Abstract
Among proteins in the c-Myc/Max/Mad/Sin3 regulatory complex, Mad4 and Sin3B are routinely detected in human glioblastoma multiforme (GBM) cell lines. In response to gamma radiation, the expression of Sin3B and Mad4 in GBM cells was upregulated in parallel over time, suggesting that Sin3B may play a role in the regulation of Mad4 stability. In agreement with this hypothesis, exogenously expressed Sin3B significantly stabilized co-transfected Mad4 and, to a lesser extent, endogenous Mad4. In addition, siRNA silencing of Sin3B induced an increase in the expression of c-Myc and Sin3A, which contributed to increased expression of Mad4. Simultaneous silencing of Sin3B, Sin3A and c-Myc decreased Mad4 stability to a greater extent than silencing of Sin3B alone. Although Mad1 was reported to be a target of c-IAP1 E3 ligase activity for degradation, the E3 ligase activity of c-IAP1 was not required for downregulation of Mad4 expression. The association of c-IAP1 with Sin3B or Mad4 suggested that Sin3B might interfere with the binding of c-IAP1 to Mad4; however, overexpression of Sin3B did not affect the interaction between Mad4 and c-IAP1. Instead, direct binding of Sin3B to c-IAP1 may protect Mad4 from degradation by c-IAP1, leading to enhanced stability of Mad4. Exogenous expression of Sin3B also inhibited c-IAP1-mediated degradation of Mad1, TRAF2, c-IAP2 and ASK1, known targets of c-IAP1 E3 ligase activity. These results indicate that Sin3B, together with other c-Myc regulatory members, maintain the steady-state level of Mad4, in part through inhibition of c-IAP1-mediated degradation of Mad4.
Introduction
The Myc gene product, c-Myc, is one of the most highly studied transcription factors, binding to at least 20% of genes in the human genome through its cognate binding site in the target promoter, the enhancer box (E-box).Citation1,Citation2 c-Myc protein possesses a transactivation domain covering approximately 1–150 amino acids residues in its N-terminus and a C-terminal DNA binding domain harboring a basic region, a helix-loop-helix domain and a leucine zipper region (bHLHZip), through which the transcription factor binds to the E-box.Citation3 However, binding of c-Myc to the E-box requires another bHLHZip-containing protein, Max. The c-Myc/Max heterodimer formed through the bHLHZip domain is the functional unit that binds to the c-Myc-specific E-box motifs in the promoter region to drive c-Myc-mediated transcription.
In addition to c-Myc, Max also interacts with a subset of bHLHZip Mad family proteins, including Mad1, Mxi1, Mad3, Mad4, Mnt and MGA.Citation4 These Mad proteins displace c-Myc to form Mad/Max heterodimers that are capable of binding the E-box motifs and repressing c-Myc-induced transcription, thus competitively inhibiting c-Myc transactivation.Citation5 Among Mad family members, Mad4 is the most abundant in the human brain and overexpression of Mad4 in human fibroblasts induces replicative senescence.Citation6
The Sin3-interacting domain (SID) in the N-terminus of Mad proteins was shown to mediate dimerization of Mad with the scaffold proteins Sin3A or Sin3B, which in turn interact predominantly with class I histone deacetylases (HDAC) HDAC1 or HDAC2, forming the Sin3-HDAC co-repressor.Citation7 HDAC1 and HDAC2 are implicated in chromatin modification and inhibition of gene expression by antagonizing histone acetyltransferase (HAT) activities at target genes. The complex formed between HDACs and Sin3/Mad/Max docks at the promoter of c-Myc-targeted genes, leading to local histone deacetylation and further repression of c-Myc-target genes.Citation4,Citation5
A growing body of evidence has revealed multiple roles of Sin3B in addition to its function in the Sin3/Mad/Max complex. In combination with HDAC, Sin3B is involved in repression of the G1 to S transition by inhibiting E2F target genes.Citation7 Overexpression of Sin3B induces senescence, confirmed by the observation that Sin3B knockout cells display resistance to replicative or oncogene-induced senescence, suggesting that Sin3B functions as a gate-keeper in the defense against cellular transformation.Citation8 Moreover, Sin3B is expressed at a high level in differentiated cells, upregulated in response to oncogenic stimuli and implicated in the downregulation of RNA polymerase II-mediated transcription through formation of a complex containing Sin3B, HDAC1, Mrg15 and the PHD finger-containing protein Pf1.Citation9
Based on our observations that Mad4 and Sin3B exhibit stable steady-state expression in human glioblastoma multiforme (GBM) cell lines and the knowledge that both proteins are involved in the regulation of c-Myc and E2F transcription factors and induce cellular replicative senescence, we investigated the interaction between Mad4 and Sin3B and their potential role in c-Myc transactivation in GBM. We found that Mad4 stability was regulated by Sin3B, Sin3A and c-Myc. Specifically, c-Myc upregulates Mad4 expression and c-Myc protein expression is modulated by Sin3B and, to a lesser extent, Sin3A. The inter-relationship among Mad4, Sin3 and c-Myc may provide insight into the inhibitory signaling pathways in human glioblastoma multiforme and through exploitation of these mechanisms, may lead to therapeutic interventions to improve the prognosis of patients with GBM.
Results
Exogenously expressed Sin3B dramatically increases Mad4 levels in GBM cells
During our studies on the effects of Sin3B and Mad4 on cell proliferation we found that overexpression of Sin3B led to increased stability of Mad4, but not vice versa. When various cell lines were transfected with Mad4 plasmid and an increasing amount of Sin3B plasmid, the expression levels of Mad4 were progressively upregulated with increasing amounts of co-transfected Sin3B (). Co-transfection of GFP with a control plasmid or Sin3B expression vector showed that overexpression of Sin3B did not lead to increased GFP protein levels in these cell lines (), confirming the specificity of Mad4 regulation by Sin3B.
Figure 1. Sin3B stabilizes exogenously expressed Mad4. (A) SF767 cells were transfected with FLAG-Mad4, FLAG-mSin3B and empty vector (EV) such that the total amount of plasmid used in each treatment was equal. After 42 to 48 h, cells were lysed in sample buffer and protein expression was measured by western blot analysis using the indicated antibodies. (B) U251 or U373 cells were transfected with the indicated plasmids and lysed in sample buffer after 42–48 h. Protein expression was determined by immunoblotting. (C) U251, U373 and SF767 cells were transfected with 8 µg Sin3B or empty vector in addition to 1 µg GFP expression vector. After 72 h, protein expression was assessed by immunoblotting as indicated.
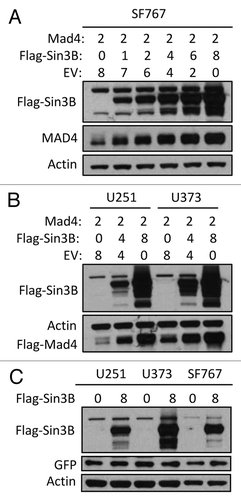
Exogenously expressed Sin3B stabilizes endogenous Mad4 protein in GBM cell lines
We next investigated whether Sin3B could also alter the steady-state expression level of endogenous Mad4 in GBM cells. We found that exogenously expressed Sin3B enhanced endogenous Mad4 expression in U251 and U373 cells () and surprisingly, both Sin3A and Sin3B enhanced the accumulation of endogenous Mad4 protein in SF767 cells (). In our previous studies, we determined that ionizing radiation induced differential modulation of Mad4 and Sin3B in various GBM cell lines. In this study, we showed that a continuous increase or a switch from increasing to decreasing Sin3B expression following irradiation was associated with a similar and concomitant change in Mad4 expression in H460 (), A172 (), U373 (, lower panel) and SF767 (, upper panel) cells.
Figure 2. Sin3B stabilizes endogenous Mad4 protein. (A) The same samples used in were assessed for endogenous Mad4 expression. (B) SF767 cells were transfected with 4 µg of empty plasmid vector or plasmids carrying human Sin3A and Sin3B. After 48 h, cells were assessed by immunoblotting as indicated. (C) H460 cells were radiated with 5 Gy and harvested at the times indicated. The expression levels of Sin3B and Mad4 over time were examined by immunoblotting. (D) Expression of Sin3B and Mad4 expression after irradiation of A172 cells with 5 Gy (C, control). (E) SF767 or U373 cells in 60-mm dishes were irradiated twice with 5 Gy each treatment over 2 d and subsequently harvested every 24 h. The expression levels of different proteins were measured as indicated (C, control). (F) Expression of Sin3B and Mad4 expression in A172 cells treated with temozolomide at increasing concentration for 43 h.
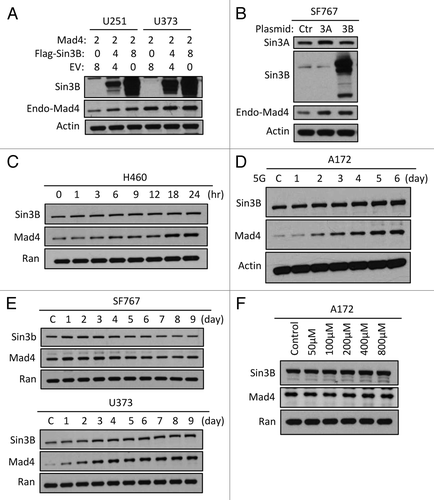
Notably, although H460 is a non-small cell lung cancer cell line it responded to ionizing radiation (IR) with an increase in Sin3B as well as Mad4, suggesting that enhanced Sin3B as an acute response to IR may be a generalizable phenomenon across tumor types. Compared with GBM cell lines, the change in Sin3B in response to IR was not as prominent in the time course of 24 h post-irradiation examined for H460 cells. When A172 or SF767 cells were treated and examined in a similar manner, the increase in Sin3B was relatively attenuated (data not shown). These observations suggest that Sin3B responds to IR with a continuous increase in expression resulting in the accumulation of Mad4 protein over days, post-irradiation.
To compare with a drug-induced DNA damage, we treated A172 cells with increasing concentrations of temozolomide (a standard alkylating agent used in the treatment of GBM). In contrast to our irradiation experiments, we did not detect an appreciable change in the expression levels of either Sin3B or Mad4 (). Taken together our data suggest that Sin3B enhances the expression of Mad4 in human GBM cell lines.
The relationship between Sin3, Mad4 and c-Myc
Although overexpression of Sin3B increased the stability of endogenous Mad4, siRNA knockdown of Sin3B did not reduce the expression of endogenous Mad4 in GBM cell lines (data not shown and ), suggesting a more complex regulatory network determining Mad4 expression in addition to the contribution of Sin3. Because the Sin3/Mad complex is implicated in the regulation of c-Myc transcriptional activity, we further investigated the relationship between Sin3, c-Myc and Mad4. siRNA knockdown of either Sin3A or Sin3B in GBM cells induced upregulation of c-Myc as well as enhanced the stability of Sin3B or Sin3A, respectively (). It has been reported that mouse Sin3A negatively regulates the transcription of Sin3B.Citation10 Therefore, reduction of Sin3A resulted in increased expression levels of c-Myc and Sin3B and depletion of Sin3B induced upregulation of c-Myc and a slight increase in Sin3A expression (). Our findings that knockdown of either Sin3B or Sin3A resulted in upregulation of c-Myc expression, lead us to speculate that c-Myc may play a role in the regulation of Mad4 expression.
Figure 3. Sin3B, Sin3A and c-Myc influence Mad4 stability. (A) Sin3A or Sin3B expression was silenced in four GBM cell lines by siRNA and c-Myc, Mad4 and β-actin expression was determined by immunoblotting. (B) SF767 cells were transfected with siRNA to knock down c-Myc expression. After 3 d, protein expression was assessed by immunoblotting. (C) Cells were transfected with siRNA oligos (30 pmol/well in 24-well plates) to knock down expression of Sin3B. After 2 d, cells were transfected with Flag-Mad4 plasmids at 2 μg per well. After 24 h, cells were assessed by immunoblotting. (D) Upper panel: SF767, U251 or U373 cells were transfected with 4 μg of Flag-Sin3A or c-Myc plasmid and 2 μg of Flag-Mad4 plasmid. After 48 h, the cell lysates were assessed by immunoblotting. Lower panel: Cells from four cell lines were lysed and assessed by immunoblotting as indicated. (E) SF676 cells were transfected with siRNA to knock down Sin3A and Sin3B expression and protein expression was assessed as indicated. (F) Left, expression of Sin3B in Sin3B wild-type or knockout MEFs; right, Sin3B wild-type or knockout MEFs were transfected with siRNA to deplete Sin3A expression and Mad4 expression was measured by immunoblotting. (G) U373 cells were transfected with siRNA to knockdown expression of Sin3A and Sin3B (AB) or Sin3A, Sin3B and c-Myc (ABM) (20 pmol each in 24-well plates) and protein expression level was determined as indicated. (H) TGR1 (c-Myc+/+) or HO15.19 (c-Myc−/−) cells were transfected with siRNA to silence expression of both Sin3A and Sin3B and the expression of Mad4 protein was assessed by immunoblotting.
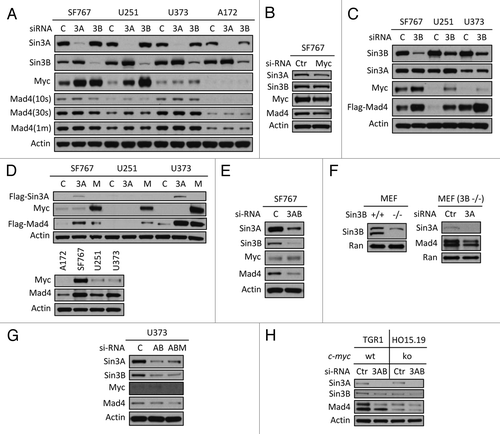
Knockdown of c-Myc expression in SF767 cells reduced expression of Sin3A and Mad4 but not expression of Sin3B, suggesting that c-Myc regulates Mad4 and/or Sin3A expression likely either transcriptionally or through a protein-protein interaction (). This was corroborated by the positive correlation observed between the levels of c-Myc protein and Sin3A or Mad4 in SF767, U251, U373 and A172 cell lines (data not shown or described below). After siRNA-induced knockdown of Sin3B, increased expression of c-Myc and Sin3A was associated with the enhanced expression of exogenously expressed Mad4 protein in SF767, U251 and U373 cells (). In addition, overexpression of Sin3A or c-Myc led to increased expression of ectopic Mad4 protein (, upper panel), consistent with the positive correlation between steady-state expression levels of c-Myc and Mad4 (, lower panel). These data suggest that, in addition to Sin3B, both c-Myc and Sin3A can also modulate Mad4 expression.
Simultaneous knockdown of Sin3A and Sin3B in SF767 cells induced a significant reduction in Mad4 expression (), but c-Myc upregulation was unexpectedly impaired compared with the levels shown in after knockdown of either gene alone. siRNA-mediated knockdown of Sin3A in Sin3B-null MEF cells resulted in a reduced level of Mad4 () (c-Myc expression in normal MEFs and Sin3B knockout MEF cells was undetectable, data not shown). Together, these results further suggest that Sin3A may also be implicated in the stabilization of Mad4 expression.
Knockdown of c-Myc in addition to knockdown of Sin3A and Sin3B led to a more pronounced reduction of Mad4 expression in U373 cells (). We also tested the effect of silencing Sin3A and Sin3B expression in c-Myc wild-type and knockout rat fibroblast cells. There was a marked reduction in Mad4 expression in HO15.19 (c-Myc−/−) cells compared with that in TGR1 (c-Myc+/+) cells () and additional knockdown of Sin3A and Sin3B in HO15.19 cells resulted in reduced Mad4 expression, further implicating c-Myc in the regulation of Mad4 expression in GBM cells.
The complex interplay among Sin3A, Sin3B, c-Myc and Mad4 indicates that Sin3B may play a dominant role in stabilizing endogenous Mad4. Although increased Sin3A and c-Myc can lead to upregulation of Mad4 expression, Sin3B expression induced by silencing of Sin3A stabilized Mad4 more effectively than the increased Sin3A or c-Myc expression resulting from the knockdown of Sin3B.
Mad4 degradation is regulated by c-IAP1
There are few reports on the regulation of degradation of Mad family members and to date only one Mad family member, Mad1, has been shown to be targeted for degradation by c-IAP1.Citation11 The inhibitors of apoptosis (IAPs) were first identified in baculovirus through the inhibition of apoptosis in infected insect cells. The function of cellular homologs of baculovirus IAPs, including c-IAP1, c-IAP2 and XIAP, is dependent upon their distinctive motifs: three tandem N-terminal baculoviral IAP repeat (BIR) domains involved in inhibition of apoptosis by binding to caspases and one C-terminal really interesting new gene (RING) domain implicated in the degradation of caspases as a result of their polyubiquitination by IAP E3 ligase activities.Citation12,Citation13 In addition to its role in inducing Mad1 degradation, c-IAP1 also targets other proteins, including TRAF2, c-IAP2 and ASK1, for degradation through its E3 ligase activity.Citation14-Citation16
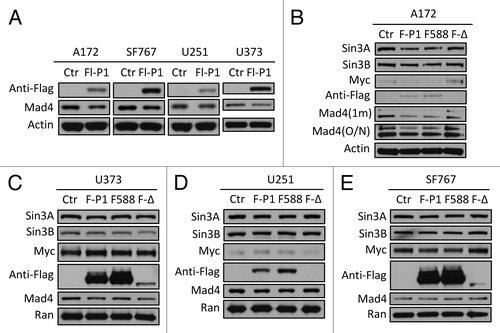
Since exogenously expressed Sin3B significantly enhanced the expression of exogenous Mad4 (), it is possible that Sin3B stabilizes Mad4 by negatively regulating Mad4 degradation. To examine the mechanism by which Mad4 degradation is regulated, we measured endogenous Mad4 protein levels after overexpression of c-IAP1, c-IAP2 or XIAP. Only c-IAP1 downregulated Mad4 expression levels in the four GBM cell lines tested (data not shown) and the degree of reduction by c-IAP1 was modest (). The moderate reduction in endogenous Mad4 by exogenously expressed c-IAP1 in these cell lines suggested that c-IAP1 may not play an important role in the degradation of Mad4 () even though Mad1 is targeted by c-IAP1 for ubiquitination and subsequent degradation.Citation11 To investigate whether the E3 ligase activity of c-IAP1 is required for Mad4 degradation, we transfected GBM cells with wild type, RING domain point mutant or deletion mutant of c-IAP1. As expected, a reduction in Mad4 expression was induced by wild-type c-IAP1 in all cell lines, but surprisingly also by the point mutant of c-IAP1 in A172 and U251 cells and by the RING deletion mutant of c-IAP1 in U373 cells (), whereas Mad4 expression was not obviously reduced by either mutant c-IAP1 in SF767 cells (). These findings indicate that the E3 ligase activity may not be essential for c-IAP1-mediated degradation of Mad4 in most of the GBM cell lines tested. It is intriguing that the point mutant and wild-type c-IAP1 caused a reduction in Sin3A, Sin3B and c-Myc expression in A172 and SF767 cells (), while these proteins were previously shown to stabilize Mad4 (). Finally, transfection with the RING deletion mutant of c-IAP1 led to reduced expression of Mad4 in U373, as well as a concomitant reduction in Sin3A and Sin3B and a slight increase in c-Myc (). Those observations suggest that c-IAP1 might affect expression of Sin3A, Sin3B and c-Myc and thus indirectly alter Mad4 levels in GBM cells.
Figure 4. Mad4 is targeted by c-IAP1 for degradation. (A) GBM cell lines were transfected with 6 µg of Flag-tagged c-IAP1 expression vector (Flg-P1) or control vector. After approximately 40 h, the expression level of endogenous Mad4 was determined by immunoblotting. (B) A172 cells were transfected with 8 µg of Flag-cIAP1 or H588A mutant or 12 µg of RING domain deletion mutant. After approximately 2 d, the expression levels of Sin3A, Sin3B, c-Myc and Mad4 were determined by immunoblotting (1 min, 1 min exposure; O/N, film exposed overnight). (C) U373 cells were transfected with 5 µg of Flag-tagged c-IAP1 or c-IAP1-H588A or 10 µg of RING domain deletion mutant and after 40–46 h cell lysates were analyzed by immunoblotting as indicated. (D) U251 cells were transfected with 6 µg of Flag-tagged c-IAP1 or c-IAP1-H588A or 10 µg of RING domain deletion mutant and after 40–46 h cell lysates were analyzed by immunoblotting as indicated. (E) SF767 cells were transfected with 6 µg of Flag-tagged c-IAP1 or c-IAP1-H588A or 12 µg of RING domain deletion mutant and after 40–46 h cell lysates were analyzed by immunoblotting as indicated.
IAPs are not only implicated in the regulation of apoptosis by inhibiting caspase catalytic activity through their BIR domains, but also transduce TNFα mediated initiation of NFκB transcription activity via poly-ubiquitination of many components of the TNR-I-mediated complex 1, including RIPK1 and TRADD.Citation13 Upon poly-ubiquitination, TAK1 activates IKKβ, which in turn phosphorylates NFκB inhibitors, leading to release of sequestrated NFκB transcription factors their translocation into the nucleus. c-IAP1/TRAF2 plays an important role in the process of poly-ubiquitination as an E3 ligase and an adaptor respectively. In addition to activation of the NFκB pathway, c-IAP1 has also been shown to degrade c-IAP2 as an E3 ligase and the ubiquitination of c-IAP2 by c-IAP1 is enhanced by TRAF2, which stabilizes c-IAP1 through its effect on c-IAP1 autoubiquitination.Citation15,Citation17 Even though c-IAP1 is able to induce degradation of Mad4, this activity does not appear to require its E3 ligase activity (). Because a complex of c-IAP1 and TRAF2 was reported to have enhanced E3 ligase activity that targets c-IAP2 for ubiquitination and proteasomal degradation, we examined whether TRAF2 plays a role in inducing Mad4 ubiquitination and degradation. Overexpression of wild-type TRAF2 or a RING domain deletion TRAF2 mutant with or without c-IAP1 in U251 or U373 cells suggested that ectopic expression of TRAF2 in addition to c-IAP1 did not further enhance Mad4 degradation by c-IAP1, although c-IAP1 was stabilized by either TRAF2 or ΔTRAF2 (data not shown).
Sin3B interferes with c-IAP1-mediated degradation of Mad4
To further investigate the interaction between c-IAP1 and Sin3B in the regulation of Mad4 stability we examined the effect of overexpressing Sin3B on c-IAP stability. Exogenously expressed Flag-Sin3B did not significantly affect the steady-state expression level of c-IAP1 in four human GBM cell lines () and conversely knockdown of Sin3B did not enhance endogenous c-IAP1 expression levels (). In fact, when the expression of endogenous Sin3B was reduced, endogenous c-IAP1 became unstable, especially in SF767 cells, in contrast to the observation that silencing of Sin3B enhanced the stability of exogenously expressed Flag-c-IAP1 and facilitated reduction of Mad4 (). These data suggest that homeostatic Sin3B levels are tightly related to c-IAP1 stability. Even if exogenous Sin3B does not directly modify c-IAP1 turnover, we speculated that Sin3B might interfere with the interaction between Mad4 and c-IAP1 in human GBM cells. To test this hypothesis, we first investigated whether c-IAP1 interacts with Mad4 or Sin3B in human GBM cells. Co-immunoprecipitation (co-IP) of c-IAP1 in four human GBM cell lines showed that endogenous c-IAP1 not only bound to Mad4, but also to Sin3B (). In the reciprocal IP, Mad4 was shown to interact with c-IAP1 in addition to Sin3B (). Second, we overexpressed Flag-tagged human Sin3B and examined changes in the interaction between endogenous c-IAP1 and Mad4. Exogenously expressed Flag-Sin3B was found to interact with c-IAP1 () in U373 cells; however, overexpression of Sin3B did not sequestrate the fraction of Mad4 protein that interacted with c-IAP1, as Mad4 bound c-IAP to a similar extent with or without exogenously expressed Sin3B () in U251 and U373 cells. These results suggested that Sin3B does not interfere with the interaction between c-IAP1 and Mad4, but rather enhances Mad4 stability by binding to c-IAP1 and inhibiting its activity, resulting in an increase in Mad4 expression.
Figure 5. Sin3B interferes with c-IAP1-mediated degradation of Mad4. (A) The plasmid Flag-Sin3B expressing Sin3B of human origin was transfected into four GBM cell lines and after 2 d cell lysates were analyzed for c-IAP1 expression by immunoblotting. (B) Knockdown of Sin3B in four cell lines was performed and c-IAP1 levels were examined by immunoblotting. (C) U251 cells were transfected with anti-human Sin3B siRNA and 2 d later the same cells were transfected with Flag-c-IAP1. After 40–44 h, cells were assessed by immunoblotting. (D) Cells from four cell lines were lysed in immunoprecipitation (IP) buffer and the lysates were pre-cleared by rabbit-IgG agarose and immunoprecipitated by rabbit anti-human c-IAP1 antibody. Immune complexes were captured by Protein G agarose. The co-precipitated proteins were assessed by immunoblotting as indicated (SE/LE, short/long exposure). (E) Cell lysates from SF767 or U373 cells in IP buffer were immunoprecipitated with goat anti-human Mad4 polyclonal antibody and analyzed with immunoblotting as indicated. Mad4 was analyzed with goat anti-human Mad4 antibody for input and with rabbit anti-human Mad4 antibody for IP. (F) U373 cells were transfected with Flag-Sin3B plasmids and lysed in IP buffer after 2 d. IP was performed using rabbit anti-human c-IAP1 polyclonal antibody and co-immunoprecipitated proteins were assessed as indicated. (G) U251 or U373 cells were transfected with Flag-Sin3B plasmids and treated with IP buffer after 2 d. Co-immunoprecipitated proteins were assessed by immunoblotting. (H) SF767/U373 cells were transfected with mouse Flag-Sin3B and U373/U251 cells were transfected with human Flag-Sin3B plasmids. After 2 d, protein expression was assessed by immunoblotting as indicated.
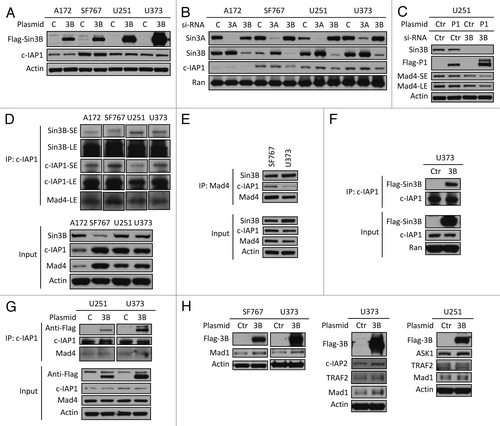
To finally validate that binding of Sin3B to c-IAP1 inhibits c-IAP1-mediated degradation of its targets, we examined reported substrates of the ubiquitin ligase c-IAP1. Flag-Sin3B was overexpressed in human GBM cell lines and expression of four known targets of c-IAP1 (Mad1, TRAF2, c-IAP2 and ASK1) was examined.Citation11,Citation14-Citation16 Exogenously expressed Sin3B significantly stabilized these four targets of c-IAP1 in the human cell lines, especially in SF767, U251 and U373 cells (). These results suggest that binding of Sin3B to c-IAP1 inhibits the activity of c-IAP1 and helps to stabilize Mad4 in human GBM cell lines.
Discussion
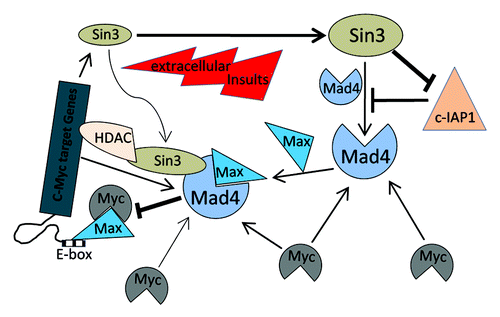
In the present study, we demonstrate that Sin3B, Sin3A and c-Myc may play a collective role in maintaining the steady-state level of Mad4 protein in GBM cells. Enhanced Mad4 expression may competitively inhibit the formation of the c-Myc/Max transcriptional complex, leading to the disruption of the c-Myc signaling pathway. Once c-Myc expression is deregulated, Mad4 as well as Sin3A may be correspondingly accumulated to help neutralize enhanced c-Myc activities. The defectiveness of this feedback loop may define an event in the progression of de novo GBM ().
Figure 6. Schematic representation of the regulation of Mad4 in GBM cells by Sin3 and c-Myc. Sin3B (as well as Sin3A) maintains the steady-state level of Mad4 partly by counteracting c-IAP1-mediated degradation of Mad4. Mad4 competitively sequesters c-Myc from its binding partner Max, thus suppressing c-Myc transactivities. Furthermore, the unbound c-Myc molecules may facilitate Mad4 protein accumulation. Upon extracellular insults, such as gamma radiation, Sin3B and Mad4 are increased, which may compromise c-Myc stability and thus interfere with c-Myc-mediated cell proliferation or further silence c-Myc targeted genes through Sin3B-linked HDAC activity. (→, enhancement; ⊣, inhibition).
Among the Mad family members, Mad4 is highly expressed in human brainCitation6 and was also more readily observed in GBM cell lines than Mad1, Mxi1, Mad3, Mnt and MGA (data not shown). We previously showed that Sin3B is also highly expressed in GBM cells and here we demonstrate that gamma irradiation treatment induces parallel changes in the expression of Mad4 and Sin3B (). Moreover, enhanced expression of Mad4 in fibroblasts triggers replicative senescence similar to overexpression of Sin3B,Citation6,Citation8 leading us to investigate whether there is a functional relationship between Mad4 and Sin3B proteins in GBM.
Although it was obvious that exogenously expressed Sin3B significantly stabilized co-transfected Mad4 in GBM cells, knockdown of Sin3B did not cause an easily distinguishable reduction in endogenous Mad4, suggesting strongly that multiple proteins may contribute to the regulation of Mad4 protein turnover in vivo. Within the c-Myc regulatory complex, c-Myc expression was increased in response to a reduction in endogenous Sin3B, especially in SF767 and U251 cell lines, which clinically represent classic and mesenchymal subgroups of GBM respectively and are both susceptible to aggressive treatments.Citation18 We also found that silencing of c-Myc led to a reduction in Mad4 and Sin3A expression, whereas overexpression of c-Myc or its increase by knockdown of Sin3B increased expression of exogenous Mad4. This novel finding is in contrast to the reported observation that c-Myc transcriptionally represses Mad4 expression through a Miz-1 mediated mechanism by binding to an initiator element (Inr) in the promoter of Mad4 in serum-stimulated quiescent young human WI38 lung embryonic fibroblasts.Citation19
The finding that the alterations in Mad4 expression described in our study were observed in highly malignant grade IV glioma cells as opposed to human WI38 lung embryonic fibroblasts suggests that high-grade gliomas may develop a novel association between c-Myc and Mad4. This is supported by the positive correlation between c-Myc and Mad4 protein expression levels in the studied GBM cell lines A172, SF767, U251 and U373 (). In the classic subgroup of GBM (including SF767 cells), the EGFR gene is amplified in 95% of patients, possibly leading to a higher c-Myc expression level via the Ras/Raf/ MEK/ERK signaling pathway.Citation18,Citation20 One could speculate that the increased c-Myc moieties in the early developing glioma milieu may facilitate the accumulation of Mad4 proteins, thus attenuating the adverse effects elicited by c-Myc-mediated cell cycle arrest and cell death before additional genetic mutations are generated in the genome (). Furthermore, higher levels of Mad4 in GBM cells may enhance their survival by causing them to enter senescence in response to DNA damage induced by radio- and chemotherapies. In addition, overexpression of Sin3A also induced an increase in exogenously expressed Mad4. It is interesting to note that the knockdown of Sin3A resulted in a more obvious increase in Sin3B expression than vice versa, suggesting that Sin3A and Sin3B each affect the stability of each other, but to differing degrees and both contribute to maintenance of the endogenous Mad4 protein level. Therefore, in the c-Myc regulatory loop the regulation of Mad4 stability involves many effectors and exactly how c-Myc is associated with Mad4/Sin3 with respect to transcription, protein-protein interaction and post-translational modification is currently being studied. The results of these studies will advance our perspectives regarding the molecular underpinnings of GBM.
The regulation of Mad/Sin3 proteins through degradation has not been extensively studied. Previous reports have shown that RING finger protein 220 (RNF220), functioning as an E3 ligase, is responsible for targeting Sin3B for degradation, although the RING-deleted RNF220 mutant retains some activity in the degradation of Sin3B.Citation21 In addition, Mad1 is targeted by the E3 ligase c-IAP1.Citation11 Interestingly, overexpression of c-IAP1 protein with a mutation in the RING domain in 293T or GBM cell lines resulted in a reduction in Mad1 levels (data not shown), which is in contrast to a previous report but is consistent with c-IAP1-mediated degradation of Mad4 independent of the RING domain. It is possible that other regions of the protein may functionally compensate for the loss of RING domain integrity in the c-IAP1 RING mutants because the p300-CBP-associated factor (PCAF), which lacks a RING, HECT or U-box domain, possesses an intrinsic E3 ligase activity for HDM2 ubiquitination and subsequent proteasomal degradation. This activity is lost if amino acids 121–242 of PCAF are deleted,Citation22 indicating that E3 activity may be conferred by functional motifs other than the canonical domains. Further experiments are required to clarify whether c-IAP1 contains a specific motif that contributes to its E3 activity.
c-IAP1 has been reported to efficiently degrade Mad1 and there is convincing evidence that the BIR domains of c-IAP1 are responsible for binding to Mad1.Citation11 However, because the BIR domains in c-IAP1 are not as efficient as those in XIAP for the inhibition of caspases,Citation13 binding of Mad1 to the c-IAP1 BIR domains would not interfere with the inhibition of apoptosis by IAPs unless it can be demonstrated that Mad1 also interacts with XIAP via its BIR domains. A report on c-IAP1-mediated degradation of Mad1 showed that overexpression of c-IAP1 wild-type or RING point mutant (H588A) increases, albeit slightly, the levels of both endogenous and exogenous c-Myc (see in ref. Citation11). Similar results were obtained when c-IAP1-wt or –H588A was overexpressed in GBM cells and c-IAP1-H588A induced a slightly stronger increase in c-Myc expression than c-IAP1-wt (data not shown). Because c-Myc appears to upregulate Mad4 expression (), the reduction of Mad4 by overexpression of c-IAP1 may be partly offset by increased c-Myc protein levels. This may be one reason why c-IAP1 is relatively inefficient in directing Mad4 degradation.
Although our study does not fully elucidate how Mad4 is regulated, it provides a new layer of complexity to the c-Myc regulatory unit and presents the novel finding that Mad4 is regulated simultaneously by several different effectors. If the expression of any of these proteins is modulated, the expression of other interrelated components will be altered accordingly. Unless the stability of Mad4 is predominantly regulated by Sin3B, the knockdown of Sin3B expression will not necessarily lead to a significant modulation of Mad4 expression levels. Further studies on the effects of Sin3 on Mad4 or other Mad family members such as Mad1 () will provide a novel insight into the regulation of GBM cell viability and cell cycle progression.
Increased expression of Sin3B followed by Mad4 in GBM cells not only represents a response to extracellular insults (such as gamma irradiation and chemotherapy), but may also represent a specific biomarker of glioma as has been published previouslyCitation23 (). Before additional genetic mutations affect the homeostatic surveillance system, such as p53 mutations, aberrantly increased c-Myc often induces cell cycle arrest, senescence or death.Citation24-Citation26 Dysregulated Sin3/Mad4 expression during gliomagenesis or in response to genomic instability induced by carcinogens or irradiation, may contribute to the abrogation of c-Myc-mediated proliferation, cell cycle exit or apoptosis. Sin3/Mad4 expression profiling in patient biopsy samples should help elucidate the role of these signaling molecules and potentially begin to define a therapeutic approach. Further studies are needed to confirm these hypotheses.
Materials and Methods
Cells
The GBM cell lines A172, SF767, U251 and U373 were purchased from American Type Culture Collection and were maintained at 37°C in DMEM with 4.5 g/ml glucose, L-glutamine and sodium pyruvate (Mediatech, MT10–013-CV or MT10–017-CM), plus 10% FBS, 100 U/ml penicillin and 100 µg/ml streptomycin in a humidified atmosphere with 5% CO2. Other cell lines, including H460 and MEFs (Sin3B+/+ or −/−), were grown under conditions described in reference Citation7. Cell lines TGR1 (c-Myc+/+) and HO15.19 (c-Myc−/−) were kindly provided by Dr. John M Sedivy (Department of Molecular Biology, Cell Biology and Biochemistry, Brown University, Providence, RI) and cultured as described previously.Citation27
Plasmids
The mouse Sin3B mammalian expression vector, Flag-Sin3B was constructed as described previously.Citation9 Human Mad4 (RC209651), Sin3A (RC213240) and Sin3B (RC216859) mammalian expression vectors were purchased from OriGene (Rockville, MD) and human c-Myc mammalian expression vector (ID#, 16011) was purchased from Addgene (Cambridge, MA). Human Flag-tagged c-IAP1, c-IAP1-H588A and c-IAP1-ΔRING mammalian expression vectors were previously describedCitation28 and were kindly provided by Dr. Junying Yuan (Department of Cell Biology, Harvard Medical School). Mammalian expression vectors for human TRAF2, pcDNA3-HA-TRAF2 and pcDNA3-HA-ΔTRAF2 (RING domain deletion mutant) were generous gifts from Dr. Jonathan D. Ashwell (National Institutes of Health, Bethesda, MD).
Reagents
Antibodies used for immune assays were as follows: Mad4 (sc-771; sc-1221), Sin3B (sc-768), Sin3A (sc-5299), c-Myc (sc-40), β-actin (sc-47778) and Mad1 (sc-222) from Santa Cruz Biotechnology; Ran (610340) from BD Biosciences PharMingen; Anti-Flag m2 (200472) from Stratagene; c-IAP1 (4952 and 7065), c-IAP2 (3130), TRAF2 (4712) and ASK1 (3762) from Cell Signaling Technology. The siRNA oligonucleotides used for knockdown experiments were: human Sin3A (sc-35973, Santa Cruz Biotechnology), Sin3B (s23529, Ambion; sc-35975, Santa Cruz) and c-Myc (6341, Cell Signaling Technology). Temozolomide (TMZ) was purchased from Sigma-Aldrich.
Cell treatment and western blot analysis
Cells were seeded in 6-well plates and treated the next day at 30–50% confluence with radiation at a dose rate of 1.6 Gy/min using a Mark I cesium irradiator (J.L. Shepherd, San Fernando, CA). The total dosage used to irradiate cells was 5–10 Gy in 24 or 48 h. Cells were used at 30–50% confluence for siRNA oligo transfection and at 90–100% confluence for plasmid transfection using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s manual. For western blotting, cells were lysed in 2X Laemmli sample buffer (125 mM TRIS-HCl, pH 6.8, 10% β-mercaptoethanol (v/v), 4% SDS, 20% glycerol, 0.05% bromophenol blue) followed by heat denaturation. Cells were lysed for analysis on different days after irradiation in 6-well plates or 60-mm dishes or 2 d (40–48 h) after transfection. Proteins were resolved in 4–12% NuPAGE using a Novex® NuPAGE® Gel Electrophoresis Systems (Invitrogen). Standard immunoblotting was performed with the indicated antibodies and visualized by enhanced chemiluminescence (Amersham Biosciences).
Immunoprecipitation
Cells were washed with cold PBS on ice three times before addition of immunoprecipitation (IP) buffer (30 mM Tris, pH7.5, 150 mM NaCl, 10% glycerol and 1% Triton X-100 supplemented with 1X complete protease inhibitors (Roche) and 1 mM phenylmethylsulfonyl fluoride). Cell lysates were rotated for 1 h at 4°C with high-speed vortexing every 20 min and then centrifuged at maximum speed in a microcentrifuge for 20 min. The supernatant was pre-cleared with mouse, rabbit or goat IgG-conjugated agarose for 1 h, followed by overnight incubation at 4°C with antibodies against the proteins of interest. The protein/antibody complex was captured by Protein G agarose (Invitrogen). The Protein G agarose was washed on ice with IP buffer six times before elution of immunoprecipitated proteins in 1X Laemmli sample buffer and heat-denaturation. The co-immunoprecipitated (co-IP) proteins were analyzed by western blotting as described above.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by a Burroughs Wellcome Career Award for Medical Scientists (1006792 to J.F.D.) and the National Institutes of Health (K08 NS076548–01 to J.F.D.). We are very grateful to Dr. Junying Yuan in the Department of Cell Biology, Harvard Medical School for sharing c-IAP1 plasmids and to Dr. Jonathan D. Ashwell in the National Institutes of Health, Bethesda, MD for sharing mammalian expression vectors for TRAF2 of human origin, pcDNA3-HA-TRAF2 and pcDNA3-HA-ΔTRAF2 (RING domain deletion mutant). In addition, we are obliged to Dr. John M. Sedivy in the Department of Molecular Biology, Cell Biology and Biochemistry, Brown University, Providence, RI for providing rat cell lines TGR1 (c-Myc+/+) and HO15.19 (c-Myc−/−). We thank Drs. Costas Koumenis and Gary Kao for helpful discussions. Finally, we acknowledge Dr. Stephen M. Hahn whose encouragement and support has helped make this research possible.
References
- Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc Natl Acad Sci U S A 2003; 100:8164 - 9; http://dx.doi.org/10.1073/pnas.1332764100; PMID: 12808131
- Perna D, Fagà G, Verrecchia A, Gorski MM, Barozzi I, Narang V, et al. Genome-wide mapping of Myc binding and gene regulation in serum-stimulated fibroblasts. Oncogene 2012; 31:1695 - 709; http://dx.doi.org/10.1038/onc.2011.359; PMID: 21860422
- Lüscher B, Larsson LG. The basic region/helix-loop-helix/leucine zipper domain of Myc proto-oncoproteins: function and regulation. Oncogene 1999; 18:2955 - 66; http://dx.doi.org/10.1038/sj.onc.1202750; PMID: 10378692
- Hurlin PJ, Huang J. The MAX-interacting transcription factor network. Semin Cancer Biol 2006; 16:265 - 74; http://dx.doi.org/10.1016/j.semcancer.2006.07.009; PMID: 16908182
- Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol 2005; 6:635 - 45; http://dx.doi.org/10.1038/nrm1703; PMID: 16064138
- Marcotte R, Qian JF, Chen J, Wang E. hMad4, c-Myc endogenous inhibitor, induces a replicative senescence-like state when overexpressed in human fibroblasts. J Cell Biochem 2003; 89:576 - 88; http://dx.doi.org/10.1002/jcb.10517; PMID: 12761891
- David G, Grandinetti KB, Finnerty PM, Simpson N, Chu GC, Depinho RA. Specific requirement of the chromatin modifier mSin3B in cell cycle exit and cellular differentiation. Proc Natl Acad Sci U S A 2008; 105:4168 - 72; http://dx.doi.org/10.1073/pnas.0710285105; PMID: 18332431
- Grandinetti KB, Jelinic P, DiMauro T, Pellegrino J, Fernández Rodríguez R, Finnerty PM, et al. Sin3B expression is required for cellular senescence and is up-regulated upon oncogenic stress. Cancer Res 2009; 69:6430 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-09-0537; PMID: 19654306
- Jelinic P, Pellegrino J, David G. A novel mammalian complex containing Sin3B mitigates histone acetylation and RNA polymerase II progression within transcribed loci. Mol Cell Biol 2011; 31:54 - 62; http://dx.doi.org/10.1128/MCB.00840-10; PMID: 21041482
- Dannenberg JH, David G, Zhong S, van der Torre J, Wong WH, Depinho RA. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev 2005; 19:1581 - 95; http://dx.doi.org/10.1101/gad.1286905; PMID: 15998811
- Xu L, Zhu J, Hu X, Zhu H, Kim HT, LaBaer J, et al. c-IAP1 cooperates with Myc by acting as a ubiquitin ligase for Mad1. Mol Cell 2007; 28:914 - 22; http://dx.doi.org/10.1016/j.molcel.2007.10.027; PMID: 18082613
- Srinivasula SM, Ashwell JD. IAPs: what’s in a name?. Mol Cell 2008; 30:123 - 35; http://dx.doi.org/10.1016/j.molcel.2008.03.008; PMID: 18439892
- Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer 2010; 10:561 - 74; http://dx.doi.org/10.1038/nrc2889; PMID: 20651737
- Li X, Yang Y, Ashwell JD. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature 2002; 416:345 - 7; http://dx.doi.org/10.1038/416345a; PMID: 11907583
- Conze DB, Albert L, Ferrick DA, Goeddel DV, Yeh WC, Mak T, et al. Posttranscriptional downregulation of c-IAP2 by the ubiquitin protein ligase c-IAP1 in vivo. Mol Cell Biol 2005; 25:3348 - 56; http://dx.doi.org/10.1128/MCB.25.8.3348-3356.2005; PMID: 15798218
- Zhao Y, Conze DB, Hanover JA, Ashwell JD. Tumor necrosis factor receptor 2 signaling induces selective c-IAP1-dependent ASK1 ubiquitination and terminates mitogen-activated protein kinase signaling. J Biol Chem 2007; 282:7777 - 82; http://dx.doi.org/10.1074/jbc.M609146200; PMID: 17220297
- Csomos RA, Brady GF, Duckett CS. Enhanced cytoprotective effects of the inhibitor of apoptosis protein cellular IAP1 through stabilization with TRAF2. J Biol Chem 2009; 284:20531 - 9; http://dx.doi.org/10.1074/jbc.M109.029983; PMID: 19506082
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al, Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010; 17:98 - 110; http://dx.doi.org/10.1016/j.ccr.2009.12.020; PMID: 20129251
- Marcotte R, Chen JM, Huard S, Wang E. c-Myc creates an activation loop by transcriptionally repressing its own functional inhibitor, hMad4, in young fibroblasts, a loop lost in replicatively senescent fibroblasts. J Cell Biochem 2005; 96:1071 - 85; http://dx.doi.org/10.1002/jcb.20503; PMID: 16167342
- Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell 1999; 3:169 - 79; http://dx.doi.org/10.1016/S1097-2765(00)80308-1; PMID: 10078200
- Kong Q, Zeng W, Wu J, Hu W, Li C, Mao B. RNF220, an E3 ubiquitin ligase that targets Sin3B for ubiquitination. Biochem Biophys Res Commun 2010; 393:708 - 13; http://dx.doi.org/10.1016/j.bbrc.2010.02.066; PMID: 20170641
- Linares LK, Kiernan R, Triboulet R, Chable-Bessia C, Latreille D, Cuvier O, et al. Intrinsic ubiquitination activity of PCAF controls the stability of the oncoprotein Hdm2. Nat Cell Biol 2007; 9:331 - 8; http://dx.doi.org/10.1038/ncb1545; PMID: 17293853
- Noerholm M, Balaj L, Limperg T, Salehi A, Zhu LD, Hochberg FH, et al. RNA expression patterns in serum microvesicles from patients with glioblastoma multiforme and controls. BMC Cancer 2012; 12:22; http://dx.doi.org/10.1186/1471-2407-12-22; PMID: 22251860
- Wang Y, Engels IH, Knee DA, Nasoff M, Deveraux QL, Quon KC. Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway. Cancer Cell 2004; 5:501 - 12; http://dx.doi.org/10.1016/S1535-6108(04)00113-8; PMID: 15144957
- Nieminen AI, Partanen JI, Hau A, Klefstrom J. c-Myc primed mitochondria determine cellular sensitivity to TRAIL-induced apoptosis. EMBO J 2007; 26:1055 - 67; http://dx.doi.org/10.1038/sj.emboj.7601551; PMID: 17268552
- Larsson LG, Henriksson MA. The Yin and Yang functions of the Myc oncoprotein in cancer development and as targets for therapy. Exp Cell Res 2010; 316:1429 - 37; http://dx.doi.org/10.1016/j.yexcr.2010.03.025; PMID: 20382143
- Mateyak MK, Obaya AJ, Adachi S, Sedivy JM. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ 1997; 8:1039 - 48; PMID: 9342182
- Mei Y, Hahn AA, Hu S, Yang X. The USP19 deubiquitinase regulates the stability of c-IAP1 and c-IAP2. J Biol Chem 2011; 286:35380 - 7; http://dx.doi.org/10.1074/jbc.M111.282020; PMID: 21849505