Abstract
Abstract An inactivating germline mutation in BRCA2 is the most common known genetic basis for familial pancreatic cancer (FPC), accounting for 5-10% of inherited cases. A genetically engineered mouse model of pancreatic ductal adenocarcinoma (PDAC) arising on the backdrop of Brca2 deficiency is likely to elucidate valuable diagnostic and therapeutic insights for FPC. Both Brca2 alleles were conditionally deleted during development within the pancreatic epithelium by generating Pdx1-Cre; Brca2f/f (“CB”) mice; in addition, triple transgenic Pdx1-Cre; Brca2f/f; LSL-Trp53R172H (“CBP”) mice were generated, in order to determine the impact of p53 deregulation on Brca2-deficient carcinogenesis. Both “CB” and “CBP” mice developed non-invasive ductal precursor lesions (murine pancreatic intraepithelial neoplasia or mPanIN), although these were observed at an earlier time point (5 versus 8 months) and with higher prevalence in “CBP” mice. A minority of “CB” mice (15%) developed invasive and metastatic PDAC at a latency of 15 months or greater; in contrast, “CBP” mice of comparable age uniformly developed PDAC with variable histological features. Mortality in the absence of neoplasia in "CB" and "CBP" mice was associated with profound loss of pancreatic parenchyma, consistent with progressive elimination of Brca2-deficient cells. Widespread DNA damage, as evidenced by overexpression of the phosphorylated histone H2AXSer139, was observed in the non-neoplastic exocrine pancreas, as well as in the mPanIN and PDAC lesions of Brca2-deficient mice, independent of p53 status. Loss of Brca2 function predisposes the exocrine pancreas to profound DNA damage, and the frequency of invasive neoplasia is accentuated by the concomitant deregulation of p53.
See commentary:
Brca2 deficiency and Trp53 deregulation in pancreatic cancer: Implications for therapeutic targeting
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an almost uniformly lethal disease and is among the most dismal of human malignancies to date.Citation1 In recent years, genetically engineered mouse models that recapitulate the multistep progression of the cognate human disease have been successfully created.Citation2,Citation3 These models have been crucial for a better understanding of the cellular and molecular pathogenesis of PDAC, and for the preclinical development of diagnostic and therapeutic targets that can be translated into the clinic.Citation4 Most of the existing models recapitulate the genetics of sporadic (i.e., non-familial) PDAC, largely because the genetic basis for familial pancreatic cancer (FPC) remains a matter of investigation. Till date, germline mutations that predispose an individual to developing PDAC in their lifetime have been identified in only a minority (∼20%) of FPCs overall, and include most commonly, inactivating mutations of BRCA2 and other members of the Fanconi Anemia gene family such as PALB2 (∼5–10%), as well as STK11/LKB1, CDKN2A/p16, PRSS1 and the DNA mismatch repair genes.Citation5–Citation7 An interest in understanding the genetic basis for FPC arises not only because it provides an avenue for developing a tangible gene test for unaffected family members, but also as a strategy to implement molecularly targeted therapies. In fact, such synthetic lethal pharmacological approaches have been used with considerable success in familial breast and ovarian cancers that arise on the backdrop of BRCA1/2 deficiency,Citation8 and as recently demonstrated from one of the institutions in this study (JHSOM), this synthetic lethal approach can also be successfully applied to familial PDAC, with striking remission of advanced disease.Citation9
The motivation for this study was to develop a mouse model that recapitulated the genetics of the most common known alteration in human FPC, i.e., bi-allelic loss of BRCA2 in the pancreas, in a patient with an existing hemizygous germline deletion. Apriori studies in human pancreata have confirmed that loss of the second BRCA2 allele is, in fact, present in the precursor lesions and cancers that arise in the setting of germline BRCA2 deficiency.Citation10 We were able to conditionally delete both Brca2 alleles in the murine pancreatic epithelium by Cre-mediated recombination during development within the Pdx1 expression domain. The resulting Pdx1-Cre; Brca2f/f (henceforth called CB) mice develop invasive and metastatic adenocarcinomas in a minority of animals (∼15%) at relatively long latencies (at or greater than 15 months of age). Notably, the disease penetrance at the comparable ages is markedly enhanced (100% of cohort) when p53 function is concomitantly deregulated in the setting of Brca2 deficiency in Pdx1-Cre; Brca2f/f; LSL- Trp53R172H (hence-forth called CBP) mice. Both CB and CBP mice that succumb in the absence of tumorigenesis demonstrate profound loss of pancreatic parenchyma, suggesting exocrine pancreatic insufficiency as the basis for their mortality. Both CB and CBP mice also demonstrate evidence of widespread DNA damage in the pancreas, consistent with the role of Brca2 as a genome maintenance protein.Citation11 Our data indicate that elimination of DNA-damage prone Brca2-deficient cells is the preferred course of action in the mammalian pancreas, but deregulation of p53 function in CBP mice likely attenuates this process, and allows surviving cells to progress to invasive cancer at a higher frequency. The relatively long latency for tumor formation in both CB and CBP mice are in accordance with the late-onset PDAC occurring in patients with germline BRCA2 mutations, and underscore the distinction between a genome maintenance gene and a traditional tumor suppressor like CDKN2A or SMAD4. These findings have important ramifications for FPC arising in individuals with germline BRCA2 mutations, and provides an animal model for testing various therapeutic modalities for disease prevention or elimination in FPC.Citation12
Results
Abrogation of Brca2 function leads to depletion of pancreatic parenchyma and early mortality in the absence of invasive neoplasia.
Both CB and CBP cohorts were associated with early mortality in subsets of mice, with death occurring as early as two months, and continuing with variable latencies throughout the period of observation. Overall, the median survival of the CB mice (N = 25) and CBP mice (N = 33) were 454 and 375 days, respectively, a difference that did not reach statistical significance (p = 0.085) (). Although logistically it was not possible to perform timely necropsy on all mice that experienced spontaneous mortality, on multiple occasions when the pancreata were retrieved in a condition suitable for histopathology, we did not find evidence of invasive neoplasia, but rather observed a profound loss of exocrine pancreatic parenchyma, with near total depletion of acinar elements (). The pancreata in such mice often demonstrated extensive adipose (fatty) replacement (), with residual islets prominently embedded in the fat. On occasion, a large portion of the pancreas was replaced by hemorrhagic cysts, the lining of which were comprised of low cuboidal epithelium (). These are likely degenerated cysts, arising on the backdrop of pancreatic exocrine dropout, as suggested by the innocuous nature of the lining epithelium as well as the absence of oncogenic signaling pathways observed in neoplastic tissues (see below). In occasional CB mice that survived for relatively longer periods of time (>1 year or beyond), we observed a remarkable replacement of the pancreatic parenchyma by confluent non-invasive precursor lesions (murine pancreatic intraepithelial neoplasia or mPanIN), with essentially no discernible acinar tissue within these mPanIN strips (). Rigorous histopathological examination in these mice with spontaneous mortality failed to reveal evidence of invasive neoplasms or other organ system pathologies, suggesting that the cause of death was likely to be exocrine insufficiency caused by the striking depletion of acinar tissue.
Murine pancreatic intraepithelial neoplasia (mPanINs) develop in the absence of Brca2 function and their onset is accelerated by p53 deregulation.
In addition to the survival cohort described above, we generated separate observation cohorts of CB and CBP mice that underwent euthanasia and necropsy at timed intervals (monthly, beginning at 2 months, up to 10 months of age). Pancreata were harvested in these mice with the specific intent of cataloging ductal precursor lesions or invasive neoplasia, and an effort was made to have at least five mice for each genotype at each of the monthly harvests. As seen in , both CB and CBP mice demonstrated the full spectrum of mPanIN lesions, although these were initiated at an earlier time point (5 vs. 8 months, respectively) in the absence of p53 function (). In addition to full-fledged mPanINs, lesions of acinar ductal metaplasia (ADM) were also observed in the surrounding pancreata ( and D), a not unexpected finding given these are considered “precursors to the precursors” in the multistep progression model of murine PDAC. Of the 45 mice in each cohort undergoing histopathological examination, none of the CB mice and only one (2%) of the CBP mice (aged 10 months) had evidence of invasive cancer.
An immunohistochemical stain for Brca2 confirmed absence of expression in the mPanIN epithelium, as well as in the epithelium lining of the cystic lesions described above (), consistent with epithelial specific developmental Pdx1 expression. In contrast, robust expression was retained in the pancreatic mesenchyme, where Cre-mediated recombination is not expected.
PDAC development occurs in CB and CBP mice at comparable latencies but deregulation of p53 enhances the frequency of invasive neoplasia.
No invasive neoplasms were seen in either the CB or CBP mice (with a single exception in the observation cohort) up to the age of 15 months. Of 13 CB mice that were followed between 15 months and 24 months, two (15%) developed invasive and metastatic PDAC. At necropsy, widespread intra-abdominal metastases were evident (), and explants from the tumors readily grew as subcutaneous xenografts in immunocompromised mice (). Histologically, the neoplasms were comprised of moderately differentiated adenocarcinomas, with evidence of intracellular mucin vacuoles within neoplastic cells, a feature often observed in human PDACs ( and D).Citation13 Notably, we did not find evidence of acceleration in the onset of invasive cancers in CBP mice, although there was a sharp increase in their frequency between 15 and 17 months of age, with five of five (100%) of mice succumbing to widely metastatic tumors during this time frame (). In addition to local invasion into neighboring tissues, the tumors also metastasized to the liver, diaphragm and kidneys. At the terminal stages, it was not uncommon to find associated cachexia and malignant ascites, both features observed in advanced human PDAC. Hemorrhagic cysts were observed in two mice. Histologically, a variety of morphological features were observed (). Thus, two of the tumors were comprised of moderately differentiated adenocarcinomas, wherein the neoplastic glands were embedded in a robust desmoplastic response, akin to what is observed in human PDAC ( and B). Two cases had a biphenotypic appearance, with both adenocarcinoma and sarcomatoid carcinoma components (); areas of sharp transition between the two components were readily visible (). The poorly differentiated sarcomatoid carcinoma was comprised of pleomorphic cells with brisk mitotic activity (). Finally, one of the tumors was an anaplastic carcinoma with bizarre giant cells, also a subtype that is occasionally seen in human PDAC ().Citation13
Brca2 depletion leads to increased DNA damage.
The Brca2 protein functions in “genome maintenance”, mainly due to its central role in DNA double-strand break repair through homologous recombination.Citation11 Phosphorylation of histone H2AX at the Ser139 residue represents an early response to DNA damage.Citation14,Citation15 Therefore, in order to assess the extent of DNA damage we stained pancreatic tissues obtained from Brca2-deficient pancreata for pH2AXSer139 (). In age-matched control mice that retained hemizygous Brca2 function (Pdx1-Cre; Brca2,f/wt), we observed minimal pH2AXSer139 expression (). In contrast, profound upregulation of pH2AXSer139 was observed in CB mice, specifically within the acinar component ( and C). Unexpectedly, we found minimal expression of pH2AXSer139 in the ductal epithelium (), which suggests that, despite the absence of Brca2 function, this epithelial compartment might be recalcitrant to DNA damage, or be exposed to fewer genotoxic stressors than the acinar compartment. In invasive cancers arising in these mice (a CBP mouse tumor is illustrated in ), there was continued elevated expression of pH2AXSer139, consistent with ongoing DNA damage.
Neoplastic lesions arising in the setting of Brca2 deficiency lack secondary Kras mutations but express Hedgehog (Hh) ligand.
Somatic mutations of the KRAS gene are one of the most common genetic alterations in PDAC, and their presence even in non-invasive precursor lesions renders them as an “early” abnormality.Citation16 Biologically relevant mouse models of mPanINs and PDAC were first generated by expression of a mutant KrasG12D allele from its endogenous locus,Citation17,Citation18 further underscoring the importance of this oncogene to ductal neoplasia. Therefore, we examined whether the mPanIN and PDACs arising in CB and CBP mice might harbor secondary Kras mutations. Sanger sequencing of microdissected lesions from a subset of mPanINs and PDAC failed to demonstrate mutations of codons 12, 13 or 61 of the Kras gene (data not shown).
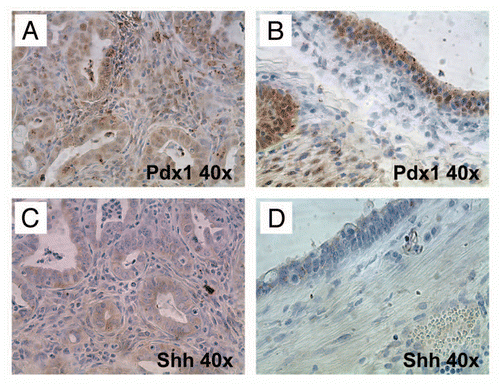
Finally, we assessed whether the neoplastic epithelium expressed the Hh ligand, sonic Hh (Shh), which is a feature of most human PDACs,Citation19,Citation20 using the epithelium lining of the aforementioned degenerated cysts as controls (). We observed that while nuclear Pdx1 was expressed by both epithelial types, only the neoplastic epithelium expressed Shh ligand.
Discussion
Germline mutations of BRCA2 account for ∼5–10% of cases with an inherited predisposition to PDAC.Citation7,Citation21 In addition, while somatic BRCA2 mutations are uncommon in sporadic PDAC (∼1%), this frequency rises to ∼10% of cancers within certain ethnic backgrounds, such as individuals of Ashkenazi-Jewish heritage. Besides the obvious implications vis-à-vis genetic counseling and cancer screening for BRCA2 germline mutation carriers,Citation22 it is now established that tumors with bi-allelic loss of BRCA2 (or related Fanconi Anemia pathway genes) also respond dramatically to certain classes of anti-neoplastic agents, such as mitomycin C, cisplatin and the poly (ADP-ribose) (PARP) inhibitor, Olaparbib.Citation8,Citation23 In fact, the utility of this “synthetic lethal” strategy has recently been demonstrated in a rather dramatic manner in a case of advanced PDAC.Citation9 The creation of a genetically engineered model of BRCA2 deficiency in the pancreas would provide an in vivo platform for designing improved diagnostic and therapeutic strategies for this genetically defined cancer subset.
Herein, we describe a mouse model of “familial pancreatic cancer” generated on the backdrop of conditional Brca2 deficiency, with or without concomitant deregulation of p53 function. Our studies demonstrate that the most prevalent response to bi-allelic deletion of Brca2 (and the secondarily accrued DNA damage) might be elimination of the affected epithelial cells, which accounts for the profound loss of exocrine pancreatic parenchyma observed in mice that succumb without developing invasive cancer. Loss of pancreatic parenchyma is observed in both CB and CBP mouse cohorts, suggesting that a p53-independent cell death pathway, possibly associated with mitotic catastrophe, triggers this response.Citation24,Citation25 In a mouse pancreas, pan-epithelial Cre-mediated recombination during development within the Pdx1 expression domain results in a large pool of Brca2-deficient cells. Needless to add, this scenario is not recapitulated in an individual with a germline BRCA2 defect, wherein the somatic “second hit” occurs in a more restricted nidus of cells. Therefore, unlike in the current mouse model, pancreatic insufficiency is not a phenotype commonly observed in humans with a germline BRCA2 mutation. Nonetheless, a BRCA2-deficient cell faces considerable selection pressure for elimination in vivo, which likely explains the relatively low-penetrance (no more than 5–10%) for lifetime PDAC incidence in BRCA2 mutation carriers.Citation21,Citation26
In mice with sufficient preservation of pancreatic function, we observed emergence of mPanIN lesions, beginning at 5 months in CBP mice and in 8 months in the CB cohort. Eventually, invasive and metastatic PDAC developed in both genotypes, and was observed at comparable latencies (beginning at ∼15 months of age), although the CBP mice had a greater frequency of tumor formation beyond 15 months. The relatively long latency to tumorigenesis is also observed in germline BRCA2 carriers, whose median age of onset for PDAC is no different from that in sporadic (non-familial) patients.Citation7,Citation21 The comparable latencies between the two cohorts underscore the role of Brca2 as a genome maintenance protein, rather than a “classic” tumor suppressor controlling cell cycle parameters, and the need for accumulation of additional genetic events that must occur prior to neoplastic transformation. What role does the Trp53R172H allele play in neoplastic progression? One possibility is that the elimination of Brca2-deficient cells that occurs with intact p53 function is attenuated (albeit not blocked) with p53 deregulation, such that a larger pool of cells with damaged DNA is available for propagation. Another postulate, not mutually exclusive, is that the “gain-of-function” p53 protein encoded by the Trp53R172H allele facilitates genomic instability, and adds to the genotoxic burden of Brca2 loss.Citation27,Citation28 For example, the karyotype of PDACs arising in compound heterozygous Pdx1-Cre;LSL-KrasG12D; LSL-Trp53R172H mice (“KPC” mice) is consistent with widespread chromosomal instability,Citation29 a feature not typically observed in other genetic cooperation models for PDAC.Citation18,Citation30 Irrespective of the precise mechanism, the concomitant deregulation of p53 and Brca2 results in a greater frequency of invasive PDAC than what is observed in CB mice alone.
In the absence of Brca2 function, we find robust expression of the phosphorylated histone pH2AXSer139, a surrogate measure of DNA damage, within the exocrine pancreata, as well as in the resulting neoplastic alterations. Surprisingly, while acinar cells demonstrate widespread pH2AXSer139 expression, the native ductal epithelium shows minimal labeling. This leads one to speculate whether the major, if not entire, contribution to the neoplastic phenotype is derived from non-ductal elements, most likely the acinar tissues. Notably, prior studies, including from our group,Citation31,Citation32 have shown the ability of mature acinar cells to spontaneously develop mPanINs and/or PDAC in the appropriate oncogenic context, and therefore, the development of “ductal” adenocarcinoma from non-ductal elements in the pancreas is not entirely unexpected. A better understanding of the cell-of-origin of Brca2-deficient pancreatic neoplasia will require rigorous lineage tracing experiments and the use of compartment-specific Cre “driver” lines. Another feature of interest was the absence of secondary Kras gene mutations at codons 12, 13 or 61 in the resulting mPanINs and PDACs of CB and CBP mice. The emerging consensus has been that expression of a mutant Kras allele might be a requirement for developing PDACs along the mPanIN multistep progression cascade, with any cooperating genetic lesion (such as loss of Cdkn2a/p16 or Smad4, or misexpression of Gli2 oncoprotein etc.),Citation18,Citation30,Citation33–Citation35 essentially altering the natural history and penetrance of the “Ras-induced” invasive neoplasia phenotype. However, we were unable to demonstrate Kras mutations in microdissected mPanIN or PDAC lesions, suggesting that, on occasion, mutations of this oncogene may be dispensable (see further discussion below). It is important to note that we do not have the information to exclude somatic mutations in downstream Ras effector genes, such BRAF, PI3KCA, AKT, etc., which are altered in minor subsets of human PDACs,Citation36,Citation37 in the murine lesions.
Finally, this discussion would not be complete without an acknowledgement of two elegant reports on Brca2-deficient pancreatic neoplasia that were recently published, while our own work was nearing completion.Citation38,Citation39 In the study by Skoulidis et al. the authors generated a model with expression of a truncated Brca2 (Brca2Tr) allele in all somatic tissues, and found that, irrespective of p53 status, pancreatic haploinsufficiency of Brca2 was sufficient to cooperate with mutant KrasG12D and induce PDAC in mice. The impact of bi-allelic Brca2 deletion, on the other hand, was largely influenced by the functional status of p53. Thus, in the presence of intact p53 function, most of the recombined epithelial cells are eliminated leading to pancreatic insufficiency, and PDAC is only observed in surviving mice after a relatively long latency, a finding that essentially mimics our own observation in the CB cohort. On the contrary, bi-allelic Brca2 loss of function, when occurring concomitantly with mutant Kras expression plus p53 deregulation, results in rapid progression to PDAC. This latter observation is in contrast to our own study, where the latency of tumor onset is not significantly affected by p53 status, and almost certainly reflects the absence of ongoing “oncogene-induced DNA damage” in the CBP mice that would be caused by co-expression of mutant Kras.Citation40,Citation41 As stated above, germline BRCA2 mutation carriers are not prone to develop PDAC at an earlier age of onset than sporadic cancers, a phenomenon that contrasts with well known “Knudsonian” syndromes like familial retinoblastoma or familial adenomatous polyposis, amongst others. The basis for this observation has not been fully examined, but one possibility could be the later onset acquisition of, or even absence, of cooperating KRAS mutations in this subset of PDACs.
The second study, by Rowley et al. also reported a mouse model of Brca2-deficient PDAC, with either Trp53 or KrasG12D as cooperating mutations, utilizing a Cre-expressing driver line identical to the current manuscript, and a floxed p53 allele, in contrast to the gain-of-function R172H allele in our study. In mice with bi-allelic deletions of both Brca2 and Trp53 (the functional equivalent of our CBP mice), they observed invasive PDAC of variable histological features, occurring with relatively long latency, essentially identical to our own findings. Another notable commonality between the studies was the low frequency of secondarily acquired Kras gene mutations in the resulting PDAC lesions (only 2 of 13, 15%, examined in the Rowley report), while none were present in our smaller cohort of CBP mice. This again begs the question as to the status of the KRAS gene in human PDACs arising on the backdrop of BRCA2 deficiency, and if their absence or low frequency could account for the later onset tumors seen in such individuals. A putative “antagonistic” relation between Brca2 deficiency and mutant Kras is also bolstered by the observation that, in the presence of intact p53 function, the incidence of mPanINs and PDAC was actually lower compared to mice expressing mutant Kras alone, when both genes were altered in the pancreatic epithelium.Citation39 The one discordance between our study and that of Rowley et al. was the observation that CB mice also developed mPanINs, albeit at a later time point than CBP mice, and in a minority of instances (15%), the lesions progressed to full blown PDAC. In the previous report, mice with loss of Brca2 function alone had no preneoplastic ductal lesions or cancers. While the exact basis for this discordance remains unclear, it is possible that the later onset mPanINs and PDAC in CB mice result from secondary abrogation of p53 function, or dysfunction of other DNA damage checkpoint proteins like ATM and Chk2, thus allowing surviving DNA damaged cells to bypass mitotic catastrophe.Citation15
In conclusion, the current report “dovetails” with two recent publicationsCitation38, Citation39 on successfully generating a mouse model of familial pancreatic cancer (FPC). These reports establish that biallelic loss of Brca2, possibly alone and certainly in conjunction with p53 deregulation, are capable of inducing the entire spectrum of ductal neoplasia of the pancreas, and that this multistep progression can occur without oncogenic mutations of Kras itself. These models provide an in vivo approach for testing new therapeutic strategies that might be synthetic lethal to a Brca2 defect, as well as for developing serum based “signatures” of Brca2 deficiency as an early detection biomarker for FPC.
Materials and Methods
Generation of Pdx1-Cre; Brca2flox/flox (CB) and Pdx1-Cre; Brca2flox/flox; LsL-Trp53R172H (CBP) cohorts.
Heterozygous Brca2flox/wt breeder mice were obtained from the National Cancer Institute's Mouse Models of Human Cancer Consortium (MMHCC) repository. LoxP sites are inserted into introns surrounding exon 11 in the Brca2 locus.Citation42 The LsL-Trp53R172H and Pdx1-Cre mice used in this study have been previously described in reference Citation29. Brca2flox/wt, LsL-Trp53R172H and Pdx1-Cre mice were interbred to generate Brca2flox/flox; LsL-Trp53R172H and Pdx1-Cre; Brca2flox/wt mice, respectively. Next, Brca2flox/flox; LsL-Trp53R172H and Brca2flox/wt; Pdx1-Cre mice were utilized as breeders to generate Pdx1-Cre; Brca2flox/flox (referred to as CB) and Pdx1-Cre; Brca2flox/flox; LsL-Trp53R172H (referred to as CBP) cohorts. For genotyping, genomic DNA was extracted from tail snips by means of the REDExtract-N-Amp™ Tissue PCR kit (Sigma-Aldrich, Saint Louis, MO), following the standard protocol provided by the manufacturer. For each mouse three independent PCR reactions were carried out to test for the presence of Trp53R172H, floxed Brca2 alleles (hetero- or homozygote) and Pdx1-Cre (using Cre-specific primers along with Gabra as positive control), respectively.
Primer sequences for these three reactions were as follows: Trp53R172H: mutant_forward-5′-AGC TAG CCA CCA TGG CTT GAG TAA GTC TGC A-3′; reverse-5′-CTT GGA GAC ATA GCC ACA CTG-3′;Citation27 Brca2: B012-5′-GGC TGT CTT AGA ACT TAG GCT G-3′, B013-5′-CTC CAC ACA TAC ATC ATG TGT C-3′, B014-5′-CTC ATC ATT TGT TGC CTC ACT TC-3′, B015-5′-TGTTGGATAcAAGGcATGTAcAc-3′;43 Pdx1-Cre: Cre26-5′-CCT GGA AAA TGC TTC TGT CCG-3′, Cre36-5′-CAG GGT GTT ATA AGC AAT CCC-3′, GABRA12-5′-CAA TGG TAG GCT CAC TCT GGG AGA TGA TA-3′, GABRA70-5′-AAC ACA CAC TGG CAG GAC TGG CTA GG-3′.
Tissue sample preparation.
Mice were euthanized and tissue samples obtained at necropsy were either snap-frozen in liquid nitrogen and stored at −80°C until further use, or directly fixed in 10% neutral buffered formalin solution for paraffin embedding. H&E staining of formalin-fixed paraffin-embedded tissue sections were performed by the Johns Hopkins Reference Histology Laboratory following established procedures.
Immunohistochemistry.
Immunohistochemistry was performed as previously described in reference Citation43. In brief, formalin-fixed paraffin-embedded tissue sections were deparaffinized using established procedures and quenched with 3% H2O2 for 10 min. Next, slides were heated in a steamer in 10 mM citrate buffer (pH 6.0) to unmask the epitopes for 20 min at 95°C, after which they were allowed to cool down to room temperature for 20 min. Slides were blocked with 10% fetal bovine serum solution (Invitrogen, Carlsbad, CA) for 30 min. Anti-Brca2 (Abcam) was used as primary antibody at a dilution of 1:100. Anti-phospho H2AXSer139 (Upstate/Millipore, Millerica, MA) was used as primary antibody at a dilution of 1:200. Anti-Pdx1 (Chemicon, Temecula, CA) was used at a primary dilution of 1:100, and anti-Shh (R&D Systems, Minneapolis, MN) was used at a dilution of 1:50. Immunolabeling was detected using the PowerVision+ Poly-HRP IHC kit (Immunovision Technologies, Norwell, MA) following the standard protocol. Slides were counterstained with Harris hematoxylin solution. Negative controls (primary antibody replaced by serum from appropriate species) were used in each run.
Direct sanger sequencing.
For confirmation of Kras codons 12, 13 and 61 somatic mutation status, tissue samples from two invasive PDAC and adjacent normal tissues, respectively, were collected by means of stereomicroscope microdissection. In addition, tissue samples from two mPanIN lesions were collected using laser capture microdissection. DNA extraction and direct Sanger sequencing were done using standard techniques as described elsewhere in reference Citation31. Primer sequences for exons 12, 13 and 61 of murine Kras are described elsewhere in reference Citation44.
Statistical analysis.
Kaplan-Meier analysis was performed using GraphPad Prism for MS Windows, version 5.02.
Abbreviations
| mPanIN | = | murine pancreatic intraepithelial neoplasia |
| ADM | = | acinar-to-ductal metaplasia |
| PDAC | = | pancreatic ductal adenocarcinoma |
| LSL | = | lox-STOP-lox |
Figures and Tables
Figure 1 Kaplan-Meier survival analysis of CB and CBP mice. Kaplan-Meier survival analysis of CB and CBP mice cohorts reveals a trend towards shorter survival in the latter, but it is not statistically significant.
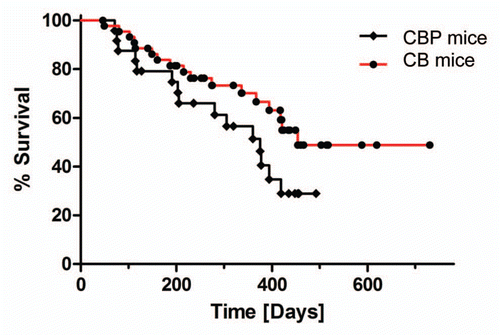
Figure 2 Histopathology of exocrine pancreatic insufficiency in CB and CBP mouse pancreata in the absence of invasive neoplasia. (A) Profound loss of exocrine pancreatic parenchyma in a CB mouse at 6 months age demonstrating extensive adipose (fatty) replacement and residual islets with minimal functioning exocrine tissue (H&E stain, 10× magnification). (B) Minimal residual exocrine parenchyma (acinar tissue) in a six month old CBP mouse (H&E stain, 20× magnification). (C) Degenerated cystic lesions observed in both CB and CBP mice, lined by flattened cuboidal epithelium and often containing hemorrhagic fluid (H&E stain, 20× magnification). (D) Strips of murine pancreatic intraepithelial neoplasia (mPanIN) are observed in a background of adipose (fatty) replacement and near total absence of viable acinar tissue in a 15-month-old CB mouse (H&E stain, 10× magnification).
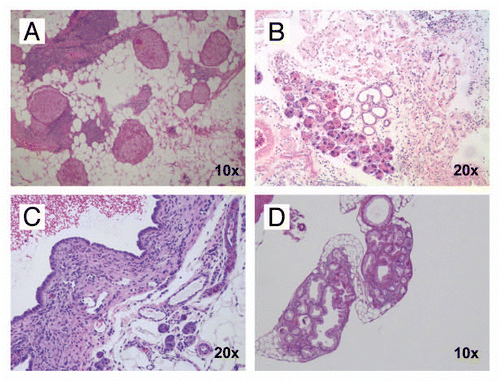
Figure 3 Histopathology of non-invasive precursor lesions in CB mice. Examples of low-grade (A) and high-grade (B) mPanIN lesions arising in the pancreas of a CB mouse at 11 months of age. Several areas with acinar-to-ductal metaplasia were also found (C); higher magnification (D). (H&E stains). (E) CBP mice (red columns) have an earlier onset of mPanIN compared to CB mice (blue columns). By 10 months of age, both cohorts have comparable frequency of mPanIN lesions.
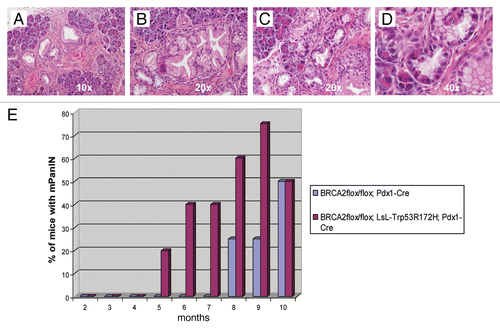
Figure 4 Brca2 immunohistochemistry confirms loss of protein expression in lesional pancreatic epithelium. (A and B) Brca2 immunohistochemistry confirms robust cytoplasmic expression in granulosa cells of the ovary, a known positive control (10× and 40″ magnification). (C and D) Minimal to absent Brca2 expression is observed within the lining epithelium of the degenerated cysts, a prototype of which is illustrated in (10× and 40× magnification). Note that the subjacent mesenchymal cells retain Brca2 expression. (E and F) Minimal to absent Brca2 expression is observed within mPanIN lesions arising in a “CPB” mouse (10× and 40× magnification).
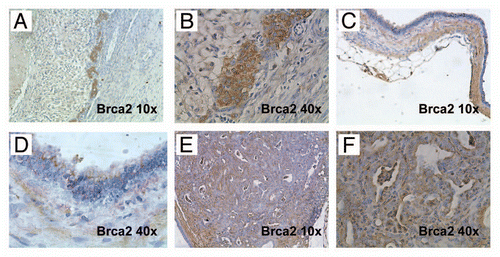
Figure 5 A minority of CB mice develop invasive and metastatic PDAC at relatively long latency (15 months or greater). (A) Example of a metastatic PDAC arising in a 24-month-old CB mouse. Multiple metastatic lesions in liver and spleen are readily visible. (B) Serial transplantation of tumor tissue into athymic mice resulted in growth as subcutaneous xenografts, a property of cancer cells. (C) Histologically, the primary and metastatic tumors were comprised of moderately differentiated PDAC (H&E stain, 10× magnification). (D) Moderately differentiated PDAC, at a higher magnification (H&E stain, 20× magnification). Note mucin vacuoles within neoplastic glands.
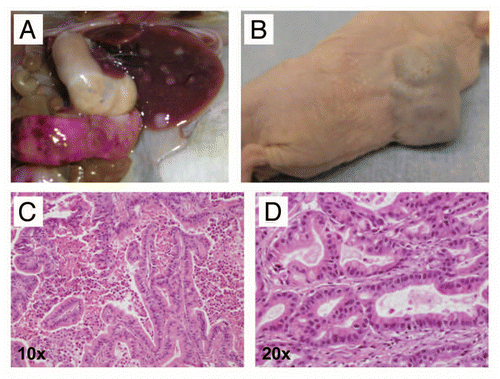
Figure 6 Macroscopic images of metastatic pancreatic cancers arising in CBP mice. (A) A primary pancreatic cancer (white dotted line) and numerous grossly visible metastatic lesions (white arrows) to distant organ sites, including peritoneum, diaphragm (B), lungs (C) and liver (D). At these advanced stages, malignant ascites was observed in some of the affected mice (E). In addition, some animals developed hemorrhagic intra-pancreatic cystic lesions, likely due to bleeding (F).
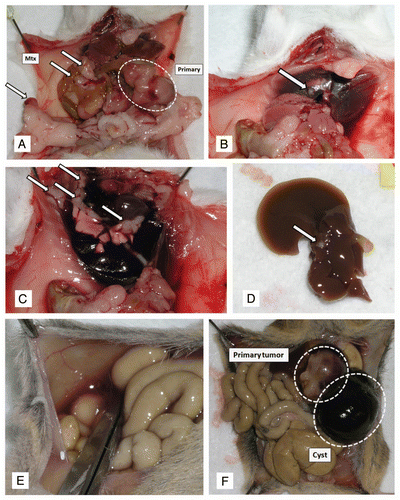
Figure 7 Variable histopathological features of PDAC arising in CBP mice. (A) Primary PDAC with extensive stromal desmoplasia, resembling the cognate human disease is observed in this CBP mouse (H&E stain, 2× magnification). (B) At higher magnification, infiltrating neoplastic glands in a bed of desmoplastic stroma can be seen (H&E stain, 20× magnification). (C) An example of a biphasic tumors with both adenocarcinoma and sarcomatoid components (H&E stain, 4× magnification). (D) At higher magnification of the tumor in (C), the confluence between the two components is highlighted (H&E stain, 20× magnification). (E) Higher magnification of the sarcomatoid carcinoma component also confirms the presence of atypical pleomorphic nuclei and mitotic figures (arrow) (H&E stain, 40× magnification). (F) An anaplastic carcinoma with prominent giant cells arising in a CBP mouse (H&E stain, 20× magnification).
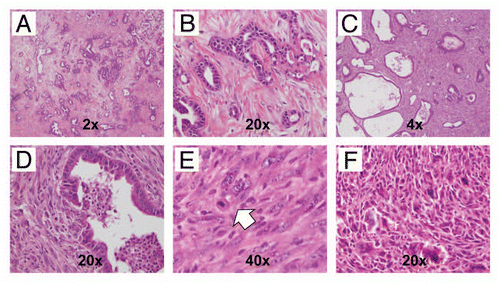
Figure 8 Inactivation of Brca2 promotes DNA damage in the exocrine pancreas. (A) Immunohistochemical staining for pH2AXSer139 shows minimal labeling within acinar cells in pancreatic tissue sections derived from hemizygous Pdx1-Cre; Brca2flox/wt mice (negativ function in CB mice results in markedly increased pH2AXSer139 immunolabeling in pancreatic acinar cells (B and C), but not in the ductal epithelia (red arrows). Note robust expression of pH2AXSer139 in two highly atypical acinar cell nuclei (white arrowheads). (D) Marked upregulation of pH2AXSer139 labeling in the sarcomatoid carcinoma component of a PDAC arising in a CBP mouse.
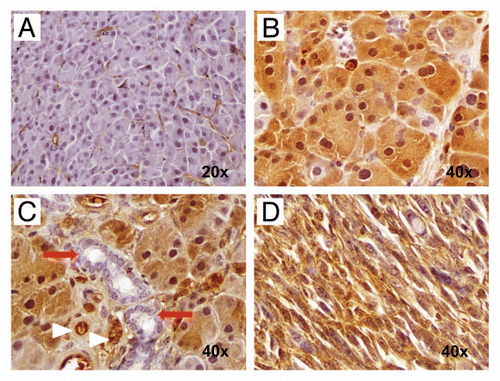
Figure 9 Hedgehog ligand is expressed by neoplastic glandular epithelium but not by the epithelium of non-neoplastic cysts. (A) and (B) Nuclear Pdx1, a marker of pancreatic progenitor cells not expressed in the mature ductal epithelium, is expressed within both neoplastic glands (A) as well as the lining epithelium of non-neoplastic cysts (B). (C and D) In contrast, Hh ligand, known to be overexpressed in most human PDAC, is expressed only by neoplastic glands (C) and not by the non-neoplastic cystic epithelium (D).
Acknowledgments
A.M. was supported by the Sol Goldman Pancreatic Cancer Research Center, the Michael Rolfe Foundation for Pancreatic Cancer Research, NIH R01CA134767, NIH P01CA134292 and NIH R01CA113669. G.F. was supported by BONFOR grant number O-111.0001.2 and by the German Cancer Foundation (Deutsche Krebshilfe) grant number 109215. V.F. was supported by a Research Grant of the University Medical Center Giessen and Marburg.
References
- Feldmann G, Maitra A. Molecular genetics of pancreatic ductal adenocarcinomas and recent implications for translational efforts. J Mol Diagn 2008; 10:111 - 122
- Hruban RH, Adsay NV, Albores-Saavedra J, Anver MR, Biankin AV, Boivin GP, et al. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res 2006; 66:95 - 106
- Tuveson DA, Hingorani SR. Ductal pancreatic cancer in humans and mice. Cold Spring Harb Symp Quant Biol 2005; 70:65 - 72
- Olive KP, Tuveson DA. The use of targeted mouse models for preclinical testing of novel cancer therapeutics. Clin Cancer Res 2006; 12:5277 - 5287
- Hruban RH, Klein AP, Eshleman JR, Axilbund JE, Goggins M. Familial pancreatic cancer: from genes to improved patient care. Expert Rev Gastroenterol Hepatol 2007; 1:81 - 88
- Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 2009; 324 217
- Couch FJ, Johnson MR, Rabe KG, Brune K, de Andrade M, Goggins M, et al. The prevalence of BRCA2 mutations in familial pancreatic cancer. Cancer Epidemiol Biomarkers Prev 2007; 16:342 - 346
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009; 361:123 - 134
- Villarroel MC, Rajesh Kumar NV, Garrido-Laguna I, De Jesus-Acosta A, Jones S, Maitra A, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther 2011; 10:3 - 8
- Goggins M, Hruban RH, Kern SE. BRCA2 is inactivated late in the development of pancreatic intraepithelial neoplasia: evidence and implications. Am J Pathol 2000; 156:1767 - 1771
- Boulton SJ. Cellular functions of the BRCA tumour-suppressor proteins. Biochem Soc Trans 2006; 34:633 - 645
- Hutchinson L. Targeted therapies: PARP inhibitor olaparib is safe and effective in patients with BRCA1 and BRCA2 mutations. Nat Rev Clin Oncol 7:549
- Hruban RH, Klimstra DS, Pitman MB. Tumors of the pancreas 2006; Washington DC American Registry of Pathology
- Fillingham J, Keogh MC, Krogan NJ. GammaH2AX and its role in DNA double-strand break repair. Biochem Cell Biol 2006; 84:568 - 577
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005; 434:864 - 870
- Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol 2008; 3:157 - 188
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003; 4:437 - 450
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 2003; 17:3112 - 3126
- Hidalgo M, Maitra A. The hedgehog pathway and pancreatic cancer. N Engl J Med 2009; 361:2094 - 2096
- Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003; 425:846 - 851
- Shi C, Hruban RH, Klein AP. Familial pancreatic cancer. Arch Pathol Lab Med 2009; 133:365 - 374
- Hruban RH, Canto MI, Griffin C, Kern SE, Klein AP, Laheru D, et al. Treatment of familial pancreatic cancer and its precursors. Curr Treat Options Gastroenterol 2005; 8:365 - 375
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434:917 - 921
- Merritt AJ, Allen TD, Potten CS, Hickman JA. Apoptosis in small intestinal epithelial from p53-null mice: evidence for a delayed, p53-independent G2/M-associated cell death after gamma-irradiation. Oncogene 1997; 14:2759 - 2766
- Vakifahmetoglu H, Olsson M, Tamm C, Heidari N, Orrenius S, Zhivotovsky B. DNA damage induces two distinct modes of cell death in ovarian carcinomas. Cell Death Differ 2008; 15:555 - 566
- Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res 1996; 56:5360 - 5364
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004; 119:847 - 860
- Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res 2005; 65:10280 - 10288
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005; 7:469 - 483
- Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA 2006; 103:5947 - 5952
- Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci USA 2008; 105:18913 - 18918
- Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 2009; 16:379 - 389
- Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 2006; 20:3130 - 3146
- Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 2007; 11:229 - 243
- Pasca di Magliano M, Sekine S, Ermilov A, Ferris J, Dlugosz AA, Hebrok M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev 2006; 20:3161 - 3173
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008; 321:1801 - 1806
- Calhoun ES, Jones JB, Ashfaq R, Adsay V, Baker SJ, Valentine V, et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am J Pathol 2003; 163:1255 - 1260
- Skoulidis F, Cassidy LD, Pisupati V, Jonasson JG, Bjarnason H, Eyfjord JE, et al. Germline Brca2 heterozygosity promotes Kras(G12D)—driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell 2010; 18:499 - 509
- Rowley M, Ohashi A, Mondal G, Mills L, Yang L, Zhang L, et al. Inactivation of Brca2 promotes Trp53-associated but inhibits KrasG12D-dependent pancreatic cancer development in mice. Gastroenterology 2011; 140:1303 - 1313; http://dx.doi.org/10.1053/j.gastro.2010.12.039
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol 11:220 - 228 (DOI: nrm2858 [pii])
- Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007; 26:7773 - 7779
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 2001; 29:418 - 425
- Fendrich V, Chen NM, Neef M, Waldmann J, Buchholz M, Feldmann G, et al. The angiotensin-I-converting enzyme inhibitor enalapril and aspirin delay progression of pancreatic intraepithelial neoplasia and cancer formation in a genetically engineered mouse model of pancreatic cancer. Gut 2009; 59:630 - 637
- Slape C, Liu LY, Beachy S, Aplan PD. Leukemic transformation in mice expressing a NUP98-HOXD13 transgene is accompanied by spontaneous mutations in Nras, Kras and Cbl. Blood 2008; 112:2017 - 2019