Abstract
Patients with serosal (pleural or peritoneal) spread of malignancy have few definitive treatment options and consequently have a very poor prognosis. We have previously shown that photodynamic therapy (PDT) can be an effective treatment for these patients, but that the therapeutic index is relatively narrow. Here, we test the hypothesis that EGFR and STAT3 activation increase survival following PDT, and that inhibiting these pathways leads to increased PDT-mediated direct cellular cytotoxicity by examining BPD-PDT in OvCa and NSCLC cells. We found that BPD-mediated PDT stimulated EGFR tyrosine phosphorylation and nuclear translocation, and that EGFR inhibition by erlotinib resulted in reduction of PDT-mediated EGFR activation and nuclear translocation. Nuclear translocation and PDT-mediated activation of EGFR were also observed in response to BPD-mediated PDT in multiple cell lines, including OvCa, NSCLC and head and neck cancer cells, and was observed to occur in response to porfimer sodium-mediated PDT. In addition, we found that PDT stimulates nuclear translocation of STAT3 and STAT3/EGFR association and that inhibiting STAT3 signaling prior to PDT leads to increased PDT cytotoxicity. Finally, we found that inhibition of EGFR signaling leads to increased PDT cytotoxicity through a mechanism that involves increased apoptotic cell death. Taken together, these results demonstrate that PDT stimulates the nuclear accumulation of both EGFR and STAT3 and that targeting these survival pathways is a potentially promising strategy that could be adapted for clinical trials of PDT for patients with serosal spread of malignancy.
Introduction
The treatment of serosal surface malignancies, including recurrent peritoneal carcinomatosis resulting from epithelial ovarian cancer (OvCa) and pleural carcinomatosis resulting from non-small cell lung cancer (NSCLC), is typically palliative in nature. Thus, the development of effective and safe novel therapies to address this pattern of cancer spread would be highly significant. One such therapy is photodynamic therapy (PDT). PDT involves the transfer of energy from a photosensitizer that absorbs visible light to molecular oxygen resulting in the creation of excited state singlet oxygen.Citation1 In combination with surgical debulking and systemic chemotherapy, both pleural and intraperitoneal PDT have shown promise in phase I and II clinical trials of PDT using the first generation photosensitizer porfimer sodium.Citation1-Citation6 While PDT can be an effective treatment for patients with malignant involvement of serosal surfaces, the therapeutic index of this therapy is frequently limited by modest and heterogeneous tumor to normal tissue uptake ratios and by dose-limiting toxicities involving tumor selectivity.Citation1-Citation6
In addition to heterogenous photosensitizer uptake, survival signaling by cancer cells in response to PDT may be an important mechanism that narrows the therapeutic index for serosal PDT.Citation6-Citation9 While interactions between the epidermal growth factor receptor (EGFR) pathway and PDT cancer cell cytotoxicity has been studied by several investigators, the mechanism by which EGFR signaling affects PDT cancer cell cytotoxicity remains incompletely understood. EGFR is a receptor tyrosine kinase that regulates important cellular functions including cell cycle progression and survival mediated through phosphatidylinositol 3′kinase (PI3K)/AKT, proliferation through Mitogen Activated Protein Kinases (MAPK) and protection from apoptosis through STAT3. EGFR inhibition using Cetuximab has been shown to synergistically enhance the therapeutic efficacy of benzoporphyrin derivative (BPD)-mediated PDT in a mouse model of ovarian cancer with serosal malignant involvement.Citation10 The mechanism for this effect could involve an increase in PDT-mediated direct cytotoxicity to cancer cells, enhancement of PDT-mediated indirect/microenvironmental effects (e.g., tumor vascular damage) or a combination of both.Citation9 In addition, the effect of PDT on EGFR remains controversial, with some studies showing upregulation of EGFR signalingCitation11 and other studies showing degradation of cell surface receptors (including EGFR).Citation12-Citation14 Here, we have sought to better define the roles of EGFR signaling in PDT mediated direct cancer cell cytotoxicity and the mechanism(s) by which EGFR pathway inhibition could lead to increased direct cell cytotoxicity.
Results
Manipulation of molecular targets involved in the cellular PDT response is a potentially promising new approach to increasing the therapeutic index of serosal PDT, however a greater understanding of the linkage between molecular and cellular PDT responses will greatly facilitate the rational introduction of these novel modalities into clinical trials. We hypothesized that EGFR inhibitors enhance PDT efficacy by downregulating PDT-initiated EGFR signaling and thereby increasing the direct cancer cell cytotoxicity of PDT. To test the linkage between EGFR signaling and direct cytotoxicity of PDT for serosal malignancies, we used human OvCa cells (OVCAR-5) and NSCLC cells (H460). These cells lines were chosen for modeling of OvCa and NSCLC for a number of reasons. The EGFR signaling pathway has beenCitation15-Citation17 studied extensively in both cell lines and both cell lines are thought to express wild-type EGFR . OVCAR-5 cells have a relatively high level of EGFR expression and are sensitive to the growth inhibitory effects of EGFR inhibitors,Citation18 while H460 have a lower level of EGFR expression and are less sensitive to the growth inhibitory effects of EGFR inhibitors.Citation17 Finally, EGFR inhibition by the antibody type inhibitor cetuximab increases the response to serosal PDT of an orthotopic OVCAR-5 murine model of peritoneal carcinomatosis.Citation10
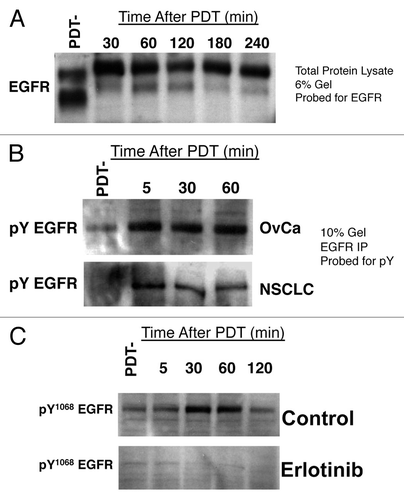
To determine the effect of BPD-mediated PDT on EGFR signaling, we began by analyzing total cellular levels of EGFR in OvCa and NSCLC cells before and after BPD-mediated PDT. However, we found that BPD-mediated PDT does not promote degradation of EGFR as others have reported for other photosensitizers,Citation12-Citation14 but rather levels of EGFR remain unchanged in the first 4h following PDT (). This effect is not cell line specific, since we have seen similar results in multiple other human cancer cell lines, including H460 and A549 NSCLC cells, and in OVCAR-4 and SKOV-3 OvCa cells (data not shown). Moreover, the decreased mobility of EGFR on lower density (6% polyacrylamide) gels suggested the possibility that BPD-mediated PDT might lead to increased phosphorylation of EGFR. Therefore, we immunoprecipitated EGFR from OVCAR-5 and H460 cell lysates after BPD incubation either before light exposure (PDT-) or at the indicated times after PDT. The resultant immune complexes were resolved by SDS-PAGE and we detected phosphotyrosine-EGFR by western blotting with an anti-phosphotyrosine antibody. EGFR probing was used to confirm IP of EGFR (data not shown). These studies demonstrate total EGFR phosphorylation is increased within 5 min after BPD-mediated PDT (). Phohosphorylation on tyrosine 1068 (Y1068) peaks from 30–60 min and begins to decline by 2h after PDT (). We have also confirmed that this tyrosine phosphorylation of EGFR was due to autophosphorylation/activation of EGFR by PDT, since phosphorylation of EGFR at Y1068 was absent following PDT in the presence of the small molecule EGFR inhibitor erlotinib ().
Figure 1. BPD-mediated PDT induces EGFR tyrosine phosphorylation. (A) OVCAR-5 cells were incubated with BPD at 100 ng/ml liposomal BPD and PDT using 0.5 J/cm2 of 690 nm light. At the indicated time following PDT, cells were lysed in denaturing lysis buffer and western blot analysis was performed using EGFR antibodies. PDT-indicates sham treatment with 0 J/cm2 light exposure. (B) OVCAR-5 cells (top panel) or H460 cells (bottom panel) were treated as above and lysed in ice cold RIPA buffer by sonication using a micro-tip sonicator for 15 10s cycles at 33% duty. EGFR was immunoprecipitated using a polyclonal antiserum and Protein-G sepharose, the resultant immune complexes washed extensively and protein complexes were resolved by SDS-PAGE. Tyrosine phosphorylated EGFR was detected by western blotting with an anti-phosphotyrosine antibody. (C) OVCAR-5 cells were treated as above with overnight pretreatment with 4µM erlotinib or carrier (1:100,000 DMSO) and lysed in denaturing, reducing buffer at the indicated time. Phosphorylated EGFR was detected using an antibody specific for EGFR tyrosine phosphorylation at residue Y1068. All results in this figure are representative of experiments performed in triplicate.
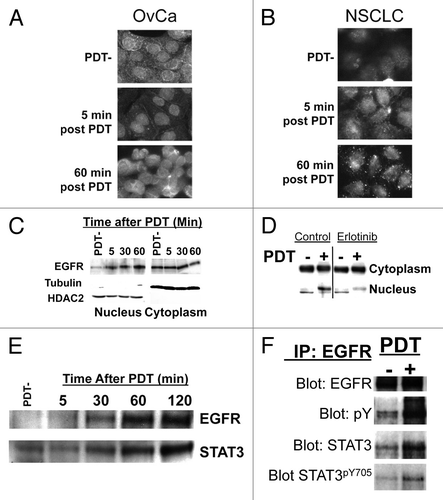
Nuclear signaling of EGFR and STAT3 is thought to promote cancer cell survival following cytotoxic agents, and nuclear EGFR signaling may trigger resistance of cancers to EGFR inhibitor therapy.Citation19,Citation20 Therefore, we hypothesized that EGFR might be translocating to the nucleus following activation by PDT. Studies using western blots of nuclear and cytoplasmic subcellular fractions and immunocytochemistry of fixed, permeabilized cells confirm increased presence of EGFR in the nucleus of BPD-PDT treated OvCa and NSCLC cells (). Prior to PDT, EGFR is seen dispersed throughout the cells. Following PDT, EGFR can be seen localizing in the nucleus as early as five minutes post-PDT and is heavily concentrated within the nuclei by one hour after treatment. To confirm this finding using a biochemical method, we prepared nuclear and cytoplasmic extracts from OVCAR-5 cells after BPD incubation but before light treatment (PDT-) and at time points after PDT. Western blot analysis of these nuclear and cytoplasmic fractions confirms translocation of EGFR to the nucleus following PDT (). In addition, inhibition of EGFR tyrosine phosphorylation by erlotinib dramatically reduced PDT-dependent nuclear translocation, suggesting that EGFR phosphorylation is important for this effect (). STAT3 and EGFR both promote survival from apoptosis-inducing stimuli and at high PDT doses formation of non-functional oligomeric complexes that contain and presumably inhibit STAT3 may be a biomarker for effective PDT treatment,Citation14,Citation21 but the effects or interactions of EGFR and STAT3 have not been studied in PDT-mediated cytotoxicity. Therefore, we also analyzed the impact of PDT on STAT3 signaling. We found that interestingly, the kinetics of STAT3 translocation to the nucleus following PDT are similar to those of EGFR () and that PDT induces association of EGFR with STAT3 ().
Figure 2. BPD-mediated PDT stimulated nuclear co-localization of EGFR-STAT3. (A) OVCAR-5 (OvCa)or (B) H460 (NSCLC) cells were incubated with BPD as in . Prior to light exposure (PDT-) or the indicated time after delivery of 0.5 J/cm2 at 30 mW/cm2 690 nm light, cells were fixed/permeabilized in 4%paraformaldehyde, 0.1% triton (added to permeablize the nucleus for staining). EGFR was visualized using a poly-clonal anti-EGFR antibody and fluorescent immunocytochemistry secondary antibodies. (C) OVCAR-5 cells treated as above and fractionated into nuclear and cytoplasmic fractions using a hypotonic, non-denaturing lysis buffer and passage through a 28Ga syringe. After greater than 95% lysis of cells and retention of unlysed nuclei were verified using light microscopy, nuclei were recovered by centrifugation and the supernatant fraction was retained. Proteins were solubilized in both fractions using a denaturing, reducing buffer and resolved by SDS PAGE. Western blotting was performed using the indicated antibody. Tubulin and HDAC2 were used as a marker to confirm the cytoplasmic and nuclear fractions, respectively. (D) OVCAR-5 cells were treated as in C) following overnight pre-treatment with 4µM erlotinib or carrier (DMSO) and western blotting was used to detect EGFR in nuclear vs cytoplasmic fractions. (E) OVCAR-5 cells were treated as in C) and nuclear fractions were analyzed for EGFR or STAT3 by western blotting. (F) OVCAR-5 cells were treated as in and EGFR was immunoprecipitated. Resultant immune complexes were washed extensively to remove non-specifically adherent proteins, resolved by SDS-PAGE under denaturing conditions and western blot analysis was performed using the indicated antibody. All results in this figure are representative of experiments performed in triplicate.
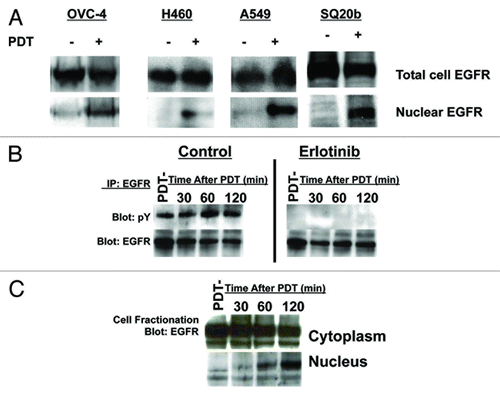
To determine whether nuclear translocation of EGFR by PDT can be seen in a variety of cancer cell types, we performed BPD-mediated PDT followed by nucleocytoplasmic fractionation and western blotting using OVCAR-4 OvCa cells and H460 and A549 NSCLC cells. We also used SQ20b head and neck cancer cells as PDT is an emerging therapy in head and neck cancersCitation1 and these cancers show a strong dependence on EGFR pathway signaling for growth and survival. These experiments revealed BPD-mediated PDT leads to EGFR nuclear translocation in all cell lines tested (). To determine whether this effect is also seen using porfimer sodium, another FDA approved photosensitizer that we have used in trials of PDT for serosal malignancies, we analyzed the impact of porfimer sodium mediated PDT on EGFR signaling using SKOV-3 OvCa cells. These studies demonstrate that porfimer sodium mediated PDT stimulates erlotinib-inhibitable tyrosine phosphorylation and nuclear translocation of EGFR ().
Figure 3. Response of OvCa, NSCLC and head and neck cancer (HNC) cell lines to BPD or porfimer sodium-mediated PDT. (A) OVCAR-4 (OvCa), H460 (NSCLC), A549 (NSCLC) or SQ20b (HNC) cells were treated with BPD-mediated PDT and total cell vs nuclear EGFR was analyzed as in . (B) SKOV-3 (OvCa) cells were treated with porfimer-sodium PDT using 0.5 J/cm2 of 630 nm light and EGFR was immunoprecipitated as in . The resultant immune complexes were analyzed by western blotting with anti-phosphotyrosine (pY) or EGFR antibodies. (C) SKOV-3 cells were treated as above and nucleocytoplasmic fraction and western blotting were performed as in . All results in this figure are representative of experiments performed in triplicate.
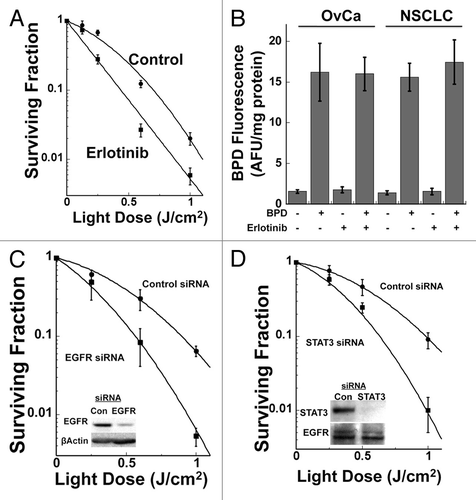
To establish whether inhibiting PDT-dependent EGFR signaling also promotes increased direct cell cytotoxicity, cells were treated with erlotinib and in vitro clonogenic cell survival assays were performed to measure cellular reproductive viability following BPD-mediated PDT. In these experiments, cells were pretreated for 24h prior to BPD-mediated PDT with either vehicle (DMSO) or 4 µM erlotinib. After PDT, cells were initially plated in medium with or without erlotinib and then this medium was replaced 24h after PDT with inhibitor-free medium. We found that in vitro treatment of OvCa cells with erlotinib results in increased direct cell cytotoxicity of BPD-mediated PDT in clonogenic cell survival assays (). H460 NSCLC cells showed similar results with a surviving (clonogenic) fraction at a 1 J/cm2 light dose of 14.9 ± 0.6% for control cells vs. 1.7 ± 0.1% for erlotinib treated cells. For some photosensitizers, the off-target effects of small molecule kinase inhibitors on drug efflux pumps can increase the intracellular photosensitizer levels and result in increased PDT cytotoxicity.Citation22 To determine whether this explained the ability of erlotinib to increase BPD-mediated PDT cytotoxicity, we measured intracellular BPD levels with and without erlotinib treatment. In these experiments, OVCAR-5 or H460 cells were pre-treated with or without 4 µM erlotinib for 21 h prior to addition of BPD for 3h. Cellular photosensitizer levels were measured on extracts using fluorometry and values were corrected for total protein concentration for each extract. The results of these experiments demonstrate that erlotinib has no effect on the total intracellular levels of BPD in either cell line (). To confirm that the effect of erlotinib was due to inhibition of EGFR signaling, we used EGFR siRNA transfection to inhibit EGFR expression. We have used this approach successfully to study the effect of EGFR signaling on the radiation response of cancer cells that express mutant Ras.Citation23 These studies show that siRNA-mediated inhibition of EGFR expression similarly results in enhancement of direct cell cytotoxicity of BPD-mediated PDT (). Clinically useful agents to inhibit STAT3 signaling are being evaluated in clinical trials. To determine whether inhibition of STAT3 also leads to increased PDT-medicated direct cell cytotoxicity, siRNA knock-down experiments were performed. These studies show that inhibiting STAT3 expression leads to increased direct cell cytotoxicity of BPD-mediated PDT as measured by clonogenic cell survival assays of H460 cells (). OVCAR-5 cells showed similar results with STAT3 knockdown, with a surviving (clonogenic) fraction at a 1 J/cm2 light dose of 11.7 ± 0.6% for control siRNA transfected cells vs 2.3 ± 0.3% for STAT3 siRNA transfected cells.
Figure 4. Inhibiting EGFR enhances BPD-mediated PDT direct tumor cell cytotoxicity. (A) OVCAR-5 cells were incubated 24h prior to PDT with either carrier (1:100,000 DMSO) or 4 µM erlotinib and 100 ng/ml liposomal BPD as in . PDT was performed to the indicated dose of 690 nm light and clonogenic survival was determined. (B) OVCAR-5 or H460 cells were treated as above without light exposure. Photosensitizer was extracted, quantified by fluorescence spectrometry and corrected for total protein concentration in each lysate. (C) OVCAR-5 cells were transfected with control (non-specific) or EGFR-specific siRNA 48h prior to BPD-mediated PDT as above and clonogenic survival assays were performed. Results are presented as mean +/−standard deviation for a minimum of 4 replicate plates per data point. Inset: western blot analysis demonstrating knock-down of EGFR at the time of PDT. (D) H460 cells were transfected with either non-specific control or STAT3 specific siRNA and clonogenic cell survival assays were performed. Results are presented as mean +/−standard deviation for a minimum of 4 replicate plates per data point. Inset: western blot analysis demonstrating knock-down of STAT3 at the time of PDT. All results in this figure are representative of experiments performed in triplicate.
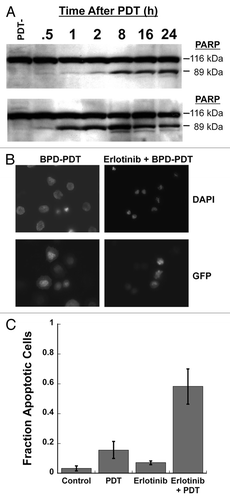
The function(s) of nuclear EGFR and STAT3 remains incompletely understood, but may involve resistance to apoptosis induced by cytotoxic agents. Therefore, we hypothesized that EGFR inhibition would lead to increased apoptotic cell death following BPD-mediated PDT. Consistent with the hypothesis, erlotinib accelerated the kinetics and lead to increased PARP cleavage following BPD-mediated PDT (). To verify that EGFR inhibition increased apoptosis following BPD-mediated PDT, H460 cells transfected with GFP were treated with PDT with and without erlotinib and analyzed for the presence of apoptotic bodies using DAPI staining. Representative fluorescent images show increased apoptotic phenotype after treatment with erlotinib prior to BPD-mediated PDT (). We have used visualization and direct enumeration as an effective means of confirming apoptotic phenotype in previous studies.Citation24 Counting a minimum of 10 high power fields per experiment for 3 experiments confirms that erlotinib increases apoptotic phenotype after BPD-mediated PDT (). Taken together with the clonogenic survival studies, these studies suggest that the mechanism for EGFR-inhibitor dependent increases in the direct cancer cell cytotoxicity of BPD-mediated PDT is through increased apoptotic cell death.
Figure 5. Inhibiting EGFR promotes increased apoptosis following BPD-mediated PDT. (A) OVCAR-5 cells were treated as in and western blotting was performed for PARP cleavage. Results are representative of experiments performed in triplicate. (B) H460 cells expressing GFP were treated as in and counterstained with DAPI. GFP fluorescence was used to identify the cytoplasm of cells and DAPI was used to identify cells with apoptotic bodies in the nucleus. Results are representative of experiments performed in triplicate. (C) Enumeration of results in (B), minimum 10 high power fields counted from 3 separate experiments.
Discussion
PDT can be an effective treatment for patients with malignant involvement of serosal surfaces, but the therapeutic index of this therapy is limited by the modest and heterogeneous tumor to normal tissue uptake ratios that are found in many patients. Recent evidence suggests inhibition of EGFR signaling synergistically enhances the therapeutic efficacy of BPD-mediated PDT in a mouse model of serosal malignancy. In this study, we have examined the impact of BPD-mediated PDT on direct cell cytotoxicity and EGFR signaling in OvCa and NSCLC cells. We found that the mechanism for EGFR-inhibitor mediated increases in BPD-mediated PDT cytotoxicity involves, at least in part, inhibition of PDT-initiated survival signaling through the EGFR pathway. In addition, BPD-mediated PDT resulted in EGFR-STAT3 association and nuclear translocation of EGFR and STAT3. Moreover, we have shown that PDT-initiated survival signals lead to increased apoptotic cell death following BPD-mediated PDT. Since nuclear translocation of EGFR and STAT3 is thought to mediate anti-apoptotic signals, these results suggest that BPD-mediated PDT initiates nuclear signaling of EGFR and STAT3 that results in decreased cancer cell cytotoxicity following PDT. These studies also may help explain why the formation of inactive STAT3 covalent oligomeric complexes is a positive prognostic sign for tumor control by PDT.Citation14,Citation21
In contrast to our findings of EGFR phosphorylation/activation after PDT, others have found EGFR degradation by western blotting. The reason for this discrepancy is unclear, but may relate to photosensitizer specific differences, since these studies were performed with HPPH and ALA as photosensitizers,Citation13,Citation14 as opposed to the BPD and porfimer sodium studies of the present report. Indeed, the affinity of hypericin analogs for EGFR binding correlates directly with the ability of these analogs to photodynamically inhibit EGFR signaling.Citation25 Additionally, or alternatively, these differences may be related to experimental protocol since different types of lysis buffers were employed in these studies. Our studies use a denaturing lysis buffer that more efficiently extracts large nuclear proteins, while investigators observing a decrease in EGFR after PDT have used a non-denaturing lysis buffer.Citation13,Citation14 Without sonication, as is used in our immunoprecipitation studies, cell lysis with a non-denaturing lysis buffer does not achieve nuclear lysis and larger nuclear proteins may be underrepresented in the resultant protein extracts. The results of the present study suggest that this would be relevant because EGFR was found to translocate to the nucleus after PDT. Finally, this could be a dose-dependent phenomenon, with low doses of PDT that are subtherapeutic leading to activation of EGFR/STAT3 and cell survival and high doses leading to formation of oligimeric complexes with signaling proteins such as EGFR and STAT3 that lead to inactivation of survival signaling.
Other investigators have found that small molecule kinase inhibitors may lead to increased intracellular photosensitizer concentrations by inhibiting efflux through membrane pumps such as ABCG2.Citation22 Indeed, ABCG2 expression protects cells from PDT damage when chlorophyll-or porphyrin-related photosensitizers are used. We found that EGFR inhibition increases direct cell cytotoxicity of BPD-mediated PDT in both cell lines. We confirmed EGFR as the target for these effects by performing EGFR knockdowns in both cell lines and obtaining similar results. Furthermore, we have found that EGFR inhibition via treatment with Erlotinib has no effect on the cellular uptake of BPD. Finally, the inhibitor effects of erlotinib for ABCG2 function have been found at concentrations 6–25 times higher than the concentrations used in this study.Citation26 Thus, the increased cytotoxicity seen with EGFR inhibition in our studies is unlikely to be simple alterations in photosensitizer cellular pharmacokinetics, although we have not analyzed whether EGFR inhibition leads to altered subcellular distribution of photosensitizer.
We have demonstrated that PDT activates both EGFR and STAT3 through phosphorylation. Furthermore, activated EGFR translocates to the cell nucleus immediately following PDT, and is accompanied by nuclear accumulation of STAT3. These results suggest that PDT stimulates the nuclear accumulation of both EGFR and STAT3, and that their activation may promote the increased survival of cancer cells following PDT. Conversely, by targeting these specific survival pathways, we may be able to increase the therapeutic index of serosal PDT in human cancer patients.
Materials and Methods
Cell lines, culture, inhibitors and antibodies
The human cancer cell lines OVCAR-5 (epithelial OvCa) and H460 (NSCLC) were obtained from the National Cancer Institute and American Type Culture Collection, respectively. Multiple replicate vials were frozen for use as a primary lab repository for each cell line and designated “passage 2.” Cells were passaged no more than 20 times prior to discarding cells and bringing up a fresh passage 2 stock. All experiments presented in this manuscript are performed with cells less than 3 mo from these authenticated stocks. Both cell lines were cultured in Gibco brand RPMI 1640 medium, H460 in 10% FBS with 1% Pen/Strep, OvCar5 in 20% FBS, 1% Pen/Strep, 10mM Hepes, 1mM Sodium pyruvate and 4.5g/L glucose. The liposomal formulation of the photosensitizer BPD (Visudyne) was obtained from Novartis Pharmaceuticals Corporation and erlotinib (Tarceva) was obtained from OSI Pharmaceuticals. siRNA for transfections were purchased from Dharmacon, Inc., EGFR siRNA (M-00311403) and STAT3 siRNA (M-003544–00–0010). Transfection reagents optiMEM and oligofectamine were purchased from Invitrogen Corp. Western blots antibodies: EGFR (cat#2232), STAT3 (cat#9132) and Phospho-Stat3 (Tyr705) (cat#9145) were purchased from Cell Signaling Technology. Phosphotyrosine (cat#05–321), Phospho-EGFR (Tyr1069) (cat#07–715) were purchased from Upstate. Secondary Mouse and Rabbit HRP linked antibodies were purchased form Jackson ImmunoResearch Laboratories, Inc.
siRNA transfection
Cells were transfected as previously described.Citation23,Citation27 Briefly, transient knockdown was performed in media containing 10% FBS without antibiotics. Cells were transfected with 150nM of either EGFR or STAT3 siRNA (Dharmacon RNA Technologies) and Oligofectamine (Invitrogen Corp.). Cells were incubated in transfection media overnight, with fresh growth media added the following day. All assays were performed on the cells 48hrs after the transfection procedure.
Photosensitizers studies
Cell dishes were treated, in triplicate, overnight with 4µM erlotinib or DMSO (1:100,000 final DMSO concentration) and fresh media containing 2% FBS and 25mM Hepes, +/−Erlotinib and 100ng/ml BPD was added. After 3hrs, the dishes were washed 2 times each with media containing 2% FBS, 25mM Hepes and Erlotinib where appropriate, with the second wash remaining on the dishes for 30min, in the incubator. For studies with porfimer sodium, cells were treated in either DMSO or erlotinib containing medium as above and porfimer sodium was added after 1h for the remainder of the overnight incubation and then treated as above. For the uptake studies, after the 30min wash, the dishes were rinsed 2 times each in Earle’s Buffer, collected in lysis buffer (1% SDS, 0.1N NaOH) and sheared 6 to 10 times in a 1cc syringe. Total protein quantification was performed using a Lowry Assay and photosensitizer was quantified using fluorimetry with an activation wavelength of 403 nm light and an emission wavelength of 690 nm. Results are given as calculated arbitrary fluorescence units per mg protein. For PDT studies, after the 30min wash, the wash media was replaced with 2ml of fresh media (2% FBS, 25mM Hepes, +/inhibitor) and plates exposed to the indicated dose of 690nm light, generated by a B&W TEK Inc. laser, model BWF2–690–1.0–400 and delivered via microlens tipped fiber with a fluence rate of approximately 30 mW/cm2.
Cell lysis and immunoprecipitation
Cell lysis and immunoprecipitation were performed as previously described.Citation28,Citation29 At the indicated time points following treatment, the samples collected for whole cell lysates were rinsed 2 times in ice cold PBS, lysed in clear 1X LDS sample (Invitrogen Technologies) buffer with 100mM DTT, sheared 6 to 10 times in a 1cc syringe and heated for 5min at 90°C. Total protein concentration was determined using amido black staining samples spotted onto nitrocellulose as previously described.Citation27 For immunoprecipitation, plates were rinsed 2 times in ice cold PBS and lysed in ice-cold RIPA buffer (50mM Tris-Cl, 150mM NaCl, 25mM NaF, 1mM EDTA, 1% NP-40, 1% SDC, 0.1% SDS, with 1mM DTT, 1mM Activated Sodium Orthovanidate, 1mM Sodium Pyruvate, 1mM PMSF and 1:1000 Protease Inhibitor Cocktail). The samples were sonicated on ice for 15 10s cycles with 30s cooling in between cycles using a micro-tip sonicator and clarified by centrifugation at 15,000 g for 10 min and the resultant supernate fractions were incubated overnight with a 1:300 dilution of either EGFR primary antibody (Santa Cruz Biotechnology, polyclonal) or STAT3 primary antibody (Cell Signaling Technology, polyclonal) at 4°C with gentle mixing. The following morning the antibodies were bound to Protein G Agarose beads for 2hrs at 4°C. The agarose beads were pelleted, rinsed extensively in ice-cold RIPA buffer and resuspended in 2X LDS buffer. The proteins were released from the antibody-bead complex by heating the samples for 5min at 90°C.
Nucleocytoplasmic fractionation
Fractionation was performed as previously described.Citation30 At the indicated time point, samples were collected into a hypotonic lysis buffer. Intact nuclei were pelleted and cytosolic fraction was separated and fully lysed in 1X LDS with 100mM DTT. Nuclear pellet was rinsed in ice cold PBS, resuspended in the cytoplasmic buffer and then fully lysed in 1X LDS with 100mM DTT. Samples were heated for 5min at 90°C, passed 5–10 times through a 1cc syringe and clarified by centrifugation at 15,000xg for 10 min. Total protein concentration was determined using amido black staining samples spotted onto nitrocellulose as previously described.Citation27
Western blotting
Western blotting was performed as previously described.Citation28,Citation29 Briefly equal amounts of protein were resolved on 10% Bis-Tris SDS-Page precast gels (Invitrogen Corp.), except for , where 6% gels were used. Proteins were transferred to nitrocellulose and non-specific binding was blocked using 3% BSA and 3% Protein Hydrolysate Amicase in PBS with 1% tween (PBSt). After probing with primary and secondary antibodies, protein bands were visualized using enhanced chemiluminescence (Amersham, GE Healthcare).
Immunocytochemistry
Immunostaining was performed as previously described.Citation31,Citation32 Briefly, cells were plated onto poly-l-lysine Hydrobromide treated, flamed, glass coverslips in 4cm dishes at 5 × 10^5 cells per dish and allowed to attach overnight. BPD-PDT was performed as described elsewhere in this paper, with dishes fixed at 5 and 60min post illumination. Cells were fixed in 4% paraformaldehyde with 0.1% Triton and incubated in blocking buffer (0.01M Tris pH7.5, 0.15M NaCl and 0.1% BSA) until probing. Cells were probed with EGFR antibody (Santa Cruz) at 1:50, and incubated in secondary antibody and DAPI at 1ug/ml for nuclear staining. Coverslips were mounted and imaged with a 60X lens on an inverted oil immersion fluorescence microscope.
Clonogenic survival assays
Clonogenic survival assays were performed as previously described.Citation27,Citation33 Briefly, 24h prior to PDT (24h after transfection if indicated), cells were plated in 6cm dishes, allowed to attach and (where indicated) either 1:100,000 DMSO or 4 µM Erlotinib was added overnight. The following morning, the media in the dishes were replaced with fresh media containing 2% FBS and 25mM Hepes, the previously treated dishes again received 4µM Erlotinib (made fresh, these dishes remain in Erlotinib until 24hrs after final plating) and 100ng/ml BPD and allowed to incubate for 3hrs. After 3hrs, the dishes were trypsinized, brought up into a single cell suspension and counted. The cells were treated to the indicated light dose in aliquots of suspended cells and then re-plated at specific cell numbers for clonogenic cell survival. The following day, all dishes received fresh, inhibitor free growth medium. Cells were typically fixed and stained simultaneously, approximately 10 d later using a 0.5% Crystal violet solution in methanol.
| Abbreviations: | ||
| BPD | = | benzoporphyrin derivative |
| EGFR | = | epidermal growth factor receptor |
| STAT3 | = | Signal Transducer and Activation of Transcription-3 |
| PDT | = | Photodynamic Therapy |
| OvCa | = | Ovarian Cancer |
| NSCLC | = | Non-small cell lung cancer |
Acknowledgments
This work was funded by National Institutes of Health/National Cancer Institute CA87971 and T32CA009677.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Cengel KA, Hahn SM. Photodynamic Therapy. Cancer 2008; 767 - 74; PMID: 18098271
- Busch TM, Hahn SM, Wileyto EP, Koch CJ, Fraker DL, Zhang P, et al. Hypoxia and Photofrin uptake in the intraperitoneal carcinomatosis and sarcomatosis of photodynamic therapy patients. Clin Cancer Res 2004; 10:4630 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-04-0359; PMID: 15269134
- Hahn SM, Fraker DL, Mick R, Metz J, Busch TM, Smith D, et al. A phase II trial of intraperitoneal photodynamic therapy for patients with peritoneal carcinomatosis and sarcomatosis. Clin Cancer Res 2006; 12:2517 - 25; http://dx.doi.org/10.1158/1078-0432.CCR-05-1625; PMID: 16638861
- Hahn SM, Putt ME, Metz J, Shin DB, Rickter E, Menon C, et al. Photofrin uptake in the tumor and normal tissues of patients receiving intraperitoneal photodynamic therapy. Clin Cancer Res 2006; 12:5464 - 70; http://dx.doi.org/10.1158/1078-0432.CCR-06-0953; PMID: 17000681
- Friedberg J, Cengel K. Pleural Malignancies. Semin Radiat Oncol 2009; 20:208 - 14; PMID: 20685584
- Cengel KA, Glatstein E, Hahn SM. Intraperitoneal photodynamic therapy. Cancer Treat Res 2007; 134:493 - 514; PMID: 17633077
- Solban N, Rizvi I, Hasan T. Targeted photodynamic therapy. Lasers Surg Med 2006; 38:522 - 31; http://dx.doi.org/10.1002/lsm.20345; PMID: 16671102
- Almeida RD, Manadas BJ, Carvalho AP, Duarte CB. Intracellular signaling mechanisms in photodynamic therapy. Biochim Biophys Acta 2004; 1704:59 - 86; PMID: 15363861
- Cengel KA, Hahn SM, Glatstein E. C225 and PDT combination therapy for ovarian cancer: the play’s the thing. J Natl Cancer Inst 2005; 97:1488 - 9; http://dx.doi.org/10.1093/jnci/dji360; PMID: 16234556
- del Carmen MG, Rizvi I, Chang Y, Moor AC, Oliva E, Sherwood M, et al. Synergism of epidermal growth factor receptor-targeted immunotherapy with photodynamic treatment of ovarian cancer in vivo. J Natl Cancer Inst 2005; 97:1516 - 24; http://dx.doi.org/10.1093/jnci/dji314; PMID: 16234565
- Fontanini G, De Laurentiis M, Vignati S, Chinè S, Lucchi M, Silvestri V, et al. Evaluation of epidermal growth factor-related growth factors and receptors and of neoangiogenesis in completely resected stage I-IIIA non-small-cell lung cancer: amphiregulin and microvessel count are independent prognostic indicators of survival. Clin Cancer Res 1998; 4:241 - 9; PMID: 9516978
- Ahmad N, Kalka K, Mukhtar H. In vitro and in vivo inhibition of epidermal growth factor receptor-tyrosine kinase pathway by photodynamic therapy. Oncogene 2001; 20:2314 - 7; http://dx.doi.org/10.1038/sj.onc.1204313; PMID: 11402326
- Wong TW, Tracy E, Oseroff AR, Baumann H. Photodynamic therapy mediates immediate loss of cellular responsiveness to cytokines and growth factors. Cancer Res 2003; 63:3812 - 8; PMID: 12839978
- Liu W, Oseroff AR, Baumann H. Photodynamic therapy causes cross-linking of signal transducer and activator of transcription proteins and attenuation of interleukin-6 cytokine responsiveness in epithelial cells. Cancer Res 2004; 64:6579 - 87; http://dx.doi.org/10.1158/0008-5472.CAN-04-1580; PMID: 15374971
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129 - 39; http://dx.doi.org/10.1056/NEJMoa040938; PMID: 15118073
- Helfrich BA, Raben D, Varella-Garcia M, Gustafson D, Chan DC, Bemis L, et al. Antitumor activity of the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor gefitinib (ZD1839, Iressa) in non-small cell lung cancer cell lines correlates with gene copy number and EGFR mutations but not EGFR protein levels. Clin Cancer Res 2006; 12:7117 - 25; http://dx.doi.org/10.1158/1078-0432.CCR-06-0760; PMID: 17145836
- Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res 2005; 65:9455 - 62; http://dx.doi.org/10.1158/0008-5472.CAN-05-1058; PMID: 16230409
- Sewell JM, Macleod KG, Ritchie A, Smyth JF, Langdon SP. Targeting the EGF receptor in ovarian cancer with the tyrosine kinase inhibitor ZD 1839 (“Iressa”). Br J Cancer 2002; 86:456 - 62; http://dx.doi.org/10.1038/sj.bjc.6600058; PMID: 11875715
- Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009; 28:3801 - 13; http://dx.doi.org/10.1038/onc.2009.234; PMID: 19684613
- Wang SC, Hung MC. Nuclear translocation of the epidermal growth factor receptor family membrane tyrosine kinase receptors. Clin Cancer Res 2009; 15:6484 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-08-2813; PMID: 19861462
- Henderson BW, Daroqui C, Tracy E, Vaughan LA, Loewen GM, Cooper MT, et al. Cross-linking of signal transducer and activator of transcription 3--a molecular marker for the photodynamic reaction in cells and tumors. Clin Cancer Res 2007; 13:3156 - 63; http://dx.doi.org/10.1158/1078-0432.CCR-06-2950; PMID: 17545518
- Liu W, Baer MR, Bowman MJ, Pera P, Zheng X, Morgan J, et al. The tyrosine kinase inhibitor imatinib mesylate enhances the efficacy of photodynamic therapy by inhibiting ABCG2. Clin Cancer Res 2007; 13:2463 - 70; http://dx.doi.org/10.1158/1078-0432.CCR-06-1599; PMID: 17438106
- Cengel KA, Voong KR, Chandrasekaran S, Maggiorella L, Brunner TB, Stanbridge E, et al. Oncogenic K-Ras signals through epidermal growth factor receptor and wild-type H-Ras to promote radiation survival in pancreatic and colorectal carcinoma cells. Neoplasia 2007; 9:341 - 8; http://dx.doi.org/10.1593/neo.06823; PMID: 17460778
- Godbout JP, Cengel KA, Cheng SL, Minshall C, Kelley KW, Freund GG. Insulin activates caspase-3 by a phosphatidylinositol 3′-kinase-dependent pathway. Cell Signal 1999; 11:15 - 23; http://dx.doi.org/10.1016/S0898-6568(98)00024-2; PMID: 10206340
- Agostinis P, Donella-Deana A, Cuveele J, Vandenbogaerde A, Sarno S, Merlevede W, et al. A comparative analysis of the photosensitized inhibition of growth-factor regulated protein kinases by hypericin-derivatives. Biochem Biophys Res Commun 1996; 220:613 - 7; http://dx.doi.org/10.1006/bbrc.1996.0451; PMID: 8607812
- Shi Z, Peng XX, Kim IW, Shukla S, Si QS, Robey RW, et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res 2007; 67:11012 - 20; http://dx.doi.org/10.1158/0008-5472.CAN-07-2686; PMID: 18006847
- Brunner TB, Cengel KA, Hahn SM, Wu J, Fraker DL, McKenna WG, et al. Pancreatic cancer cell radiation survival and prenyltransferase inhibition: the role of K-Ras. Cancer Res 2005; 65:8433 - 41; http://dx.doi.org/10.1158/0008-5472.CAN-05-0158; PMID: 16166322
- Cengel KA, Kason RE, Freund GG. Phosphatidylinositol 3′-kinase associates with an insulin receptor substrate-1 serine kinase distinct from its intrinsic serine kinase. Biochem J 1998; 335:397 - 404; PMID: 9761740
- Cengel KA, Freund GG. JAK1-dependent phosphorylation of insulin receptor substrate-1 (IRS-1) is inhibited by IRS-1 serine phosphorylation. J Biol Chem 1999; 274:27969 - 74; http://dx.doi.org/10.1074/jbc.274.39.27969; PMID: 10488146
- Horan AD, Giandomenico AR, Koch CJ. Effect of oxygen on radiation-induced DNA damage in isolated nuclei. Radiat Res 1999; 152:144 - 53; http://dx.doi.org/10.2307/3580087; PMID: 10409323
- Dowling M, Voong KR, Kim M, Keutmann MK, Harris E, Kao GD. Mitotic spindle checkpoint inactivation by trichostatin a defines a mechanism for increasing cancer cell killing by microtubule-disrupting agents. Cancer Biol Ther 2005; 4:197 - 206; http://dx.doi.org/10.4161/cbt.4.2.1441; PMID: 15753652
- Harris EE, Hwang WT, Lee EA, Cengel KA, Feldman MD, Demichele A, et al. The impact of HER-2 status on local recurrence in women with stage I-II breast cancer treated with breast-conserving therapy. Breast J 2006; 12:431 - 6; http://dx.doi.org/10.1111/j.1075-122X.2006.00297.x; PMID: 16958961
- Cengel KA, Deutsch E, Stephens TC, Voong KR, Kao GD, Bernhard EJ. Radiosensitizing effects of the prenyltransferase inhibitor AZD3409 against RAS mutated cell lines. Cancer Biol Ther 2006; 5:1206 - 10; http://dx.doi.org/10.4161/cbt.5.9.3172; PMID: 16969121