Abstract
The X-linked deubiquitinase USP9X affects the stability and activity of numerous regulatory proteins that influence cell survival. Recent studies suggest that decreased USP9X expression can confer a selective advantage onto developing cancer cells and thereby promotes disease progression. To examine the effect of USP9X on the cellular responses to anticancer therapies, we derived cancer cell lines in which the USP9X locus was disrupted by homologous recombination. The resulting USP9X-deficient cancer cells exhibited increased activation of apoptotic pathways and markedly decreased clonogenic survival in response to 5-fluorouracil, a chemotherapeutic drug that is widely used for treatment of gastrointestinal malignancies. These unexpected results suggest that cancers with low USP9X expression might be specifically sensitized to some conventional therapeutic agents.
Introduction
During tumorigenesis, evolving cancer cell clones tend to acquire resistance to death-inducing stimuli.Citation1 As the efficacy of many widely used anticancer agents is thought to depend at least in part on their ability to induce apoptosis and/or anoikis, the downregulation of these programmed cell death pathways during tumorigenesis could represent a significant obstacle to effective therapy. Non-apoptotic forms of cell death can also impair clonogenic survival and thus contribute to optimal therapeutic responses, but little is known about their relative importance.
One mediator of cell survival that has recently been the focus of considerable attention is the ubiquitin-specific protease 9x (USP9X), a deubiquitinase encoded on the X chromosome. There are several substrates through which USP9X has been shown to control cell death pathways. In cancer cells of lymphocyte lineage, USP9x binds, deubiquitinates and stabilizes the pro-survival BCL2 family member MCL1 and thereby downregulates apoptosis.Citation2 In the acinar cells of the pancreas, USP9X can function in a pro-survival pathway that affects autophagy.Citation3 In other cellular contexts, USP9X can promote cell death. USP9X deubiquitinates and stabilizes Itch,Citation4 an E3 ligase that mediates the degradation of several proteins that support cell survival.Citation5 The transcriptional activity of SMAD4, a tumor suppressor protein that is inactivated in the majority of pancreatic cancersCitation6 can be activated by USP9X.Citation7
The ability of USP9X to control both pro-death and pro-survival proteins in cultured cells has complicated efforts to understand its potential role in cancers. The ability of USP9X to stabilize the anti-apoptotic MCL1 oncoprotein in lymphoma and myeloma cells suggested that USP9X might be a promising therapeutic target.Citation8 A recent study has challenged the generality of this conclusion. Disruption of Usp9x by transposon-mediated mutagenesis was found to accelerate tumorigenesis in a mouse model of pancreatic ductal neoplasia, suggesting that USP9X might actively suppress tumor formation in some epithelial tissues.Citation9 In murine cells, knock down of Usp9x suppressed anoikis and promoted anchorage-independent growth, suggesting that Usp9x is required for tissue homeostasis in pancreatic ducts. Interestingly, MCL1 was not found to be affected by Usp9x levels in this system. Based upon this mouse data, an analysis of human pancreatic cancers revealed that USP9X mRNA and protein were reduced in cancer tissues, and that decreased expression in tumors correlated with shortened patient survival.Citation9 These studies suggest that in the epithelia of the pancreatic ducts, and perhaps in other tissues, USP9X primarily functions to inhibit tumorigenesis by preventing the survival of cells that detach from the basement membrane.
The direct correlation between USP9X levels and patient outcomes indicates that cancer cells with low USP9X expression are particularly dangerous. In an effort to better understand how such cells might be more effectively killed, we used gene targeting methods to develop human cancer cell lines deficient in USP9X. In vitro assessment of these novel cell lines demonstrated a significant role for USP9X in resistance to 5-fluorouracil, an antimetabolite that is a mainstay of adjuvant therapy for colorectal cancer and pancreatic cancer.
Results
USP9X (alternatively known as FAM) is encoded by a 45-exon gene that spans 150 kb on Xp11.4. To evaluate the cellular phenotypes of USP9X, we designed a gene targeting construct that would delete two coding exons and disrupt the reading frame of downstream exons upon homologous integration (). This targeting construct was packaged in a recombinant adeno-associated virus and delivered to the colorectal cancer cells HCT116 and DLD-1. These mismatch repair-deficient cell lines were chosen for analysis because both are karyotypically stable and harbor a single X chromosome.Citation10 Gene-targeted derivatives of these lines have been extensively employed to study the effects of individual genes and pathways on cellular responses to therapeutic agents.Citation11-Citation16 Such studies have revealed unambiguous phenotypes in knockout cells that in many cases were not apparent after RNAi-mediated knockdown.Citation17
Figure 1. Targeted inactivation of human USP9X. (A) A targeting construct with two regions of homology flanking a selectable marker cassette (Neor) was designed to delete a 0.8 kb region containing exons 7 and 8 upon homologous integration. Primers that anneal outside the region of homology, within the region of deletion and within the selectable marker were used to identify recombinant clones. Following infection of target cells with the targeting rAAV and screening, multiple homologous recombinant clones were identified. (B) Shown are representative PCR products obtained with the indicated primer pairs. In this experiment, template DNAs were derived from one nontargeted clone (NT) and two homologous recombinants (1 and 2), all derived from HCT116 cells.
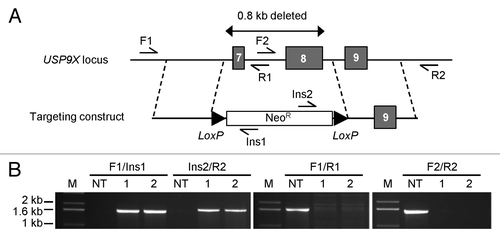
Following drug selection, transgenic clones were evaluated by PCR. In several positive clones, integration of the targeting vector was confirmed by amplification across both homology arms, and also by a loss of amplification within the intended region of deletion (). These PCR-positive cell lines exhibited a complete loss of USP9X protein expression ().
Figure 2. Levels and stability of MCL1 protein. (A) Cells of the indicated genotypes were treated for 2h with 10 μM MG132 (+MG132) or with DMSO (-MG132), and then harvested for protein analysis. Immunoblots were probed with the indicated antibodies. *Indicates a cross-reacting protein. (B) Wild type HCT116 cells and USP9X-deficient derivatives were incubated with the protein synthesis inhibitor cycloheximde (CHX; 20 μg/ml) for the times shown, and then harvested for protein analysis. The levels of α-tubulin were assessed as a loading control.
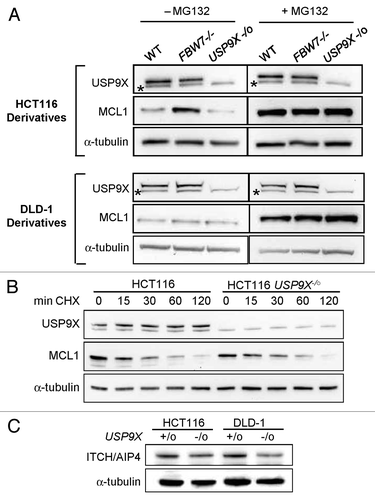
The stability of the MCL1 protein has been shown to be controlled by the net effects of the ubiquitin ligase FBW7 (also known as hCDC4) and USP9X, which respectively add and remove ubiquitin moieties.Citation2 We examined the levels of MCL1 in HCT116 and DLD-1 cells that harbor wild type USP9X and FBW7 alleles, and in isogenic derivatives in which these loci were disrupted by homologous recombination.Citation18 In the HCT116 background, an increase in MCL1 protein could be seen in FBW7-nullizygous cells, while the level of MCL1 was similar in USP9X-nullizygous cells and cells harboring wild type USP9X (). In DLD-1, the levels of MCL1 appeared to be independent of these genotypes. The levels of MCL1 in all cell lines could be increased by incubation of cells with the proteosome inhibitor MG132.
To investigate the effect of USP9X on MCL1 levels in more detail, we measured the half-life of MCL1 protein with the use of the protein synthesis inhibitor cycloheximide. In the HCT116 background, the kinetics of MCL1 degradation appeared similar in USP9X-deficient and -proficient cells, with a half-life of approximately 30 min (). Based on these experiments, we conclude that USP9X did not contribute to the stabilization of MCL1 in HCT116 or DLD-1. In contrast, USP9X-deficient cells did exhibit modestIy decreased levels of ITCH (). This result is consistent with a recent report that knockdown of murine USP9x could destabilize Itch.Citation9 ITCH protein levels are likewise decreased in human pancreatic cancers with low USP9X expression.Citation9
The chemotherapeutic drug 5-fluorouracil (5-FU) is frequently employed in the treatment of gastrointestinal malignancies. In vitro, 5-FU can trigger apoptosis in colorectal cancer cellsCitation11 and this effect has been proposed to contribute to its efficacy. We examined the induction of apoptosis in HCT116 and the USP9X-/o derivative after treatment with 5-FU. We treated a panel of isogenic HCT116 cells with two doses of 5-FU for 72 h and observed the cell nuclei for evidence of condensation, blebbing, or fragmentation, which are cardinal signs of programmed cell death. At the 5-FU doses tested, the USP9X-/o cell population exhibited a significantly increased proportion of apoptotic nuclei (). As has been previously observed,Citation19 apoptosis was suppressed in isogenic cells deficient in SMAD4; the effect of FBW7 genotype on apoptosis was variable.
Figure 3. Apoptosis induced by 5-FU. (A) HCT116 cells of the indicated genotypes were incubated with 5-FU (10 mM or 50 mM) for 72 h. Cell nuclei were scored for morphological evidence of apoptosis. (B) Cells were treated with 10 μM 5-FU for 72 h and then harvested for protein analysis. Immunoblots were probed with the indicated antibodies. (C) Cells were treated with 50 μM 5-FU for the times indicated. Subcellular fractions enriched for cytosolic (Cyto) or mitochondrial (Mito) proteins were assessed by immunoblot. The mitochondrial protein TOM20 and the cytosolic protein α-tubulin were assessed as loading controls. (*p ≤ 0.002).
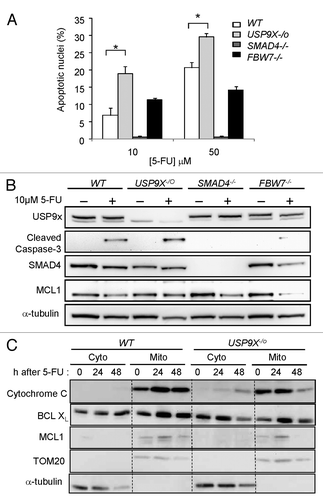
Analysis of proteins in the apoptosis pathways mirrored our quantitative analysis of cell nuclei. Cleavage of caspase-3, an early event in the apoptosis proteolysis cascade, was higher in USP9X-deficient cells, suppressed in SMAD4-deficient cells and relatively unchanged in FBW7-deficient cells (). These results are consistent with previous reports that SMAD4 is required for 5-FU-induced apoptosis.Citation19 In subcellular fractions of USP9X-deficient cells, we could observe the translocation of the pro-death BCL2 family member BCLXL from the cytosol into the mitochondria in tandem with the movement of cytochrome c from the mitochondria into the cytosol, and the degradation of mitochondrial MCL1, from 24 to 48 h after the start of 5-FU treatment (). These apoptotic changes were not induced by this dose of 5-FU (50 μM) in wild type cells.
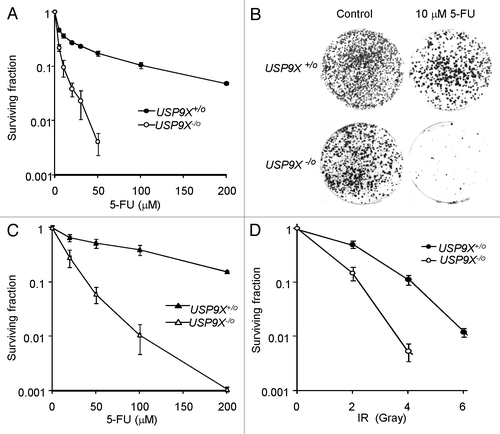
USP9X-knockout HCT116 cells exhibited a marked survival defect after 5-FU treatment, as compared with isogenic USP9X-proficient controls (). This effect was apparent over a broad dose range. DLD-1 cells were somewhat more resistant to 5-FU than HCT116, but the proportional effect of USP9X-nullizygosity was similarly striking (). As many treatment regimens involve the combination of a drug with ionizing radiation (IR), we also examined the effects of IR on clonogenic survival. We observed a more modest but still significant survival defect in USP9X-deficient cells after treatment with clinically relevant doses of IR ().
Figure 4. Effects of USP9X on clonogenic survival. (A) The survival of HCT116 cells and the USP9X-deficient derivative (USP9X -/o) was assessed following treatment with 5-FU at the indicated concentrations for 24 h. (B) Representative plates from experiment described in (A). Colonies are stained with crystal violet. (C) The survival of DLD-1 cells and the USP9X-deficient derivative (USP9X -/o) was assessed following treatment with 5-FU at the indicated concentrations for 24 h. (D) The survival of HCT116 cells and the USP9X-deficient derivative (USP9X -/o) was assessed following treatment with a single dose of ionizing radiation (IR) as indicated.
The apoptotic response to 5-FU in HCT116 cells is abrogated by genetic disruption of p53Citation11 or its transcriptional target FDXR.Citation12 While the defect in apoptosis caused by p53 deficiency is striking, clonogenic survival after 5-FU treatment is only modestly affected by p53 in this system.Citation11 This observation suggests that apoptosis alone cannot account for the loss of survival caused by 5-FU treatment. Indeed, we observed that DLD-1 cells that express only mutant p53 nonetheless exhibited sensitization to 5-FU following knockout of USP9X ().
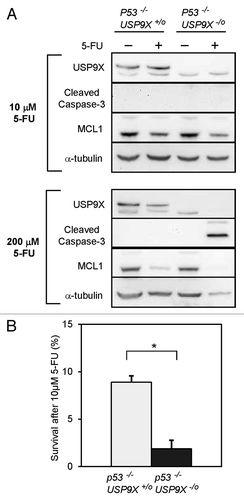
As functional p53 is frequently lost in evolving tumors of the gastrointestinal tract, we wished to confirm that USP9X can mediate apoptosis and/or survival in the context of p53-deficiency. To further explore the relationships between p53, apoptosis and survival, we derived additional USP9X -/o cell lines from p53 −/− HCT116 cells. As expected, p53-deficient cells were relatively resistant to apoptosis induced by a low dose of 5-FU, as demonstrated by a lack of caspase-3 cleavage and minimal degradation of MCL1 (). However, at a high dose of 5-FU, biochemical evidence of apoptotic activation could be observed in the double knockout cells (genotype p53−/− USP9X-/o). Interestingly, clonogenic survival was reduced in p53−/− USP9X-/o cells when treated under the conditions that produced little evidence of apoptosis (). These results suggest that even in the context of p53-deficiency, with the concomitant downregulation of apoptotic pathways, the loss of USP9X expression can sensitize cells to 5-FU.
Figure 5. Apoptosis and survival mediated by USP9X in p53-nullizygous HCT116 cells. (A) Cells of the indicated genotypes were treated with 10 μM or 200 μM 5-FU for 72 h, and then harvested for immunoblot analysis. (B) Clonogenic survival was assessed in USP9X-proficient and -deficient cells, in the p53−/− background, after treatment with 10 μM 5-FU for 72 h. (*p < 0.001).
Discussion
In this study, we have employed a simple but powerful genetic system to demonstrate a role for USP9X in resistance to 5-FU. The recent finding that a subset of particularly aggressive pancreatic cancers can be identified by their low levels of USP9X expressionCitation9 suggests that USP9X transcripts or protein might be useful as a biomarker for patient stratification. Our results extend this discovery. The exquisite sensitivity to 5-FU that we observed in cancer cells that do not express USP9X suggests that tumors with low USP9X expression might similarly exhibit increased sensitivity to therapeutic doses of 5-FU, a component of drug regimens that are widely employed for metastatic colorectal cancers and pancreatic cancers.Citation20 The effect of USP9X on evolving pancreatic ductal adenomas was discovered in a tissue-specific model system and then confirmed in human pancreatic tumor samples. It remains to be seen if USP9X plays a similar role in other types of cancer. If so, these results could be relevant to many patients. Further investigation will be needed to determine whether adjusted the dosing of 5-FU might be indicated for tumors with low USP9X expression.
The correlation between USP9X expression and 5-FU resistance was striking in our genetically defined model system, but this study raises several questions. It is interesting that differences in the induction of apoptosis between isogenic cell lines were not in proportion to the observed differences in clonogenic survival. This discordance has been observed previously. For example, p53-deficient cells exhibit a dramatic loss of 5-FU-induced apoptosis, but only a modest decrease in clonogenic survival after the same treatment.Citation11 Intuitively, apoptosis triggered in a cell population must decrease clonogenic survival to some extent, but clearly other cellular processes exert considerable influence. Here, we observed that genotype-specific differences in clonogenic survival () were greater than differences in apoptosis (). This finding implies that other cellular phenotypes, such as cell cycle arrest or senescence, might also suppress clonogenic survival in response to 5-FU. We did not observe significant differences in DNA content profiles that would point us to an obvious pathway (not shown). In addition to apoptosis, USP9X has been shown to control regulators of anoikis and TGF-β signaling. It remains to be shown whether control of these regulators by USP9X might contribute to 5-FU resistance in vitro and/or in vivo. Apoptosis triggered by 5-FU is dependent on SMAD4 (ref. Citation19; ), but our data suggest that SMAD4-deficient cancers may nonetheless be 5-FU sensitive if they have low expression of USP9X.
During tumorigenesis, evolving cell clones acquire phenotypes that allow them to successively expand their niches. In some cases, the genetic and epigenetic changes that promote the growth and spread of tumors also confer unique sensitivities that can be exploited. The decreased expression of USP9X that is observed in human pancreatic tumors confers a selective advantage that makes tumors more lethal, but may also provide an unexpected opportunity for effective therapy. It must be emphasized that the correlation between USP9X expression and 5-FU resistance remains at this point an in vitro phenomenon observed in a model system that is based on established cancer cells, and may not accurately predict the behavior of diverse tumors as they develop in situ. Nonetheless, these results suggest that further study of cancers with low levels of USP9X expression—and the effects of therapeutic agents on this subset of tumors—may be warranted.
Materials and Methods
Cell culture and gene targeting
The human colon cancer cell lines HCT116, DLD-1 and all genetically altered derivatives were cultured in 5% CO2, in McCoy’s 5A medium supplemented with 6% fetal calf serum and penicillin/streptomycin (Invitrogen). The endogenous USP9X locus was disrupted in HCT116 and DLD-1 cells using recombinant adeno-associated virus (rAAV)-based gene targeting methods. Briefly, a targeting construct (pAAV-USP9X) was assembled that contains two regions of genomic homology flanking a synthetic exon promoter trap (SEPT) cassette.Citation21 This construct was packaged into infectious rAAV that was subsequently used to generate transgenic clones. Identification and expansion of homologous recombinant cell lines was performed as described.Citation22 Details of vector construction and PCR screen are available upon request. At least two independent clones were isolated and analyzed for each derivative cell line described. All cell lines were authenticated within the past 12 mo by verification of targeted alterations by DNA sequencing.
Clonogenic survival
Clonogenic survival was assessed by a colony formation assay. Cells growing in multiwell plates were treated with drug for 24 h or a single dose of ionizing radiation delivered with a 135Cs irradiator (GammaCell) at a dose-rate of 0.5 Gy/min. Treated cells and untreated controls were washed with Hank’s Buffered Saline Solution (Invitrogen), and harvested following detachment in 0.5 ml trypsin/EDTA. Cells were transferred to 1.5 ml medium and vortexed. Approximately 104 cells were plated in triplicate to 10 cm tissue culture dishes and grown for 14 d. Colonies were stained with 0.2% crystal violet in 50% methanol. Colonies containing more than 50 cells were scored. Clonogenic survival is expressed as a proportion of the colony number on control dishes. Significance between data points was determined by the application of the Student’s t-test.
Antibodies and immunoblotting
Whole cell lysates were denatured, sonicated, and fractionated on NuPAGE gels (Invitrogen). Proteins were transferred to PVDF membranes (Millipore), which were then incubated with antibodies directed against USP9X (Novus), ITCH/AIP4 (Abcam), α−tubulin, SMAD4, TOM20 (Santa Cruz Biotechnology), MCL1, Cleaved Caspase-3, Cytochrome C, or BCL-XL (Cell Signaling Technologies) under conditions recommended by the manufacturers. Blots were developed using enhanced chemiluminescence (Amersham).
Microscopic analysis
Cells were collected by incubation in trypsin/EDTA, pelleted by centrifugation and were fixed in a solution containing 3.7% formaldehyde, 0.5% IGEPAL, and 10 μg/ml Hoechst 33258 in PBS. Apoptotic indices were determined by visual scoring of at least 300 nuclei. Significance was determined by the application of the Student’s t-test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was supported by funds from the Johns Hopkins SPORE in gastrointestinal cancer (P50CA062924) and a grant from the National Cancer Institute to F.B. (R01157535). D.R.H. was supported by a training grant from the National Cancer Institute (T32CA121937).
References
- Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer 2008; 8:121 - 32; http://dx.doi.org/10.1038/nrc2297; PMID: 18202696
- Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010; 463:103 - 7; http://dx.doi.org/10.1038/nature08646; PMID: 20023629
- Grasso D, Ropolo A, Lo Ré A, Boggio V, Molejón MI, Iovanna JL, et al. Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. J Biol Chem 2011; 286:8308 - 24; http://dx.doi.org/10.1074/jbc.M110.197301; PMID: 21173155
- Mouchantaf R, Azakir BA, McPherson PS, Millard SM, Wood SA, Angers A. The ubiquitin ligase itch is auto-ubiquitylated in vivo and in vitro but is protected from degradation by interacting with the deubiquitylating enzyme FAM/USP9X. J Biol Chem 2006; 281:38738 - 47; http://dx.doi.org/10.1074/jbc.M605959200; PMID: 17038327
- Bernassola F, Karin M, Ciechanover A, Melino G. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell 2008; 14:10 - 21; http://dx.doi.org/10.1016/j.ccr.2008.06.001; PMID: 18598940
- Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996; 271:350 - 3; http://dx.doi.org/10.1126/science.271.5247.350; PMID: 8553070
- Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell 2009; 136:123 - 35; http://dx.doi.org/10.1016/j.cell.2008.10.051; PMID: 19135894
- Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol 2011; 12:439 - 52; http://dx.doi.org/10.1038/nrm3143; PMID: 21697901
- Pérez-Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL, et al, Australian Pancreatic Cancer Genome Initiative. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 2012; 486:266 - 70; PMID: 22699621
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature 1997; 386:623 - 7; http://dx.doi.org/10.1038/386623a0; PMID: 9121588
- Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest 1999; 104:263 - 9; http://dx.doi.org/10.1172/JCI6863; PMID: 10430607
- Hwang PM, Bunz F, Yu J, Rago C, Chan TA, Murphy MP, et al. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med 2001; 7:1111 - 7; http://dx.doi.org/10.1038/nm1001-1111; PMID: 11590433
- Kim JS, Crooks H, Foxworth A, Waldman T. Proof-of-principle: oncogenic beta-catenin is a valid molecular target for the development of pharmacological inhibitors. Mol Cancer Ther 2002; 1:1355 - 9; PMID: 12516970
- Wilsker D, Bunz F. Loss of ataxia telangiectasia mutated- and Rad3-related function potentiates the effects of chemotherapeutic drugs on cancer cell survival. Mol Cancer Ther 2007; 6:1406 - 13; http://dx.doi.org/10.1158/1535-7163.MCT-06-0679; PMID: 17431119
- Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA Jr., Kinzler KW, et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A 2009; 106:3964 - 9; http://dx.doi.org/10.1073/pnas.0813333106; PMID: 19225112
- Sangster-Guity N, Conrad BH, Papadopoulos N, Bunz F. ATR mediates cisplatin resistance in a p53 genotype-specific manner. Oncogene 2011; 30:2526 - 33; http://dx.doi.org/10.1038/onc.2010.624; PMID: 21258400
- Karakas B, Weeraratna AT, Abukhdeir AM, Konishi H, Gustin JP, Vitolo MI, et al. P21 gene knock down does not identify genetic effectors seen with gene knock out. Cancer Biol Ther 2007; 6:1025 - 30; http://dx.doi.org/10.4161/cbt.6.7.4202; PMID: 17611398
- Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, et al. Inactivation of hCDC4 can cause chromosomal instability. Nature 2004; 428:77 - 81; http://dx.doi.org/10.1038/nature02313; PMID: 14999283
- Papageorgis P, Cheng K, Ozturk S, Gong Y, Lambert AW, Abdolmaleky HM, et al. Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Res 2011; 71:998 - 1008; http://dx.doi.org/10.1158/0008-5472.CAN-09-3269; PMID: 21245094
- Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011; 364:1817 - 25; http://dx.doi.org/10.1056/NEJMoa1011923; PMID: 21561347
- Topaloglu O, Hurley PJ, Yildirim O, Civin CI, Bunz F. Improved methods for the generation of human gene knockout and knockin cell lines. Nucleic Acids Res 2005; 33:e158; http://dx.doi.org/10.1093/nar/gni160; PMID: 16214806
- Rago C, Vogelstein B, Bunz F. Genetic knockouts and knockins in human somatic cells. Nat Protoc 2007; 2:2734 - 46; http://dx.doi.org/10.1038/nprot.2007.408; PMID: 18007609