Abstract
We have recently proposed a new model for understanding how tumors evolve. To achieve successful "Tumor-Stroma Co-Evolution", cancer cells induce oxidative stress in adjacent fibroblasts and possibly other stromal cells. Oxidative stress in the tumor stroma mimics the effects of hypoxia, under aerobic conditions, resulting in an excess production of reactive oxygen species (ROS). Excess stromal production of ROS drives the onset of an anti-oxidant defense in adjacent cancer cells, protecting them from apoptosis. Moreover, excess stromal ROS production has a "Bystander-Effect", leading to DNA damage and aneuploidy in adjacent cancer cells, both hallmarks of genomic instability. Finally, ROS-driven oxidative stress induces autophagy and mitophagy in the tumor micro-environment, leading to the stromal over-production of recycled nutrients (including energy-rich metabolites, such as ketones and L-lactate). These recycled nutrients or chemical building blocks then help drive mitochondrial biogenesis in cancer cells, thereby promoting the anabolic growth of cancer cells (via an energy imbalance). We also show that ketones and lactate help "fuel" tumor growth and cancer cell metastasis and can act as chemo-attractants for cancer cells. We have termed this new paradigm for accelerating tumor-stroma co-evolution, "The Autophagic Tumor Stroma Model of Cancer Cell Metabolism". Heterotypic signaling in cancer-associated fibroblasts activates the transcription factors HIF1alpha and NFκB, potentiating the onset of hypoxic and inflammatory response(s), which further upregulates the autophagic program in the stromal compartment. Via stromal autophagy, this hypoxic/inflammatory response may provide a new escape mechanism for cancer cells during anti-angiogenic therapy, further exacerbating tumor recurrence and metastasis.
Biomarker Studies: Identification of a “Lethal Tumor Microenvironment”
We recently identified that a loss of stromal caveolin-1 (Cav-1) is a powerful independent biomarker for predicting clinical outcome in human breast cancer patients.Citation1–Citation4 More specifically, a loss of stromal Cav-1 effectively predicts early tumor recurrence, lymph-node (LN) metastasis, lympho-vascular invasion (LVI) and tamoxifen-resistance and, as a consequence, is associated with poor clinical outcome in human breast cancer patients.Citation1 Importantly, the prognostic value of a loss of stromal Cav-1 is independent of epithelial marker status (ER, PR, HER2), making it a valuable biomarker in all the most common sub-types of breast cancer,Citation1 including triple negative (TN) and basal-like breast cancer.Citation2 For example, >75% of TN patients with high stromal Cav-1 have an overall survival of nearly 12 years.Citation2 Conversely, <10% of TN patients with absent stromal Cav-1 remain alive 5 years post-diagnosis.Citation2 Thus, a loss of stromal Cav-1 in human breast cancers is associated with a “lethal stromal phenotype”. Similar results were obtained with DCIS (a breast cancer precursor lesion),Citation4 and prostate cancer,Citation5 providing an indication that the predictive value of stromal Cav-1 may extend to a wide variety of other tumor types.
The “Autophagic Tumor Stroma Model of Cancer”: Co-Culture Studies with Fibroblasts and Mouse Animal Models
To understand the molecular basis of the lethality of a loss of stromal Cav-1, we developed a co-culture system to mimic tumor-stroma co-evolution.Citation6 In this co-culture system, stromal fibroblasts (hTERT-BJ1 cells) were mixed with human breast cancer cells (MCF-7) and allowed to interact for 3–5 days.Citation6 Using this simplified model system, we observed that cancer cells induce oxidative stress in adjacent stromal fibroblasts, with the over-production of reactive oxygen species (ROS) and local DNA damageCitation7,Citation8 ().
Oxidative stress in stromal fibroblasts, in turn, induced an anti-oxidant defense in neighboring cancer cells, dramatically protecting them against apoptosisCitation7,Citation8 (). In this regard, we observed that the fibroblasts specifically induce anti-oxidant (peroxiredoxin1) and anti-apoptotic proteins (TIGAR) in adjacent cancer cells.Citation7,Citation8 However, ROS over-production in cancer-associated fibroblasts also had a “Bystander Effect”, driving DNA damage (gamma-H2AX staining; a marker of DNA double-strand breaks) and aneuploidy in cancer cells.Citation7,Citation8 These are hallmarks of genomic instability. Thus, fibroblasts protect cancer cells against apoptosis and at the same time, allow cancer cells to undergo random mutagenesis, to accelerate tumor-stroma co-evolutionCitation7,Citation8 (). In accordance with this hypothesis, genomic alterations are frequently observed both in tumor cells and the surrounding stromal cells.Citation9,Citation10
Finally, we also observed that cancer cells induce a loss of Cav-1 and mitochondria in adjacent fibroblasts.Citation7,Citation8 This appears to be due to the onset of autophagy, as both anti-oxidants (such as N-acetyl-cysteine (NAC), metformin and quercetin), as well as lysosomal/autophagy inhibitors (chloroquine) can prevent this process.Citation7,Citation8 Autophagy in cancer associated fibroblasts provides adjacent tumor cells with energy-rich recycled nutrients and chemical building blocks to fuel the anabolic growth of cancer cells.Citation7,Citation8 Since these cancer-associated fibroblasts lose their mitochodria via mitophagy (the autophagic destruction of mitochondria), they are forced to undergo aerobic glycolysis, resulting in the stromal production of pyruvate, lactate and ketone bodies (3-hydroxybutyrate).Citation7,Citation8,Citation11 To describe this phenomenon, we have coined the term, “The Reverse Warburg Effect,”Citation12–Citation16 as the conventional Warburg Effect was thought to be confined to cancer cells and not to occur in stromal fibroblasts. Importantly, these energyrich metabolites can then be transferred to adjacent cancer cells, where they fuel oxidative mitochondrial metabolism, the TCA cycle and ATP production, as well as additional mitochondrial biogenesis.Citation12–Citation16 This results in a unilateral, vectorial, net energy transfer from the catabolic tumor stroma to anabolic cancer cells. Thus, autophagy in the tumor stroma results in a negative energy balance, in favor of the cancer cells.Citation11 We have termed this new mechanism underlying tumor-stroma co-evolution, “The Autophagic Tumor Stroma Model of Cancer Cell Metabolism”Citation11().
Using a variety of molecular and genetic approaches, we showed that oxidative stress drives the induction of HIF1alpha and NFκB-activation in cancer associated fibroblasts, leading to the onset of autophagy and mitophagy.Citation7,Citation8,Citation11,Citation15 For example, we see that cancer cells activate HIF1- and NFκB-responsive luciferase reporters in adjacent stromal fibroblasts.Citation8 Moreover, genetic activation of HIF1alpha or NFκB in stromal cells was sufficient to confer the cancer-associated fibroblasts phenotype, resulting in 2–3-fold increases in epithelial tumor growth,Citation17 as well as the onset of local lymph node metastasis, without any detectable increases in tumor angiogenesis.Citation17 Finally, direct administration of energy-rich metabolites (such as L-lactate and 3-hydroxy-butyrate) was sufficient to increase tumor volume and metastasis,18 without any increases in tumor angiogenesis.Citation18
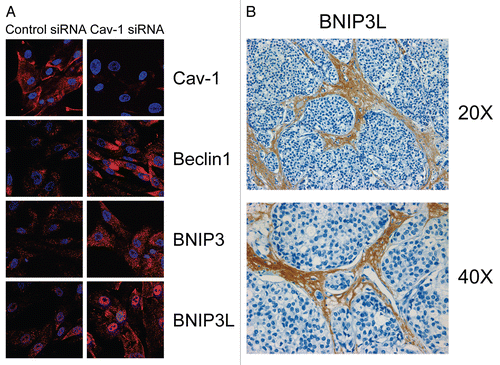
Importantly, we validated that acute loss of Cav-1, using an siRNA approach, is indeed sufficient to induce ROS production and mitochondrial dysfunction in human fibroblastsCitation7 (). Similarly, acute loss of Cav-1, using the same approach, is sufficient to activate autophagy and mitophagyCitation8 (). Under these conditions, we see the upregulation of both autophagy (Beclin1) and mitophagy (BNIP3 and BNIP3L) markersCitation8 (). Thus, oxidative stress and autophagy acutely downregulate Cav-1 by autophagic/lysosomal degradationCitation6 and a loss of Cav-1 further exacerbates these stressors, driving more oxidative stress, mitochondrial dysfunction and autophagy—in a feed-forward fashion.Citation7,Citation8 Consistent with these findings, human breast cancers that lack stromal Cav-1 show the stromal overexpression of markers for (1) aerobic glycolysis (PKM2/LDH-B)Citation12,Citation18 and (2) mitophagy (BNIP3L)Citation8 (). The selective stromal expression of PKM2 and LDH-B was also validated using a xenograft model employing GFP-tagged MDA-MB-231 cells,Citation7,Citation17 which are an aggressive metastatic triple-negative/basal breast cancer cell line.
One mechanism by which an acute loss of Cav-1 generates oxidative stress is via increased NO (nitric oxide) production,Citation7,Citation15 as Cav-1 is a natural endogenous inhibitor of NOS (nitric oxide synthase).Citation19 Increased NO production, in turn, generates ROS and oxidative stress via mitochondrial dysfunction, by inhibiting mitochondrial oxidative phosphorylation via Complex I and Complex IV. Interestingly, we have previously shown that a loss of Cav-1 induces the tyrosine nitration of mitochondrial Complex I,Citation15 consistent with our hypothesis. Furthermore, metabolic restriction with glycolysis (2-DG) and Complex I (metformin) inhibitors is synthetically lethal with a Cav-1 deficiency in mice.Citation15 Thus, Cav-1 (-/-) null mice have a severely reduced mitochondrial reserve capacity.Citation15
As predicted based on the above studies, mammary fat pads derived from Cav-1 (-/-) null mice show the upregulation of markers for hypoxia, oxidative stress and autophagy.Citation8,Citation11 Metabolomic analysis of the Cav-1 (-/-) mammary fat pad demonstrates the upregulation of nearly 100 metabolites, consistent with a major catabolic phenotype.Citation11 Their metabolic profile is consistent with constitutive oxidative stress and mitochondrial dysfunction, with the elevation of markers of oxidative stress (ADMA) and mitochondrial dysfunction (3-hydroxy-butyrate).Citation11 Thus, ADMA and ketone production are features of a lethal tumor microenvironment.Citation11 This may explain why patients with diabetes, that show increased levels of ADMA and ketones, as well as oxidative stress and autophagy, have an increased risk for the development of a variety of epithelial cancer sub-types.Citation11
Lastly, an informatics analysis of the unbiased transcriptional profiles of tumor stroma isolated from human breast cancers shows the overexpression of markers of oxidative stress, hypoxia and autophagy, as well as lysosomal enzymes.Citation11,Citation16 This provides further translational validation of the “Autophagic Tumor Stroma Model of Cancer.”Citation11 Many of these markers are also associated with breast cancer tumor recurrence and metastasis. In fact, the transcriptional profile of autophagic Cav-1 (-/-) null mesenchymal stem cells is most closely related to (1) the primary tumor stroma derived from breast cancer patients that have undergone metastasis; and (2) the brains of patients with Alzheimer disease.Citation16 Consistent with these findings, the pathogenesis of Alzheimer disease is thought to involve ROS and NO over-production (oxidative stress) and mitochondrial dysfunction, as well as autophagy.Citation16 In fact, the Alzheimer disease gene list is also most closely related to the tumor stroma isolated from patients with metastatic breast cancer,Citation16 as compared to patients without metastatic disease.
Implications for Cachexia and Effective Cancer Chemotherapy
The “Autophagic Tumor Stroma Model of Cancer” also has important implications for understanding cancer-associated cachexia and improving cancer chemotherapy. Cancerassociated cachexia, also known as systemic wasting, occurs in patients with advanced and metastatic cancer.Citation20–Citation24 Cachexia is due to increased energy expenditures and an increased metabolic rate, which results in a negative energy balance.Citation20–Citation24 Thus, we believe that autophagy in the tumor stroma represents the local microscopic equivalent of cancer-associated cachexia and may explain how cachexia could start locally and then spread systemically. In accordance with this hypothesis, others have previously suggested that cancer-associated cachexia is caused by oxidative stress and is responsive to treatment with anti-oxidants.Citation25–Citation35
Regarding cancer chemotherapy, we propose that anti-oxidants and autophagy/lysosomal inhibitors could be used systemically to halt autophagy in the tumor stroma, effectively cutting off the tumor's fuel supply. In essence, anti-oxidants and autophagy inhibitors would metabolically uncouple the autophagic tumor stroma from adjacent anabolic “parasitic” cancer cells. Thus, new clinical trials with anti-oxidants (such as N-acetyl-cysteine, metformin and quercetin), as well as lysosomal inhibitors (chloroquine) may be warranted. Importantly, these drugs are either available over-the-counter (OTC) as dietary supplements or are FDA-approved drugs.
Understanding Why Anti-Angiogenic Drugs Paradoxically Promote Tumor Progression and Metastasis
Finally, our current model of tumor-stroma co-evolution may also explain the clinical failure of anti-angiogenic drugs, such as Bevacizumab (Avastin). Effective anti-angiogenic therapies induce hypoxia in the tumor stroma.Citation36–Citation49 Hypoxia in the tumor stroma then induces autophagy. We have shown that hypoxia and autophagy in the tumor stromal microenvironment are the necessary “ingredients” for driving the development of a “lethal tumor stromal phenotype.” Thus, autophagy in the tumor stroma provides a new escape mechanism for cancer cells during anti-angiogenic therapy,Citation14 which could then further drive more aggressive tumor progression and metastasis.Citation14 This explains how anti-angiogenic therapy could convert a non-aggressive tumor into a lethally aggressive metastatic cancer, by fueling tumor-stroma co-evolution.
Figures and Tables
Figure 1 Deciphering tumor-stroma co-evolution: the autophagic tumor stroma model of cancer cell metabolism. Cancer cells induce oxidative stress in adjacent stromal fibroblasts. Then, the resulting oxidative stress in cancer-associated fibroblasts helps drive tumor-stroma co-evolution, by randomly mutagenizing cancer cells, while protecting them against apoptosis and providing them with abundant recycled nutrients and chemical building blocks via autophagy. This results in a net energy transfer from the autophagic tumor stroma to the anabolic “hungry” cancer cells. Thus, stromal oxidative stress and autophagy function(s) as a “battery” to drive tumor-stroma co-evolution and “fuel” oxidative mitochondrial metabolism in cancer cells. A+ (positive), increased autophagy/mitophagy in cancer associated fibroblasts; A− (negative), decreased autophagy/mitophagy in epithelial cancer cells. AR (resistant), denotes the successful evolution of autophagy-resistant cancer cells, due to genetic silencing or deletion of required autophagy genes, such as Beclin1.
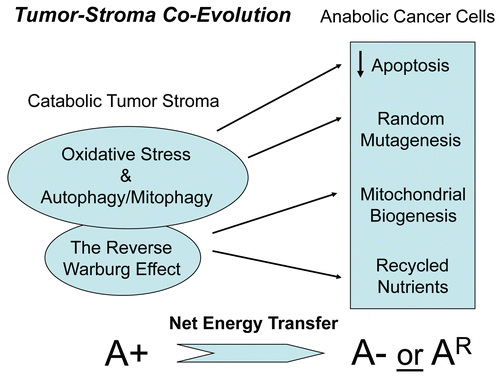
Figure 2 Acute knock-down of Cav-1 in human stromal fibroblasts increases ros production and negatively affects mitochondrial activity. (Upper) Cav-1 knock-down induces ROS production. CM-H2DCFDA staining (green) was performed on hTER T-fibroblasts treated with Cav-1 siRNA (right) or control siRNA (left). Cells were counterstained with Hoechst nuclear stain (blue). Samples were then immediately imaged using a 488 nm excitation wavelength. Note that Cav-1 knock-down greatly promotes ROS generation. Importantly, images were acquired using identical exposure settings. Original magnification, 20x. (Lower) Cav-1 knock-down decreases mitochondrial activity. hTERT-fibroblasts were treated with Cav-1 siRNA (right) or control siRNA (left). Then, functional mitochondria with active membrane potential were visualized using MitoTracker staining (red). DAPI was used to stain nuclei (blue). Note that transient Cav-1 knock-down greatly decreases mitochondrial activity. Importantly, paired images were acquired using identical exposure settings. Original magnification, 63x. Images were reproduced from references,Citation7 and Citation7 with permission.
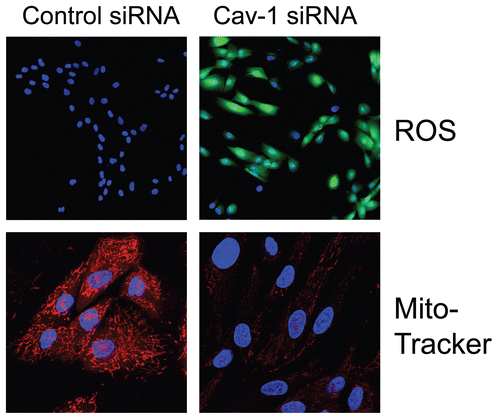
Figure 3 Acute knock-down of Cav-1 in human stromal fibroblasts activates autophagy and mitophagy: implications for human breast cancer. (A) Acute loss of Cav-1 increases the expression of autophagic markers. hTER T-fibroblasts were treated with Cav-1 siRNA or control (CTR) siRNA. Cells were fixed and immuno-stained with antibodies against Beclin1, BNIP3 and BNIP3L. DAPI was used to visualize nuclei (blue). Importantly, paired images were acquired using identical exposure settings. Original magnification, 40x. Note that acute Cav-1 knockdown is sufficient to greatly increase the expression levels of all the autophagy/mitophagy markers we examined. (B) BNIP3L is highly increased in the stroma of human breast cancers that lack stromal Cav-1. Paraffin-embedded sections of human breast cancer samples lacking stromal Cav-1 were immuno-stained with antibodies directed against BNIP3L. Slides were then counter-stained with hematoxylin. Note that BNIP3L is highly expressed in the stromal compartment of human breast cancers that lack stromal Cav-1. Original magnification, 20x and 40x, as indicated. Images were reproduced from the references Citation7 and Citation8, with permission.
Acknowledgements
F.S. and her laboratory were supported by grants from the W.W. Smith Charitable Trust, the Breast Cancer Alliance (BCA) and a Research Scholar Grant from the American Cancer Society (ACS). M.P.L. was supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660) and the Susan G. Komen Breast Cancer Foundation. A.K.W. was supported by a Young Investigator Award from Breast Cancer Alliance, Inc., and a Susan G. Komen Career Catalyst Grant. R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072 and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L. and F.S.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
References
- Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An Absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol 2009; 174:2023 - 2034
- Witkiewicz AK, Dasgupta A, Sammons S, Er O, Potoczek MB, Guiles F, et al. Loss of stromal caveolin-1 expression predicts poor clinical outcome in triple negative and basal-like breast cancers. Cancer Biol Ther 2010; 10:135 - 143
- Witkiewicz AK, Casimiro MC, Dasgupta A, Mercier I, Wang C, Bonuccelli G, et al. Towards a new “stromal-based” classification system for human breast cancer prognosis and therapy. Cell Cycle 2009; 8:1654 - 1658
- Witkiewicz AK, Dasgupta A, Nguyen KH, Liu C, Kovatich AJ, Schwartz GF, et al. Stromal caveolin-1 levels predict early DCIS progression to invasive breast cancer. Cancer Biol Ther 2009; 8:1167 - 1175
- Di Vizio D, Morello M, Sotgia F, Pestell RG, Freeman MR, Lisanti MP. An absence of stromal caveolin-1 is associated with advanced prostate cancer, metastatic disease and epithelial Akt activation. Cell Cycle 2009; 8:2420 - 2424
- Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010; 9:2423 - 2433
- Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010; 9:3256 - 3276
- Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFγB activation in the tumor stromal microenvironment. Cell Cycle 2010; 9:3515 - 3533
- Eng C, Leone G, Orloff MS, Ostrowski MC. Genomic alterations in tumor stroma. Cancer Res 2009; 69:6759 - 6764
- Holliday C, Rummel S, Hooke JA, Shriver CD, Ellsworth DL, Ellsworth RE. Genomic instability in the breast microenvironment? A critical evaluation of the evidence. Expert Rev Mol Diagn 2009; 9:667 - 678
- Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010; 9:3485 - 3505
- Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009; 8:3984 - 4001
- Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, et al. The reverse Warburg effect: Glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle 2010; 9:1960 - 1971
- Migneco G, Whitaker-Menezes D, Chiavarina B, Castello-Cros R, Pavlides S, Pestell RG, et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: Evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010; 9:2412 - 2422
- Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of Stromal Caveolin-1 Leads to Oxidative Stress, Mimics Hypoxia and Drives Inflammation in the Tumor Microenvironment, Conferring the “Reverse Warburg Effect”: A Transcriptional Informatics Analysis with Validation. Cell Cycle 2010; 9:2201 - 2219
- Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer's disease and “Neuron-Glia Metabolic Coupling”. Aging 2010; 2:185 - 199
- Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1alpha functions as a tumor promoter in cancer associated fibroblasts and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle 2010; 9:3534 - 3551
- Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010; 9:3506 - 3514
- Garcia-Cardena G, Martasek P, Siler-Masters BS, Skidd PM, Couet JC, Li S, et al. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin: Functional Significance of the NOS caveolin binding domain in vivo. J Biol Chem (Communication) 1997; 272:25437 - 25440
- Warnold I, Lundholm K, Schersten T. Energy balance and body composition in cancer patients. Cancer Res 1978; 38:1801 - 1807
- Bosaeus I, Daneryd P, Svanberg E, Lundholm K. Dietary intake and resting energy expenditure in relation to weight loss in unselected cancer patients. Int J Cancer 2001; 93:380 - 383
- Fouladiun M, Korner U, Bosaeus I, Daneryd P, Hyltander A, Lundholm KG. Body composition and time course changes in regional distribution of fat and lean tissue in unselected cancer patients on palliative care—correlations with food intake, metabolism, exercise capacity and hormones. Cancer 2005; 103:2189 - 2198
- Norton JA, Peacock JL, Morrison SD. Cancer cachexia. Crit Rev Oncol Hematol 1987; 7:289 - 327
- Barber MD, Ross JA, Fearon KC. Cancer cachexia. Surg Oncol 1999; 8:133 - 141
- Droge W, Hack V, Breitkreutz R, Holm E, Shubinsky G, Schmid E, et al. Role of cysteine and glutathione in signal transduction, immunopathology and cachexia. Biofactors 1998; 8:97 - 102
- Hack V, Schmid D, Breitkreutz R, Stahl-Henning C, Drings P, Kinscherf R, et al. Cystine levels, cystine flux and protein catabolism in cancer cachexia, HIV/SIV infection and senescence. FASEB J 1997; 11:84 - 92
- Droge W, Gross A, Hack V, Kinscherf R, Schykowski M, Bockstette M, et al. Role of cysteine and glutathione in HIV infection and cancer cachexia: therapeutic intervention with N-acetylcysteine. Adv Pharmacol 1997; 38:581 - 600
- Ushmorov A, Hack V, Droge W. Differential reconstitution of mitochondrial respiratory chain activity and plasma redox state by cysteine and ornithine in a model of cancer cachexia. Cancer Res 1999; 59:3527 - 3534
- Mantovani G, Maccio A, Madeddu C, Mulas C, Massa E, Astara G, et al. Phase II study of subcutaneously administered interleukin-2 in combination with medroxyprogesterone acetate and antioxidant agents as maintenance treatment in advanced cancer responders to previous chemotherapy. Oncol Rep 2002; 9:887 - 896
- Mantovani G, Maccio A, Madeddu C, Mura L, Gramignano G, Lusso MR, et al. Antioxidant agents are effective in inducing lymphocyte progression through cell cycle in advanced cancer patients: Assessment of the most important laboratory indexes of cachexia and oxidative stress. J Mol Med 2003; 81:664 - 673
- Mantovani G, Maccio A, Madeddu C, Mura L, Massa E, Gramignano G, et al. Reactive oxygen species, antioxidant mechanisms and serum cytokine levels in cancer patients: impact of an antioxidant treatment. J Cell Mol Med 2002; 6:570 - 582
- Mantovani G, Madeddu C, Gramignano G, Lusso MR, Mocci M, Massa E, et al. Subcutaneous interleukin-2 in combination with medroxyprogesterone acetate and antioxidants in advanced cancer responders to previous chemotherapy: phase II study evaluating clinical, quality of life and laboratory parameters. J Exp Ther Oncol 2003; 3:205 - 219
- Marin-Corral J, Fontes CC, Pascual-Guardia S, Sanchez F, Olivan M, Argiles JM, et al. Redox balance and carbonylated proteins in limb and heart muscles of cachectic rats. Antioxid Redox Signal 2010; 12:365 - 380
- Barreiro E, de la Puente B, Busquets S, Lopez-Soriano FJ, Gea J, Argiles JM. Both oxidative and nitrosative stress are associated with muscle wasting in tumourbearing rats. FEBS Lett 2005; 579:1646 - 1652
- Argiles JM. Cancer-associated malnutrition. Eur J Oncol Nurs 2005; 9:39 - 50
- Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003; 3:347 - 361
- Steeg PS. Angiogenesis inhibitors: motivators of metastasis?. Nat Med 2003; 9:822 - 823
- Grepin R, Pages G. Molecular mechanisms of resistance to tumour anti-angiogenic strategies. J Oncol 2010; 2010:835680
- Mancuso MR, Davis R, Norberg SM, O'Brien S, Sennino B, Nakahara T, et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest 2006; 116:2610 - 2621
- Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer 2002; 2:38 - 47
- Rapisarda A, Melillo G. Role of the hypoxic tumor microenvironment in the resistance to anti-angiogenic therapies. Drug Resist Updat 2009; 12:74 - 80
- Ide T, Kitajima Y, Miyoshi A, Ohtsuka T, Mitsuno M, Ohtaka K, et al. Tumor-stromal cell interaction under hypoxia increases the invasiveness of pancreatic cancer cells through the hepatocyte growth factor/c-Met pathway. Int J Cancer 2006; 119:2750 - 2759
- Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009; 15:232 - 239
- Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009; 15:35 - 44
- Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer 2009; 9:239 - 252
- Kerbel RS. Therapeutic implications of intrinsic or induced angiogenic growth factor redundancy in tumors revealed. Cancer Cell 2005; 8:269 - 271
- Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005; 8:299 - 309
- Peinado H, Cano A. A hypoxic twist in metastasis. Nat Cell Biol 2008; 10:253 - 254
- Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol 2008; 10:295 - 305