Abstract
Dioscin has been shown to promote anticancer activity against several forms of cancers. However, its detailed molecular mechanisms have not been clarified. In this study, we demonstrate that dioscin induces apoptosis in cancer cells through the induction of oxidative stress. Treatment with cancer cells in vitro with dioscin resulted in rapid generation of reactive oxygen species (ROS) and the induction of mitochondrial pathway apoptosis in human esophageal cancer cell line Kyse510. Inhibition of oxidative stress by the antioxidant N-acetylcysteine blocked the induction of apoptosis by dioscin, indicating that ROS generation is the primary mechanism responsible for the proapoptotic activity of dioscin. Proteomic analysis and protein gel blotting further revealed peroxiredoxins 1 and 6 (PRDX 1 and 6), which are implicated in ROS metabolism and apoptosis, were associated with the anticancer effects of dioscin. Meanwhile, overexpression of PRDX 1 and 6 significantly blocked the elevated ROS and apoptosis induced by dioscin. In conclusion, we suggest that PRDX1 and PRDX6 are key targets in the process of dioscin-induced apoptosis that involves intracellular elevated ROS.
Introduction
Dioscin, a glucoside saponin isolated from the roots of Dioscorea panthaica (), has been shown to possess various biological activities including anti-inflammation, hepatoprotection, as well as its anti-proliferative effects against a number of human cancer cells.Citation1,Citation2 Recent studies further indicate that dioscin induced apoptosis in human leukemia cell line HL-60 through an intrinsic mitochondrial dependent pathway. Moreover, it has been demonstrated that elevated reactive oxygen species (ROS) levels might be the main reason for triggering the apoptotic cascades by dioscin.Citation3 Despite these insights, the biochemical targets of dioscin in cancer cells have not been fully clarified, and the mechanisms underlying the anticancer properties remain poorly understood.
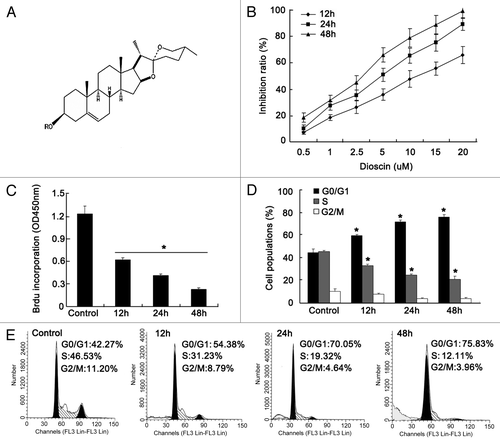
Figure 1. Proliferation inhibition effects of dioscin on human esophageal cancer cell lineKyse510. (A) Chemical structure of dioscin. (B) Cytotoxicty of dioscin on Kyse510 wasassessed using MTT assay. The results showed that dioscin inhibited proliferation of Kyse510 cells in a dose- and time-dependent manner (values represents means ± SD, n = 6). The IC50 of dioscin on Kyse510 cells for 24 h was determined as 5.4 μM. (C) Time course treatment with IC 50 dose of dioscin on Kyse510 cells and then labeled with BrdU. DNA synthesis was measured by BrdU cell proliferation enzyme-linked immunosorbent assay. The results indicated that diosicn inhibited DNA synthesis in a time-dependent manner. Data are presented as means ± SD, n = 6, *p < 0.01, vs. untreated control. (D–E) Cell cycle analysis showed that with the increasing administration time of dioscin on Kyse510 cells, the G1 population gradually increased, while S phase population decreased, indicating a G1/S arrest was induced by dioscin. In addition, a sub-G1 peak was observed on dioscin-treated cells, implying that disocin might also induce Kyse510 cells apoptosis (values represents means ± SD, n = 3, *p < 0.05 vs. normal controls).
ROS are a group of reactive, short-lived, oxygen-containing species including superoxide (O2-), hydrogen peroxide (H2O2), Hydroxyl radical (OH), singlet oxygen (1O2·) and lipid peroxyl radical (LOO).Citation4 A number of studies have indicated ROS might be a common mediator for apoptosis. Evidence in favor of such a role includes the findings that (1) elevated ROS can be detected in many forms of apoptosis, (2) antioxidants prevent most forms of apoptosis whether or not they appear to be initiated by ROS, and (3) mitochondrion, a major source of ROS, appear to be critical for apoptosis occurrence.Citation5-Citation8 Intracellular ROS are derived from two main sources: the mitochondrial oxidative phosphorylation respiration chain and other cytosolic organelles, such as endoplasmic reticulum, etc.Citation9 Under normal circumstances, cells are able to defend themselves against ROS damage with enzymes such as superoxide dismutase, catalases, lactoperoxidases, glutathione peroxidases and peroxiredoxins.Citation10 Once the redox balance state kept by these enzymes is destroyed, the elevated ROS will bring great damage on DNA synthesis, protein folding and other normal metabolic processes such as mitochondrial respiration function.
In order to further elucidate the detailed mechanisms accounting for the dioscin-induced apoptosis, we used a proteomic analysis to identify proteins potentially involved in anticancer mechanisms of dioscin in the human esophageal cancer cell line Kyse510. By using this approach, we detected two proteins, namely, peroxiredoxin 1 and 6 (PRDX1 and PRDX6), which were previously implicated in ROS metabolism and apoptosis. Protein gel blotting analysis further confirmed the expression of these proteins in Kyse510 cells after dioscin administration. In summary, we suggest that the apoptosis-induced effect of dioscin is mediated by PRDX1 and PRDX6 through mechanisms involving alterations in ROS metabolisms.
Results
Effects of dioscin on the proliferation and cell cycle in Kyse510 cells
To determine the anti-proliferation activity of dioscin upon Kyse510 cells, we first performed an MTT assay. As shown in , time- and dose-dependent cytotoxic effects for dioscin on Kyse510 cells were demonstrated. The half maximal inhibitory concentration (IC 50) of 24 h was approximately 5.4 μM. In order to determine whether the cytotoxicity effect was attributed to the proliferation inhibition caused by dioscin, we performed the BrdU experiment; the results showed that cell proliferation was significantly suppressed by dioscin in a time-dependent manner when treated with the IC 50 concentration (). Cell cycle analysis also indicated that G1/S arrest was induced after dioscin administration, presenting as an increased G0/G1 population and a decreased S phase population (). The results indicated that the proliferation inhibition effects of dioscin might be partly caused by cell cycle arrest. In addition, the appearance of sub-G1 peak after dioscin treatment suggested that apoptosis might be also an important mechanism involved in the proliferation inhibition effects.
Dioscin induces apoptosis mediated by elevated ROS level
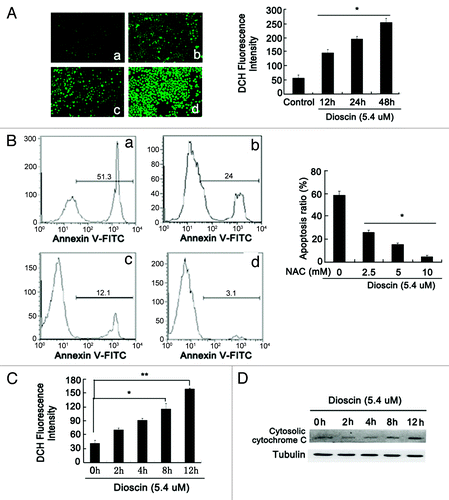
The effect of dioscin on apoptosis was further validated by flow cytometric analysis using Annexin V-PI staining. Results in showed that after dioscin treatment for 48 h, the apoptosis ratio dramatically increased from 4.68 ± 0.79 to 58.69 ± 4.3%. Meanwhile, DAPI staining also revealed typical apoptotic morphological following dioscin treatment, such as chromatin condensation and membrane blebbing (). Dioscin-induced apoptosis was also confirmed by protein gel blotting analysis. It was found that the expression of cleaved caspase-9 rather than caspase-8 was upregulated, indicating that the apoptosis induced by dioscin was in a mitochondrial dependent manner. In addition, increased expression of cytosolic Cytochrome C, BAX and downregulation of Bcl-2 further demonstrated that the mitochondrial apoptotic pathway was activated (). JC-1 staining also showed that the mitochondrial membrane potential Δψm was lowered after dioscin administration (). These observations together demonstrated that dioscin induced apoptosis in Kyse510 cells. As ROS has been reported to be an important inducer of mitochondrial-mediated apoptosis and DNA damage, DCFH-DA was applied to detect the change of intracellular ROS levels after dioscin administration. As shown in , the fluorescence intensity of ROS, presenting as green fluorescence, was highly elevated by dioscin. In order to validate the role of ROS in inducing apoptosis triggered by dioscin, the antioxidant N-acetylcysteine (NAC) was administrated together with dioscin. The results showed that with the increasing concentration of NAC, the apoptosis ratio induced by dioscin gradually decreased ().The above results suggested that interruption of intracellular oxidative balance might play an important role in mediating dioscin-induced apoptosis. In addition, it was found that the intracellular ROS accumulation occurred as early as 2 h after dioscin administration (). However, the release of Cytochrome C from mitochondrion was only significantly detected at 8 h following dioscin treatment, suggesting that ROS generation was an early event, prior to the occurrence of the mitochondrial pathway apoptosis (). In summary, these findings show that the dioscin-induced apoptosis is followed the accumulation of ROS in cells, confirming the crucial role of oxidative stress in dioscin-induced apoptosis.
Figure 2. Dioscin induces Kyse510 cells apoptosis through mitochondrial pathway. (A) Time-course detection of apoptotic Kyse510 cells in the presence of dioscin at IC50 dose by Annexin V-PI staining analysis. The results showed that dioscin could significantly induce Kyse510 cells apoptosis, the apoptosis ratio increased from 22.5 ± 3.16% at 12 h to 58.7 ± 4.3% at 48 h (a: control, b: 12 h, c: 24 h, d: 48 h; values represented as means ± SD, n = 3, *p < 0.01 vs. normal controls). (B) DAPI staining showed that typical apoptotic morphological changes including chromatin fragmentation, nucleus condensation and cell shrinkage were induced by dioscin (a: control, b: 12 h, c: 24 h, d: 48 h). (C) Protein gel blotting revealed that following dioscin treatment, the expression of cleaved-caspase-3/9, cytosolic Cytochrome C, BAX were upregulated, the expression of anti-apoptotic protein Bcl-2 was downregulated, while no significant expression of clevaed-caspase-8 was detected, suggesting that the mitochondrial pathway might be the main form of dioscin-induced apoptosis. (D) Decreased mitochondrial membrane potential Δψm was detected in dioscin treated cells. The left panel showed that after dioscin treatment, JC-1 fluorescence shifted from red-orange to green gradually with prolonged administration time (magnification × 200). The data are summarized in the right panel from six randomly chosen fields in each group (a: control, b: 12 h, c: 24 h, d: 48 h, values represents as means ± SD, n = 6, *p < 0.01 vs. normal controls).
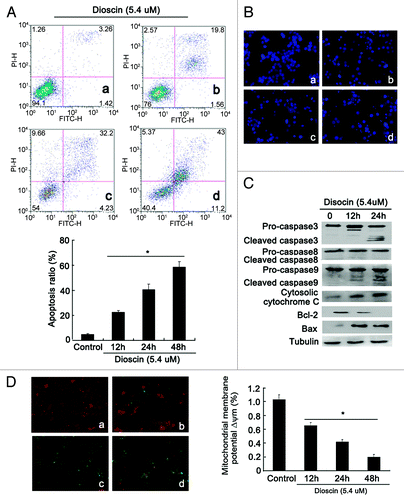
Figure 3. Crucial role of oxidative stress in dioscin-induced apoptosis. (A) DCFH-DA probe staining showed that dioscin stimulated a significant ROS accumulation in Kyse510 cells. The right panel showed that time-course ROS production stimulated by dioscin. For each group, six fields were randomly chosen and analyzed (a: control, b: 12 h, c: 24 h, d: 48 h, values represents as means ± SD, n = 6, *p < 0.01 vs. normal controls). (B) Flow cytometry analysis of Kyse510 cells indicated that the dioscin-induced apoptosis was abrogated by the antioxidant NAC (a: control, b: 12 h, c: 24 h, d: 48 h; values represents as means ± SD, n = 3, *p < 0.01 vs. normal controls). (C) Dioscin induced accumulation of ROS in Kyse510 cells as early as 2 h after dioscin treatment (values represents as means ± SD, n = 6,* p < 0.05, **p < 0.01 vs. normal controls). However, the release of Cytochrome C from mitochondrion was only significantly detected at 8 h (D), which suggested that ROS burst might be an early event accounting for the initiation of mitochondrial pathway apoptosis.
Comparsion of protein expression profiles between control and dioscin-treated Kyse510 cells 4Digure-5ts by protein gel blotting
In order to determine key proteins that responsible for triggering dioscin-induced apoptosis and elevated oxidative stress, whole-cell extracts of Kyse510 cells before and after treatment with dioscin for 24 h were separated by 2-DE. The representative images of protein extracts from control and dioscin-treated cells were depicted in . Simultaneous image comparisons between control and dioscin treated cells revealed differential expression of about 400 protein spots, with molecular masses ranging from 10–200 Kda. Thirty-nine protein spots showed more than 2-fold changes in density. Thirteen of these were subsequently selected for protein identification on the basis of consistency occurring changes in optical density in Coomassie brilliant blue-stained 2-DE gels. Detailed alteration patterns of the 13 proteins were described in .
Figure 4. Protein profiles of control and dioscin-treated Kyse510 cells. (A) Kyse510 cells were treated with 5.4 μM dioscin for 24 h. Proteins were separated by pI and molecular weight and visualized by silver staining. Arrows indicated each differentially expressed protein spots between control and dioscin-treated cells. These spots were then identified by MALDI-TOF MS. (B) Detailed alteration patterns of proteins selected for identification.
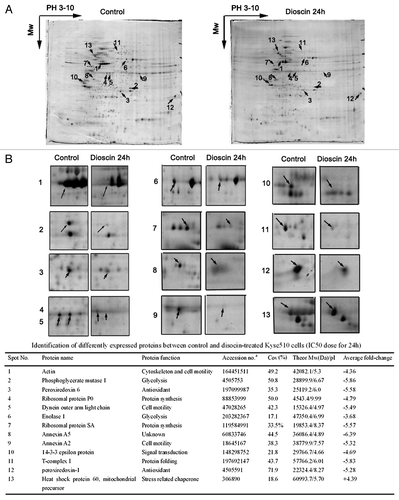
Among these spots, 12 proteins were downregulated, while 1 protein was upregulated following dioscin treatment. These spots were identified using MALDI-TOF MS followed by NCBI database searches. Proteins were then classified according to their roles in the following functions: glycolysis, protein synthesis and folding, cell motility, signal transduction and antioxidant ().
PRDX1 and PRDX6 are key targets in dioscin-induced elevated ROS and apoptosis
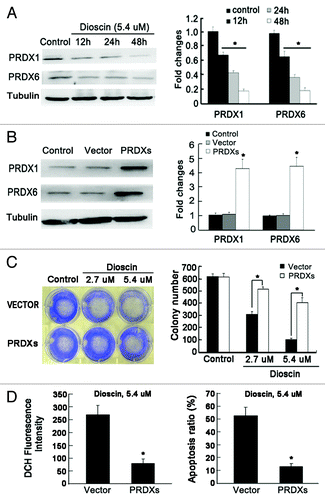
Based on the results that dioscin destroyed the balance of intracellular oxidative defense system and induced mitochondrial pathway apoptosis, it was suggested that antioxidation enzymes might be suppressed by dioscin. Combining the results of 2-DE, we identified PRDX1 and PRDX6, which are implicated in ROS metabolism and apoptosis, for further investigation. Protein gel blotting results confirmed that with increased administration time, both protein expression were downregulated (). These findings were consistent with the results of 2-DE proteomic analysis. To assess the crucial role of PRDXs in mediating elevated ROS and apoptosis induced by dioscin, both PRDX1 and PRDX6 were cloned and stably co-transfected into Kyse510 cells, and empty vector was used as a control (). Colony formation assay was applied to determine whether PRDXs overexpression could block the inhibitory effects of dioscin on colonogenic ability of cancer cells. As shown in , in the empty vector control cells, dioscin dose-dependently inhibited the colony formation. However, in PRDXs overexpression cells, the colony-number of dioscin in treated samples were significantly more than that in the vector control group, indicating that PRDX1 and PRDX6 play an important role in keeping long-term survial of cancer cells after dioscin administration. Meanwhile, overexpression of PRDXs significantly blocked the elevated ROS levels and apoptosis induced by dioscin (), suggesting the central importance of PRDXs in the mechanism of action of dioscin and implying that PRDX1 and PRDX6 might be the key targets of dioscin in inducing cancer cell apoptosis that involves oxidative stress.
Figure 5. The crucial role of PRDX1 and PRDX6 in dioscin-induced apoptosis and oxidative stress in Kyse510 cells. (A) Kyse 510 Cells were treated with dioscin at IC50 dose for 12, 24 and 48 h. The results showed that the expression of both PRDX1 and PRDX6 were downregulated with increasing administration time. The results are consistent with the 2-DE proteomic analysis. (B) PRDX1 and PRDX6 were subcloned and co-transfected into Kyse510 cells. pcDNA 3.1(+) empty vector was also stably transfected into Kyse510 cells and used as negative control. (C) Overexpression of PRDX1 and PRDX 6 significantly blocked the colonogenic inhibitory effects induced by dioscin (values represents as means ± SD, n = 3, *p < 0.05). (D) Overexpression of PRDX1 and PRDX6 abrogated the induction of ROS burst and apoptosis by dioscin administration. Values represents as means ± SD, n = 3, *p < 0.05.
Discussion
Natural plants are important sources to develop novel chemotherapeutic agents.Citation12 Dioscin is one of the best known saponins that exists in Dioscorea panthaica, a famous Chinese herb. Dioscin has been shown to exhibit potent cytotoxicity toward a number of human cancer cell lines. However, the detailed mechanisms of dioscin-induced cell death are still unclear. Apoptosis is a tightly controlled multistep process to eliminate unwanted cells in various biological systems, and is one of the key mechanisms for development of chemotherapeutic drugs.Citation13 In the present study, we showed that dioscin administration resulted in a significant increase of cell population in the Annexin V positive and PI negative region. In addition, DAPI staining revealed typical apoptotic morphological changes presenting as plasma membrane blebbing, cell shrinkage, nucleus condensation and chromatin fragmentation. All these results suggest that apoptosis is the main anticancer mechanism of dioscin.
Apoptosis can be triggered through either extrinsic pathway or intrinsic pathway. The extrinsic pathway, also referred to as cytoplasm pathway, is initiated with ligation of Fas death receptor. The activation of these receptors will recruit an adaptor protein known as Fas-associated death domain protein (FADD) and caspase-8 to form the death-inducing signal complex (DISC). Caspase-3 is then activated and initiate degradation of the cell.Citation14,Citation15 The intrinsic pathway, also known as mitochondrial pathway, is triggered by cellular stress, specifically mitochondrial stress caused by factors such as DNA damage and heat shock. In response to apoptotic stimuli, proapoptotic proteins will undergo post-translational modifications that include dephosphorylation and cleavage resulting in their activation and translocation to the mitochondrion.Citation16-Citation18 Once the balance between the pro- and anti-apoptotic factors is destroyed, the mitochondrial membrane potential will be lowered, resulting in the release of cytochrome-c, which subsequently activates caspase-9 and caspase-3 and leads to apoptosis. Our results showed that dioscin treatment resulted in a significant decrease in mitochondrial membrane potential. Meanwhile, the upregulation of BAX, Cytosolic Cytochrome C, cleaved caspase-3/9, and downregulation of the anti-apoptotic protein Bcl-2 also indicated that dioscin triggered apoptosis mainly through the mitochondrial dependent pathway.
Numerous studies have proposed ROS as common mediators for apoptosis. Many agents that induce apoptosis are either oxidants or stimulators of cellular oxidative metabolism. Conversely, many inhibitors of apoptosis have antioxidant activities or enhance cellular antioxidant defenses.Citation19-Citation21 Our results showed that oxidative stress induction by dioscin is very rapid and precedes the earliest events associated with apoptosis. Meanwhile, antioxidant NAC administration completely blocked dioscin-induced ROS generation and apoptosis, suggesting that oxidative stress is required for triggering apoptosis by dioscin. ROS generation in cells is controlled by multiple enzymes such as SODs and GSH, etc. To better understand molecular mechanisms related to elevated ROS level induced by dioscin, 2-DE proteomic analysis was performed. Thirteen spots that show dramatic changes between dioscin-treated and control cells were selected for the experiment. Interestingly, several proteins including PRDX1 and PRDX6 are regarded as ROS- and apoptosis- related proteins. The results of 2-DE proteomic analysis were also validated by protein gel blotting procedures.
Previous studies suggested that cancer cells, due to their high metabolic activity, produce higher levels of ROS than normal cells and use ROS signals to stimulate both proliferation and progression.Citation22,Citation23 This reliance on high ROS levels renders cancer cells vulnerable to further increase in oxidative stress that force the cell beyond a threshold resulting in apoptosis. Thus, agents that either increase ROS generation or decrease the expression of antioxidant enzymes have the potentiality to target cancer cells with little or no effect on normal cells. PRDX are a ubiquitous super family of thiol-antioxidant enzymes associated with cell proliferation, differentiation, and apoptosis.Citation24,Citation25 PRDX can be classified into either 1-cys or 2-cys PRDX, depending on whether the protein contains one or two conserved active-site cysteine residues. To date, six PRDX family members have been identified. PRDX 1–5 belong to the 2-cys PRDX, which uses thioredoxin as an electron donor.Citation26 The only 1-cys PRDX is PRDX6, a bifunctional protein with glutathione peroxidase and phospholipase A2 (PLA2) activities.Citation27 Both PRDX1 and PRDX6 have been found to be elevated in several human cancer cells and tissues, and to influence diverse cellular processes including cell survival, proliferation, oxidative stress and apoptosis.Citation28-Citation33 In this study, we demonstrated decreased expression of both PRDX1 and PRDX6 in Kyse510 cells following treatments with increasing time of dioscin. Meanwhile, overexpression of PRDX1 and PRDX6 significantly blocked the ROS burst, apoptosis and colonogenic inhibitory effects induced by dioscin in cancer cells, showing that PRDXs might play a crucial role in mediating the apoptosis-induction effects of dioscin. However, further research is still needed to clarify how dioscin inhibits PRDXs expression in cancer cells.
In conclusion, although some studies have reported anticancer activities of dioscin, we suggest that ROS production is an important event in dioscin-mediated apoptosis in Kyse510 cells. Meanwhile, the pro-oxidant activities of dioscin might be correlated with downregulation of PRDX1 and PRDX6. Our findings that dioscin is an inducer of ROS highlight its potential application as novel agent in clinical cancer treatment.
Materials and Methods
Cell culture
Human esophageal cell line KYSE510 was cultured in RPMI-1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin, in a humified incubator with an atmosphere of 5% CO2 at 37°C. When the cells reached approximately 80% confluency, they were harvested and plated for either passages or drug treatments. Dioscin was purchased from Aktin Chemicals, Inc. with a HPLC purity ≥ 98%. A stock solution of dioscin (100 mM) was prepared in DMSO and stored at -20°C.
Cell proliferation assay
Cells were seeded in 96-well flat-bottom plates at a density of 5 × 103 cells/well. After 24 h, dioscin was at a final concentration of 0.5, 1, 2.5, 5, 10, 15 and 20 μM was added and incubated for 12, 24 and 48 h at 5% CO2, 37°C. Then the suspension was discarded and cells were incubated with MTT (5 mg/ml) for 4 h. The resulting formazan precipitate was dissolved with 150 μl DMSO and the absorption was measured at 540 nm on a micro platereader. For proliferation analysis, the BrdU labeling solution was added and the cells were incubated further for 12 h. After the incubation, the effect of dioscin on the proliferation of cancer cells was determined by the extent of BrdU incorporation using the protocol supplied by the manufacturer (Roche Diagnostics). In brief, after the incubation of cells with the BrdU labeling solution, the medium was aspirated and the cells were fixed and incubated with anti-BrdU antibody. After washing, the cells were incubated with secondary antibody conjugated with horse radish peroxidase. Finally, the extent of BrdU incorporation was determined colorimetrically at 450 nm. Triplicate independent experiments were conducted.
Cell cycle analysis
Cells were seeded in 6-well plates and incubated with IC50 concentration of dioscin for 12, 24 and 48 h. Cells were trypsinized, harvested and fixed in 70% ice cold ethanol at 4°C overnight. Afterwards, cells were washed three times and resuspended in 500 μl PBS containing 100 μg/ml RNase A, 50 μg/ml propidium iodide (PI) and 1% Triton X-100, followed by incubation in dark for 30 min at 37°C. Cell cycle distribution was analyzed by a FACStar Plus flow cytometer.
Flow cytometric analysis of apoptosis and DAPI staining
Dioscin-induced apoptosis was determined by Annexin V-PI staining kit (Clontech) according to the manufacturer’s instructions. Briefly, 3 × 105 Kyse510 cells were seeded in 6-well plates before dioscin administration for 12, 24 and 48 h. Then cells were harvested, resuspended in ice-cold PBS solution and stained by Annexin V and PI for 15 min protected from light at room temperature. Ten thousand cells were analyzed for each sample with a FACStar Plus flow cytometer. The percentage of apoptotic cells in each treatment were analyzed using Flowjo software. For DAPI staining, after dioscin treatment, cells were washed three times with PBS and fixed in 4% paraformaldehyde for 10 min. Then cells were rinsed with PBS for three times and stained with DAPI. Stained cell samples were observed under fluorescence microscope.
Measurement of mitochondrial membrane potential (Δψm)
Mitochondrial membrane potential was assessed with the fluorescent probe JC-1 (Molecular Probes). Cancer cells were seeded on 6-well plates at a density of 1 × 105/well. After dioscin treatment for 24 h, Kyse510 cells were stained with 25 μM JC-1 at 37°C for 30 min. Fluorescent images were visualized by a fluorescent microscope (Carl Zeiss). Acquired signals were analyzed with image J software. A minimum of six fields were selected and average intensity for each region was quantified. The ratio of J-aggregate to J-monomer intensity for each region was normalized to untreated cells and calculated.
Detection of oxidative stress
2′7′-dichlorofluorescein diacetate (DCFH-DA, Sigma) was used to measure ROS formation. After exposure to different concentrations of dioscin for 12, 24 and 48 h, cancer cells were incubated in DCFH-DA containing medium (final concentration: 10 μM) at 37°C for 20 min. Cells were washed with PBS three times to remove DCFH-DA that did not entered cells. The fluorescence was visualized immediately at wavelengths of 485 nm for excitation and 530 nm for emission by inverted fluorescence microscope. Total green fluorescence intensities of each well were quantified using image-analysis software.
2-DE
2-DE was performed with 18 cm IPG strips (pH 3–10, GE healthcare), in accordance with a previously described protocol.Citation11 Protein samples (250 ug) extracted from untreated and control and 5.4 uM dioscin treatment were used for 2-DE analysis. Triplicate electrophoresis was performed to ensure reproducibility. All gels were visualized by silver staining. Image acquisition and analysis were performed with Imagemaster software (GE Healthcare). Comparisons were made between gel images of cells treated with dioscin and untreated controls. Altered protein spots with consistent and significant volume changes (> 2-fold differences) were selected for MALDI-TOF-MS spectrometer (Applied Biosystems) with delayed extraction and reflection. The resulting data were identified by searching NCBI database. Triplicate runs were made to ensure the accuracy of the analysis.
Protein gel blot analysis
Quantified protein lysates (15 µg) were resolved on SDS-PAGE gel, transferred onto PVDF membrane (Millipore), and immunoblotted with antibodies for caspase-3, caspase-8, caspase-9, Cytochrome C, Bcl-2, BAX, PRDX1, PRDX6 and tubulin (Cell Signaling). Triplicate independent experiment was repeated.
Vector construction and transfection
The full-length cDNA of PRDX1 and PRDX6 were amplified and subcloned into the pcDNA3.1 (+) vector (invitogen) respectively. After PCR and DNA sequencing validation, the positive plasmids were co-transfected in to Kyse510 cells using Lipofectamine 2000 as described by the manufacturer (Life Technologies) and empty vector was used as a control. After 48 h of co-inucbation, transfected cells were selected in primary cell culture medium containing 400 μg/ml G418. After 2–3 weeks, single independent clones were randomly isolated, and each individual clone was plated separately. After clonal expansion, cells from each independent clone were tested for PRDX1 and PRDX6 expression by protein gel blotting.
Colony formation assay
To determine long-term effects of dioscin, normal and PRDXs overexpression Kyse510 cells were seeded on a 6-well plates at a density of 300 cells per well. Cells were treated with vehicle or dioscin for 4 h. After being rinsed with fresh medium, cells were allowed to grow for 10 d to form colonies, which were then fixed with 4% paraformaldehyde for 30 min and stained by coommasie blue. Triplicate independent experiment was repeated.
Statistical analysis
Statistical analysis was performed using two-tailed Students t-test and p < 0.05 was considered significant. Data were expressed as mean ± SD in triplicate, and reproducibility was confirmed in three separate experiments.
Acknowledgments
We thank Vicki Geall of the Research Services Section of The University of Hong Kong for editorial assistance. This work was supported in part by the National Natural Science Foundation of China (Grant No. 30371761).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Li M, Han X, Yu B. Synthesis of monomethylated dioscin derivatives and their antitumor activities. Carbohydr Res 2003; 338:117 - 21; http://dx.doi.org/10.1016/S0008-6215(02)00443-3; PMID: 12526835
- Cai J, Liu M, Wang Z, Ju Y. Apoptosis induced by dioscin in HeLa cells. Biol Pharm Bull 2002; 25:193 - 6; http://dx.doi.org/10.1248/bpb.25.193; PMID: 11853164
- Wang Y, Cheung YH, Yang Z, Chiu JF, Che CM, He QY. Proteomic approach to study the cytotoxicity of dioscin (saponin). Proteomics 2006; 6:2422 - 32; http://dx.doi.org/10.1002/pmic.200500595; PMID: 16548062
- Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res 2010; 44:479 - 96; http://dx.doi.org/10.3109/10715761003667554; PMID: 20370557
- Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med 2010; 48:749 - 62; http://dx.doi.org/10.1016/j.freeradbiomed.2009.12.022; PMID: 20045723
- Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000; 5:415 - 8; http://dx.doi.org/10.1023/A:1009616228304; PMID: 11256882
- Matés JM, Sanchez-Jimenez FM. Role of reactive oxygen species in apoptosis: implications for cancer therapy. Int J Biochem Cell Biol 2000; 32:157 - 70; http://dx.doi.org/10.1016/S1357-2725(99)00088-6; PMID: 10687951
- Hildeman DA, Mitchell T, Kappler J, Marrack P. T cell apoptosis and reactive oxygen species. J Clin Invest 2003; 111:575 - 81; PMID: 12618509
- Forman HJ, Maiorino M, Ursini F. Signaling functions of reactive oxygen species. Biochemistry 2010; 49:835 - 42; http://dx.doi.org/10.1021/bi9020378; PMID: 20050630
- Lu W, Ogasawara MA, Huang P. Models of reactive oxygen species in cancer. Drug Discov Today Dis Models 2007; 4:67 - 73; http://dx.doi.org/10.1016/j.ddmod.2007.10.005; PMID: 18591999
- Fu L, Qin YR, Xie D, Chow HY, Ngai SM, Kwong DL, et al. Identification of alpha-actinin 4 and 67kDa laminin receptor as stage-specific markers in esophageal cancer via proteomic approaches. Cancer 2007; 110:2672 - 81; http://dx.doi.org/10.1002/cncr.23110; PMID: 17960614
- Wang ZY, Wang DM, Loo TY, Cheng Y, Chen LL, Shen JG, et al. Spatholobus suberectus inhibits cancer cell growth by inducing apoptosis and arresting cell cycle at G2/M checkpoint. J Ethnopharmacol 2011; 133:751 - 8; http://dx.doi.org/10.1016/j.jep.2010.11.004; PMID: 21073941
- Jana S, Paliwal J. Apoptosis: potential therapeutic targets for new drug discovery. Curr Med Chem 2007; 14:2369 - 79; http://dx.doi.org/10.2174/092986707781745569; PMID: 17896985
- Stoneman VE, Bennett MR. Role of Fas/Fas-L in vascular cell apoptosis. J Cardiovasc Pharmacol 2009; 53:100 - 8; http://dx.doi.org/10.1097/FJC.0b013e318198fe60; PMID: 19188840
- Timmer T, de Vries EG, de Jong S. Fas receptor-mediated apoptosis: a clinical application?. J Pathol 2002; 196:125 - 34; http://dx.doi.org/10.1002/path.1028; PMID: 11793363
- Sheridan C, Martin SJ. Mitochondrial fission/fusion dynamics and apoptosis. Mitochondrion 2010; 10:640 - 8; http://dx.doi.org/10.1016/j.mito.2010.08.005; PMID: 20727425
- Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci 2009; 122:437 - 41; http://dx.doi.org/10.1242/jcs.031682; PMID: 19193868
- Fulda S. Exploiting mitochondrial apoptosis for the treatment of cancer. Mitochondrion 2010; 10:598 - 603; http://dx.doi.org/10.1016/j.mito.2010.05.001; PMID: 20519148
- Engel RH, Evens AM. Oxidative stress and apoptosis: a new treatment paradigm in cancer. Front Biosci 2006; 11:300 - 12; http://dx.doi.org/10.2741/1798; PMID: 16146732
- Qi XF, Kim DH, Yoon YS, Kim SK, Cai DQ, Teng YC, et al. Involvement of oxidative stress in simvastatin-induced apoptosis of murine CT26 colon carcinoma cells. Toxicol Lett 2010; 199:277 - 87; http://dx.doi.org/10.1016/j.toxlet.2010.09.010; PMID: 20883752
- Kuo PL, Chen CY, Hsu YL. Isoobtusilactone A induces cell cycle arrest and apoptosis through reactive oxygen species/apoptosis signal-regulating kinase 1 signaling pathway in human breast cancer cells. Cancer Res 2007; 67:7406 - 20; http://dx.doi.org/10.1158/0008-5472.CAN-07-1089; PMID: 17671211
- Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res 1991; 51:794 - 8; PMID: 1846317
- Cabello CM, Bair WB, Wondrak GT. Experimental therapeutics: targeting the redox achilles heel of cancer. Curr Opin Investig Drugs 2007; 8:1022 - 37; PMID: 18058573
- Robinson MW, Hutchinson AT, Dalton JP, Donnelly S. Peroxiredoxin: a central player in immune modulation. Parasite Immunol 2010; 32:305 - 13; http://dx.doi.org/10.1111/j.1365-3024.2010.01201.x; PMID: 20500659
- Rhee SG, Kang SW, Chang TS, Jeong W, Kim K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life 2001; 52:35 - 41; http://dx.doi.org/10.1080/15216540252774748; PMID: 11795591
- Kang SW, Rhee SG, Chang TS, Jeong W, Choi MH. 2-Cys peroxiredoxin function in intracellular signal transduction: therapeutic implications. Trends Mol Med 2005; 11:571 - 8; http://dx.doi.org/10.1016/j.molmed.2005.10.006; PMID: 16290020
- Manevich Y, Fisher AB. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic Biol Med 2005; 38:1422 - 32; http://dx.doi.org/10.1016/j.freeradbiomed.2005.02.011; PMID: 15890616
- Kim JH, Bogner PN, Baek SH, Ramnath N, Liang P, Kim HR, et al. Up-regulation of peroxiredoxin 1 in lung cancer and its implication as a prognostic and therapeutic target. Clin Cancer Res 2008; 14:2326 - 33; http://dx.doi.org/10.1158/1078-0432.CCR-07-4457; PMID: 18413821
- Pak JH, Choi WH, Lee HM, Joo WD, Kim JH, Kim YT, et al. Peroxiredoxin 6 overexpression attenuates cisplatin-induced apoptosis in human ovarian cancer cells. Cancer Invest 2011; 29:21 - 8; http://dx.doi.org/10.3109/07357907.2010.535056; PMID: 21166495
- Kim YJ, Lee WS, Lp C, Chae HZ, Park EM, Park YM. Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res 2006; 66:7136 - 42; http://dx.doi.org/10.1158/0008-5472.CAN-05-4446; PMID: 16849559
- Riddell JR, Bshara W, Moser MT, Spernyak JA, Foster BA, Gollnick SO. Peroxiredoxin 1 controls prostate cancer growth through Toll-Like receptor 4-dependent regulation of tumor vasculature. Cancer Res 2011; 71:1637 - 46; http://dx.doi.org/10.1158/0008-5472.CAN-10-3674; PMID: 21343392
- Choi H, Chang JW, Jung YK. Peroxiredoxin 6 interferes with TRAIL-induced death-inducing signaling complex formation by binding to death effector domain caspase. Cell Death Differ 2011; 18:405 - 14; http://dx.doi.org/10.1038/cdd.2010.113; PMID: 20829884
- Ho JN, Lee SB, Lee SS, Yoon SH, Kang GY, Hwang SG, et al. Phospholipase A2 activity of peroxiredoxin 6 promotes invasion and metastasis of lung cancer cells. Mol Cancer Ther 2010; 9:825 - 32; http://dx.doi.org/10.1158/1535-7163.MCT-09-0904; PMID: 20354123