).Citation10 AKT2 is amplified in 10% of PDACs,Citation11 underscoring its importance in PDAC. The crucial role of the PI3K-Akt signaling cascade is also highlighted by the fact that phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a negative regulator of this pathway, and that a murine model of conditional PTEN inactivation in mice harboring a Kras mutation is associated with invasive pancreatic cancer arising from the centroacinar cell.Citation12
Carter, et al. point out that the R839C mutant of PIK3CG may function as a tumor suppressor, and the gene has been reported to be deleted, mutated or downregulated by CpG hypermethylation, depending on the tissue in which the cancer arises.Citation6 Interestingly, PI3Kγ is expressed in the pancreatic acinar cells where it transmits signals downstream of the G-protein-coupled cholecystokinin (CCK) receptor, thereby leading to enhanced calcium mobilization, digestive enzyme release and NFκB activation.Citation13 There are two CCK receptors (A and B) and a splice variant of the CCK B receptor, termed CCK-C receptor, is expressed in pancreatic cancer cells and has been implicated in enhancing their proliferation.Citation14 Is it possible that activation of CCK B receptor by gastrointestinal hormones, when occurring in the context of an initiating Kras mutation and the R839C mutant of PIK3CG, contributes to PDAC initiation and progression in some patients? Moreover, excessive activation of the CCK receptor leads to acinar cell damage and acute pancreatitis, and two episodes of acute pancreatitis in mice harboring a Kras mutation in the pancreas leads to accelerated PanIN progression of mouse PDAC.Citation15 PI3Kγ activation also promotes leukocyte chemotaxis and inflammatory responses, as well as mast cell degranulation.Citation16 Inasmuch as inflammation and mast cells may also contribute to pancreatic carcinogenesis,Citation17,Citation18 these observations suggest that a mutation in PIK3CG could have multiple consequences on PDAC initiation and progression, and underscore the need for assessing the roles of wild-type and mutated PI3Kγ in the pancreas.
The second intriguing gene identified by CHASM as a mutated driver gene is NEK8, which encodes a serine-threonine kinase that plays a role in cell cycle progression from G2 to M-phase.Citation19 As noted above, it localizes to primary cilia, which are cell surface organelles that are found in most epithelial cell types, as well as on non-epithelial cells, such as fibroblasts. The membranes of primary cilia possess ion channels and transmembrane signaling receptors implicated in linking cell function to chemical and mechanical stimuli, cytosolic calcium fluxes, and other signaling pathways that modulate mitogenesis, differentiation and motogenesis.Citation20 Moreover, primary cilia appear to be especially important for Sonic hedgehog (Shh) signaling in the pancreatic ductal cell.Citation21 Thus, in the absence of Shh, the 12-transmembrane patched receptor localizes to the ciliary membrane and represses the accumulation of the smoothened (Smo) 7-transmembrane receptor within the cilia. Smo accumulates in the cilia and is activated following suppression of the upstream inhibitory receptor, Patched, by Shh ligation.Citation20,Citation21
The presence of primary cilia is associated with a differentiated, non-proliferative state, whereas initiation of cell cycle progression mandates ciliary disassembly, which requires several kinases that are also involved in the regulation of mitosis at the G2/M transition. Thus, Nek8 disrupts ciliary microtubules and promotes cell cycle progression,Citation19 whereas Aurora A kinase phosphorylates and activates histone deacetylase 6, which then functions as microtubule deacetylase and mediates ciliary disassembly, allowing Aurora A kinase to regulate mitotic entry at the G2/M interphase.Citation22 Aurora A kinase is often overexpressed in PDAC.Citation23 It activates RalA,Citation24 and its inhibition suppresses pancreatic cancer cell proliferation and attenuates their chemoresistance.Citation25 Therefore, its proximity to the actions of Nek8 in relation to ciliary disassembly raises tantalizing questions regarding the potential interactions between NEK8 and Aurora A kinase.
While ciliary retraction is coordinated with the re-entry of the cell cycle progression and cellular proliferation, this is not a requirement in PDAC as the vast majority of the PanIN and cancer cells in PDAC do not possess primary cilia.Citation26 Indeed, for reasons that are not readily evident, the pancreatic acinar cell is also devoid of primary cilia; by contrast, duct cells, islet cells and centro-acinar cells possess primary cilia.Citation26 Inasmuch as recent evidence indicates that PDAC may arise from the progenitor cell lineage that normally gives rise to the pancreatic acinar cell,Citation27 these observations raise the possibility that the absence of cilia in the cancer cells in PDAC may reflect an acinar cell origin for most cases of PDAC. Alternatively, it is possible that Kras mutations and other alterations interfere with ciliogenesis in the cancer cells in PDAC.Citation26 Indeed, the PI3K inhibitor, LY294002, has been shown to restore ciliogenesis in RInk-1 mouse pancreatic cancer cells established from Pdx1-Cre;LSL-KrasG12D;Ink4a/Arflox/lox mice, in which oncogenic Kras is combined with deletion of the Ink4a locus that contains the p16 gene.Citation26
Irrespective of the reasons for the absence of primary cilia in pancreatic cancer cells, these observations also point to a unique mechanism for attenuating Shh-mediated autocrine effects in the cancer cells, while allowing for cancer cell derived Shh to exert paracrine effects on the stroma. Indeed, the principal effect of Shh in PDAC is on the stroma,Citation27 and this has therapeutic implications, given that targeting the stroma may improve our ability to deliver drugs into the pancreatic tumor mass.Citation28 In support of this hypothesis, the stromal cells in PDAC possess primary cilia, and it has been recently shown that oncogenic Kras switches Shh actions from an autocrine to a paracrine pathway by activating the dual specificity tyrosine phosphorylated and regulate kinase 1B (DYRK1B),Citation29 which is downstream of PI3K-Akt signaling.Citation29 NEK8 has also been implicated in modulating cell adhesion.Citation30 Thus, it is possible that mutations in NEK8, CMAS or PCDHB2 may lead to altered cell adhesion and altered cancer cell stroma interactions. These observations underscore the fact that pancreatic cancer cells can exhibit multiple defects in a given signaling pathway. An improved understanding of these defects could lead to therapies that target the specific pathway addiction of a particular person's cancer.
Figures and Tables
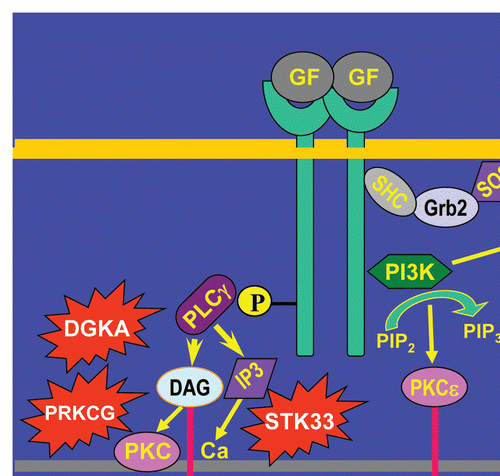
Figure 1 Pathways activated by novel driver mutations intersect with Kras driven aberrant signaling pathways in pancreatic cancer cells. In addition to harboring mutated Kras, pancreatic cancer cells overexpress tyrosine kinase receptors, an example of which is shown binding excessive levels of growth factors (GF). This leads to excessive Ras activation even when Kras is not mutated. Ras becomes active downstream of the adapter proteins SHC and GRB2 and the SOS guanine nucleotide exchange factor. There is activation of additional pathways downstream of ras, as described in the text, but only the RAF, MEK, MAPK cascade is shown. PI3K activation leads to PIP3 generation and subsequent AKT and protein kinase C (PKC) epsilon (PKCε) activation, serving to promote cancer cell survival and chemoresistance. Mutated PI3KCG, which encodes a mutated PI3Kγ, may activate aberrant pathways downstream of gastrointestinal hormones such as cholecystokin (CCK) or gastrin, leading to excessive ras activation and PIP3 generation, further promoting cancer cell survival and proliferation. Concomitantly, there is activation of phospholipase C-gamma (PLCγ), which leads to the generation of inositol-1,4,5-trisphosphate (IP3) which mobilizes intracellular calcium and diacylglycerol (DAG), which activates PKC. Mutated DAG kinase (DGKA), STK33 (which exhibits homology with calcium-calmodulin dependent kinase) and PRKCG, which encodes the gamma isoform of PKC, can in theory “join forces” to lead to enhanced cancer cell proliferation and invasion.
Acknowledgements
Supported, in part, by US Public Health Service Grant CA-75059, awarded by the National Cancer Institute to M.K.
Commentary to:
References
- Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer 2002; 2:897 - 909
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003; 4:437 - 450
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 2003; 17:3112 - 3126
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005; 7:469 - 483
- Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 2006; 20:3130 - 3146
- Carter HC, Samayoa J, Hruban RH, Karchin R. Prioritization of Driver Mutations in Pancreatic Cancer using Cancer-specific High-throughput Annotation of Somatic Mutations (CHASM). Cancer Biol Ther 2010; 10:582 - 587
- Preis M, Korc M. Kinase signaling pathways as targets for intervention in pancreatic cancer. Cancer Biol Ther 2010; 9:754 - 763
- Ramjaun AR, Downward J. Ras and phosphoinositide 3-kinase: partners in development and tumorigenesis. Cell Cycle 2007; 6:2902 - 2905
- Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta 2008; 1784:159 - 185
- Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev 2010; 20:87 - 90
- Cheng JQ, Ruggeri B, Klein WM, Sonoda G, Altomare DA, Watson DK, et al. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci USA 1996; 93:3636 - 3641
- Stanger BZ, Stiles B, Lauwers GY, Bardeesy N, Mendoza M, Wang Y, et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell 2005; 8:185 - 195
- Gukovsky I, Cheng JH, Nam KJ, Lee OT, Lugea A, Fischer L, et al. Phosphatidylinositide 3-kinase gamma regulates key pathologic responses to cholecystokinin in pancreatic acinar cells. Gastroenterology 2004; 126:554 - 566
- Smith JP, Stanley WB, Verderame MF, Zagon IS. The functional significance of the cholecystokinin-C (CCK-C) receptor in human pancreatic cancer. Pancreas 2004; 29:271 - 277
- Carriíre C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun 2009; 382:561 - 565
- Kim MS, Rådinger M, Gilfillan AM. The multiple roles of phosphoinositide 3-kinase in mast cell biology. Trends Immunol 2008; 29:493 - 501
- Esposito I, Kleeff J, Bischoff SC, Fischer L, Collecchi P, Iorio M, et al. The stem cell factor-c-kit system and mast cells in human pancreatic cancer. Lab Invest 2002; 82:1481 - 1492
- Farrow B, Evers BM. Inflammation and the development of pancreatic cancer. Surg Oncol 2002; 10:153 - 169
- Bowers AJ, Boylan JF. Nek8, a NIMA family kinase member, is overexpressed in primary human breast tumors. Gene 2004; 328:135 - 142
- Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 2010; 11:331 - 344
- Cervantes S, Lau J, Cano DA, Borromeo-Austin C, Hebrok M. Primary cilia regulate Gli/Hedgehog activation in pancreas. Proc Natl Acad Sci USA 2010; 107:10109 - 10114
- Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 2007; 129:1351 - 1363
- Li D, Zhu J, Firozi PF, Abbruzzese JL, Evans DB, Cleary K, Friess H, et al. Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin Cancer Res 2003; 9:991 - 997
- Lim KH, Brady DC, Kashatus DF, Ancrile BB, Der CJ, Cox AD, et al. Aurora-A phosphorylates, activates and relocalizes the small GTPase RalA. Mol Cell Biol 2010; 30:508 - 523
- Hata T, Furukawa T, Sunamura M, Egawa S, Motoi F, Ohmura N, et al. RNA interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res 2005; 65:2899 - 2905
- Seeley ES, Carriere C, Goetze T, Longnecker DS, Korc M. Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res 2009; 69:422 - 430
- Bailey JM, Mohr AM, Hollingsworth MA. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene 2009; 28:3513 - 3525
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009; 324:1457 - 1461
- Lauth M, Bergström A, Shimokawa T, Tostar U, Jin Q, Fendrich V, et al. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat Struct Mol Biol 2010; 17:718 - 725
- Natoli TA, Gareski TC, Dackowski WR, Smith L, Bukanov NO, Russo RJ, et al. Pkd1 and Nek8 mutations affect cell-cell adhesion and cilia in cysts formed in kidney organ cultures. Am J Physiol Renal Physiol 2008; 294:73 - 83