Abstract
The KRAS gain-of-function mutation confers intrinsic resistance to targeted anti-cancer drugs and cytotoxic chemotherapeutic agents, ultimately leading to treatment failure. KRAS mutation frequency in lung adenocarcinoma is ~15–30%. Novel therapeutic strategies should be developed to improve clinical outcomes in these cases. Deregulation of the p16/cyclin-dependent kinase (CDK) 4/retinoblastoma (Rb) pathway is frequently observed in various cancers and it represents an attractive therapeutic target. We compared the anti-tumor efficacy of genetically knocked-down CDK4 and a pharmacological inhibitor of CDK4/6, CINK4, in KRAS mutation-positive lung adenocarcinoma cells. We also investigated changes in anti-proliferative activity and downstream molecules with these treatments in combination with paclitaxel. CDK4 short interfering RNA (siRNA) significantly increased paclitaxel sensitivity in KRAS mutation-positive H23 cells. CINK4 demonstrated concentration- and time-dependent anti-proliferative activity in 5 adenocarcinoma lines. CINK4 induced G1 arrest by downregulating the p16/cyclin D1/Rb pathway, resulting in apoptotic induction via increased expression of cleaved caspase3, cleaved PARP and Bax. Combined CINK4 and paclitaxel produced synergistic anti-proliferative activity and increased apoptosis through reduced cyclin D1 and Bcl-2 in KRAS mutation-positive cancer cells. These data suggest CDK4 is a promising target for development of anti-cancer drugs and CINK4 combined with paclitaxel may be an effective therapeutic strategy for enhancing anti-tumor efficacy in KRAS mutation-positive lung adenocarcinoma.
Introduction
Mutations in the KRAS oncogene have been implicated in the development of various malignancies including non-small cell lung cancer (NSCLC).Citation1 Approximately 15–30% of lung adenocarcinoma patients possess a gain-of-function mutation in KRAS codons 12, 13, or 61Citation2,Citation3; the majority of KRAS mutations occur at codon 12.Citation4 Many clinical researchers have reported that KRAS mutation positivity confers poor prognosis with intrinsic resistance to cytotoxic chemotherapeutic drugs.Citation5 Targeted anti-cancer drugs including epidermal growth factor receptor tyrosine kinase inhibitors (EGFR TKIs) exhibit anti-tumor efficacy, causing a paradigm shift in the treatment of advanced NSCLCs. However, they are only effective in the presence of activating EGFR mutations, not KRAS mutations.Citation6 In addition, chemotherapeutic outcomes for NSCLC patients harboring mutationally activated KRAS are also disappointing.Citation6-Citation8 Currently, MEK1/2 inhibitor or sorafenib alone and in combination with cytotoxic drugs have shown promise in treatment of KRAS mutation-positive lung tumors modelsCitation9,Citation10 but these treatment schedules have not been adopted in clinical practice. An effective therapeutic strategy for KRAS mutation-positive lung adenocarcinoma is urgently needed to improve clinical outcomes.
KRAS mutations constitutively activate the RAS/RAF/ERK signaling pathway.Citation11 Activation of ERK1/2 modulates the activity of target transcription factorsCitation12 such as cyclin D1 and the cyclin D1-CDK4 complex.Citation13,Citation14
The p16/cyclin D1/CDK4/Rb signaling pathway is frequently altered in NSCLCs. The CDKN2A/p16 gene is a candidate tumor suppressor in wild-type Rb malignancies; p16 binds CDK4 and inhibits catalytic activity of the CDK4/cyclin D complex.Citation15 Rb and p16INK4A expression in lung cancer are inversely correlated; frequently, Rb-positive NSCLCs have little or no detectable p16INK4A protein due to homozygous deletions, mutations and methylation of CpG islands.Citation16,Citation17 Loss of p16INK4A frees these CDKs from inhibition, permitting constitutive phosphorylation of Rb and inactivation of its cell growth inhibitory properties. In addition, overexpression of p16 blocks tumor cells in S phase and inhibits RAS-induced cell proliferation.Citation18,Citation19
The activity of CDK4, negatively regulated by P16INK4A, is required for tumor progression in a KRASG12V induced lung adenocarcinoma model. RAS and CDK4 co-expression promotes Rb phosphorylation, ultimately resulting in human invasive neoplasm.Citation20 Ablation of CDK4 selectively decreases the number of KRASG12V-expressing cells.Citation21 Therefore, CDK4 may be an attractive drug candidate for KRAS mutation-positive NSCLC.
PD0332991, a CDK4/CDK6 selective inhibitor with significant therapeutic activity in a KRASG12V-induced NSCLC modelCitation22 and is being developed with other chemotherapeutic agents in clinical trials.Citation22-Citation24
Paclitaxel has potent cytotoxic activity as it stabilizes the microtubule cytoskeleton, thus blocking cell cycle progression. Because paclitaxel alone has a 21–24% response rate in advanced NSCLCs,Citation25,Citation26 it is commonly used in combination with other anti-cancer agents.
In this study, we investigated whether selective molecular (siRNA) or pharmacological inhibition (CINK4, a small molecule targeting CDK4 and CDK6) of CDK4 activity may enhance the anti-tumor activity of paclitaxel in KRAS mutation-positive lung adenocarcinoma cell lines.
Results
Baseline expression of p16/CDK4/Rb signaling molecules
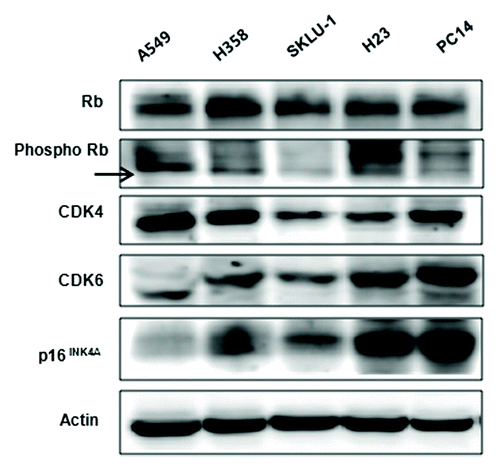
We observed different baseline expression levels for Rb, phosphorylated Rb (p-Rb), CDK4, CDK6 and p16INK4 proteins in five cell lines (). Rb and p-Rb were present in all cell lines but p-Rb expression was lowest in SKLU-1 cells. The CDK4 is highest in A549 and H358 cells, while p16INK4 is highest in H23 and PC14 cells. Similarly, CDK4 and CDK6 expression do not correlate.
Figure 1. Baseline expression of p16/CDK4/Rb signaling molecules in 5 cancer cell lines. Constitutive basal expression of Rb, p-Rb, CDK4, CDK6, and p16INK4A were detected by western blotting. Actin was used as a loading control.
siRNA knockdown of CDK4 and cell cycle distribution
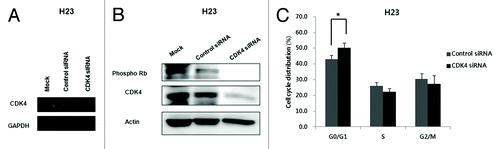
To understand the molecular function of CDK4 in KRAS mutation-positive lung cancer cells, we used siRNA to knockdown CDK4 in H23 cells. We confirmed that CDK4 siRNA efficiently abolished p-Rb as well as CDK4 mRNA and protein expression (). We also analyzed the cell cycle distribution in cells transfected with CDK4 siRNA. The cells in G0/G1 phases increased (P < 0.05) and the cell population in S and G2/M phase decreased ().
Figure 2. Efficient inhibition of CDK4 mRNA and protein and changes in cell cycle distribution in H23 cells transfected with CDK4 siRNA. (A) Expression of CDK4 mRNA examined by RT-PCR 48 h after CDK4 siRNA transfection. GAPDH was used as a loading control. (B) Expression of CDK4 and pRb protein was significantly reduced 48 h after CDK4 siRNA transfection. Actin was used as a loading control. (C) Cell cycle distribution. G0/G1 phase increased 48 h after CDK4 siRNA transfection of H23 cells. Each experiment was performed in triplicate. Data represent the mean ± SD. Statistically significant differences between the CDK4 siRNA and control are presented as *(P < 0.05).
CDK4 knockdown enhances sensitivity to paclitaxel
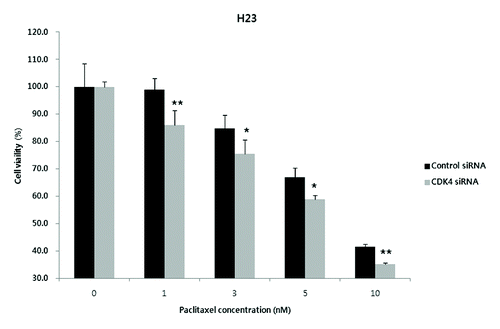
When exposed to various concentrations of paclitaxel (0–10 nM), the inhibitory effect on cell proliferation was significantly higher in CDK4 siRNA-transfected H23 cells than in control siRNA-transfected H23 cells. Thus, selective inhibition of CDK4 may increase paclitaxel sensitivity in KRAS mutation-bearing H23 cells (P < 0.05 for paclitaxel 1 and 10 nM; P < 0.01 for paclitaxel 3 and 5 nM) ().
Figure 3. Enhanced anti-proliferative effect of paclitaxel in CDK4 siRNA-transfected H23 cells. When exposed to the different concentrations of paclitaxel (1, 3, 5, and 10 nM), a concentration-dependent increase of anti-proliferative effects was observed in the CDK4 siRNA-transfected cells. Each experiment was performed in triplicate. Data represent the mean ± SD. Statistically significant differences between the CDK4 siRNA and control are presented as * (P < 0.05) and ** (P < 0.01).
Anti-proliferative efficacy of CDK4/6 inhibitor CINK4
We measured the anti-tumor efficacy of CINK4 to compare genetic silencing vs. pharmacologic inhibition of CDK4 in 4 KRAS mutant and 1 KRAS wild-type cell line. CINK4 treatment at various concentrations (~0.1–40 µM) yielded dose- and time-dependent cytotoxicity in all 5 cell lines, regardless of the presence or absence of KRAS mutation. The IC50 values of CINK4 at 72 h were 4–7 µM. Cell proliferation in all tumor cells was completely inhibited by > 20 µM CINK4 (). The IC50 values of CINK4 at 24, 48, and 72 h are summarized in .
Figure 4. Anti-proliferative effect of a CDK4/6 inhibitor, CINK4, in 5 lung adenocarcinoma cell lines. When treated with various concentrations of CINK4 (0.1–40 μM), the anti-proliferative effects of CINK4 were similar for each cell line at 72 h. Cell viability was quantitatively measured by SRB assay and presented as a percentage of the control cell population. Each experiment was performed in triplicate. Data represent the mean ± SD.
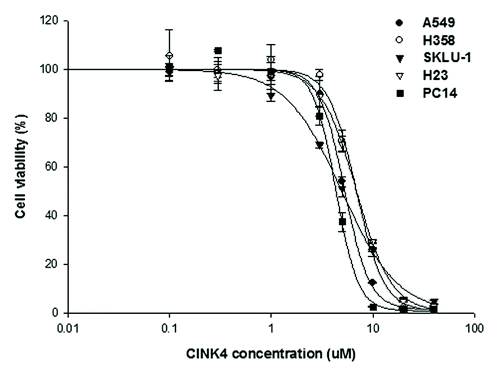
Table 1. Sensitivity of NSCLC cell lines to CINK4
CINK4 induces G0-G1 arrest through downregulation of phosphorylated Rb and induced apoptosis
We analyzed cell cycle distributions following 48 h CINK4 treatment in 5 lung cancer cell lines. In A549 and H23 cells, the cell population in G0/G1 phase increased but S and G2/M phase decreased in a concentration-dependent manner (). A similar cell cycle distribution pattern was observed in the other 3 cell lines (data not shown). In addition, the subG1 fraction of H23 cells significantly increased at 10 µM CINK4 (15.8 ± 3.8% vs. 4.6 ± 0.6% for control, P < 0.01); this was not observed in A549 cells (). The subG1 fraction of H358 and PC14 cells (but not SKLU-1) significantly increased (data not shown). Double staining with annexin V-FITC and PI was used to determine whether the increased subG1 fraction was due to induction of apoptosis. In H23 but not A549 cells, apoptotic induction occurred at 10 µM CINK4 for 72 h (27.2 ± 3.1% vs. 9.9 ± 0.5% for control, P < 0.001) (). CINK4 also induced apoptosis at same concentration in H358 and PC14 cells, but not in SKLU-1 cells (data not shown).
Figure 5. The effects of CINK4 on cell cycle distribution and apoptosis in KRAS mutation-bearing A549 and H23 cells. (A) Changes in cell cycle distribution were measured by flow cytometry after CINK4 treatment for 48 h. Each experiment was performed in triplicate. Data represent the mean ± SD. Statistically significant differences between CINK4 treatment groups and the control were presented as *(P < 0.05) and ** (P < 0.01). (B) Apoptotic cells induced by CINK treatment (5 and 10 μM) were measured by flow cytometry. Annexin V-FITC/PI double staining was performed and Annexin V-positive apoptotic cells were counted by flow cytometry. Each experiment was performed in triplicate. Data represent the mean ± SD. Statistically significant differences between CINK4 treatment groups and controls are presented as ** (P < 0.01) and *** (P < 0.001). (C) The effects of CINK4 on cell cycle regulators and apoptosis-related molecules were evaluated by western blotting. A549 and H23 cells were treated with CINK4 (3, 5, and 10 µM) for 72 h. Actin was used as a loading control.
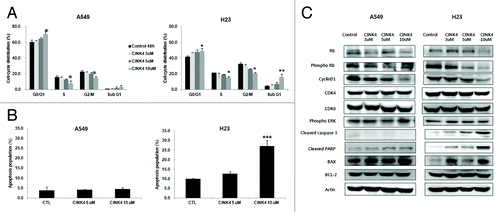
Prolonged 72 h exposure of A549 and H23 cells to CINK4 reduced phosphorylated Rb and cyclin D1 in a concentration-dependent manner. We obtained similar results in H358, SKLU-1 and PC14 cells (data not shown). Total Rb was reduced at higher concentrations (10 µM) in A549 and H23 cells. CINK4 had no inhibitory effect on expression of phosphorylated ERK, nor did CDK4 and CDK6 have an effect on A549 and H23 cells (). The remarkable increase in cleaved PARP and caspase 3 occurred at 10 µM CINK4 in H23 cells but not A549 cells. Bax expression was strongly increased but Bcl-2 expression was not significantly altered at 10 µM CINK4 in H23 and A549 ().
CINK4 enhances paclitaxel sensitivity in KRAS mutation-bearing lung cancer cells
Paclitaxel (0.1–300 nM; 72 h) induced dose-dependent cytotoxicity in 3 KRAS mutation-positive lung cancer cell lines. The IC50 value was 3 nM in A549 and H23 cells and 40 nM in SKLU-1 cells (). Even at 300 nM paclitaxel, 8.9 ± 1.6% and 9.0 ± 0.4% of A549 and H23 cells and 45.4 ± 3.2% of SKLU-1 cells remained alive.
Figure 6. Anti-proliferative effects of paclitaxel alone and combined with CINK4 in 3 KRAS mutation-positive NSCLC cell lines. (A) Anti-proliferative effect of paclitaxel (0.1–300 nM) in 3 KRAS mutation-positive NSCLC cell lines at 72 h. (B) Paclitaxel (1, 3, 5, and 10 nM) was combined with CINK4 (1, 3, 5, and 10 μM) for 72 h in individual cell line. Interactions between paclitaxel and CINK4 were analyzed by combination index (CI). The calculated CIs were divided into 3 categories: CI < 1, CI = 1, or CI > 1, indicating synergistic, additive and antagonistic effects, respectively.
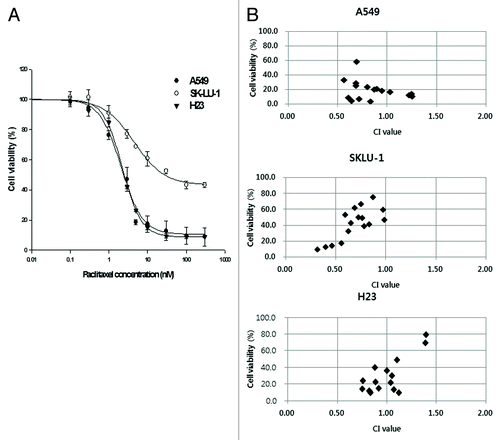
KRAS mutation-positive tumor cell growth inhibition was enhanced by a simultaneously combination of paclitaxel and CINK4, with a few exceptions (). CINK4 (1, 3, 5 and 10 μM) combined with paclitaxel (1, 3, 5 and 10 nM) yielded CI values of 0.85 ± 0.23 for A549, 0.69 ± 0.19 for SKLU-1 and 0.98 ± 0.20 for H23. We observed synergistic anti-tumor activities with 10 µM CINK4 plus paclitaxel (1, 3, 5 and 10 nM). The CI values were 0.69 ± 0.10 for A549, 0.42 ± 0.10 for SKLU-1, and 0.87 ± 0.16 for H23 cells.
Paclitaxel combined with CINK4 increases apoptosis
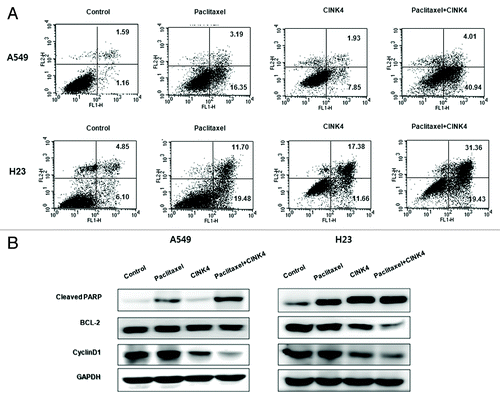
A549 and H23 cells treated with paclitaxel, CINK4 and paclitaxel+CINK4, the proportion of apoptotic cells was 19.4 ± 0.2% (P < 0.001 vs. control), 10.2 ± 0.6% (P < 0.01 vs. control) and 45.0 ± 0.1% (P < 0.001 vs. paclitaxel single) in A549 cells and 26.1 ± 7.2% (P < 0.01 vs. control), 30.0 ± 1.3% (P < 0.01 vs. control) and 50.3 ± 0.8% (P < 0.01 vs. paclitaxel single) in H23 cells (). Cleaved PARP, a marker of apoptosis, was increased in the combination vs. individual treatments. Expression of Bcl-2 and cyclin D1 decreased in the combination vs. individual treatments in A549 and H23 cells ().
Figure 7. Paclitaxel combined with CINK4 increases apoptosis in A549 and H23 cells. (A) Apoptosis induction. Annexin V-FITC/PI double staining was performed. Annexin V-positive apoptotic cells were counted by flow cytometry. Each experiment was performed in triplicate. The typical apoptosis induction by single and combination treatments are shown. (B) Apoptosis-related protein expression. Actin was used as a loading control. Cells were treated for 72 h with paclitaxel (3 nM) and CINK4 (10 μM), alone or in combination.
Discussion
Alternative signaling pathways may offer new therapeutic opportunities for treatment of KRAS mutation-positive lung adenocarcinoma. Selective inactivation of the p16/cyclin D1/CDK4/Rb signaling pathway is an important strategy for anti-cancer drug development. In this study, we investigated the anti-tumor effects of CDK4 suppression by molecular or pharmacologic inhibition in KRAS mutant cells. We also examined the impact of CINK4, a CDK4/6 inhibitor, in combination with paclitaxel.
RNA interference (RNAi) technology is a powerful approach to silence mammalian gene expression for studies of gene function; it also has therapeutic potential. In our study, transient downregulation of CDK4 with siRNA enhanced paclitaxel-induced cytotoxicity in KRAS mutation-bearing H23 cells; cytotoxic activity was statistically significant, however, potent cytotoxicity as we expected did not occur.CDK4 siRNA combined with paclitaxel may have been insufficient to induce potent anti-proliferative activity for several possible reasons. (1) Knockdown of CDK4 with transient CDK4 siRNA may not produce complete functional inhibition of CDK4. Various siRNAs targeting different sequences of the same gene can generate enormously variable inhibitory effects.Citation27 (2) Silencing of CDK4 may not be enough to enhance paclitaxel cytotoxicity because CDK6 can compensate for functional loss of CDK4. CDK4 and CDK6 are both essential for initiation of the cell cycle in response to various mitogenic stimuli. CDK4 is dispensable for proliferation in most cells, wherein CDK6 has a compensatory role.Citation28,Citation29 In addition, ablation of CDK4 and CDK6 did not affect the expression of D-type cyclins.Citation29 As cyclin D1/CDK6 also play important roles in cell cycle progression in KRAS mutant cells with functional CDK4 loss, this may explain the modest cytotoxic effect of paclitaxel in our study. We used CINK4, a dual pharmacologic inhibitor of CDK4 and CDK6 in the next experiment.
We measured the growth inhibitory activity of CINK4 in 5 lung adenocarcinoma cell lines. Growth inhibition by CINK4 (IC50) was similar in all cell lines, regardless of KRAS and p16 status. Thus, neither KRAS nor p16 status is directly correlated with sensitivity to CINK4. However, the cancer cell lines in our experiment all expressed active Rb. Our observation was consistent with previous reports that CINK4 had potent inhibitory activity on cell proliferation in the presence of Rb.Citation30
The cell cycle is a process that tightly controls cell growth and proliferation. Disruption of the cell cycle process causes an imbalance between cell proliferation and cell death, thereby resulting in cancer development.Citation31 Our study demonstrated that CINK4 efficiently reduced phosphorylated Rb with the CDK4-specific Ser807 and Ser811 residues and cyclin D1 in a dose-dependent manner, subsequently leading to the accumulation of tumor cells in G0/G1 phase. However, neither CDK4 nor CDK6 expression changed in A549 and H23 cells treated with CINK4. Previously, Rajeev et al. reported that CINK4 did not reduce total expression of CDK4, but acts on CDK4 via phosphorylation of its tyrosine residue.Citation30
An extensive list of signaling pathways regulate cyclin D1 and its transcription promoter binding sites.Citation32 ERK, a major downstream effector of RAS, is strongly associated with cell cycle entry particularly for the induction of cyclin D1 expression via downregulation of p27kip1.Citation33,Citation34 Conversely, cyclin D1 activity is independent of ERK expression.Citation34 In our experiment, CINK4 reduced cyclin D1, but did not affect its upstream regulator, phosphorylated ERK. These results suggest that the relationship between cyclinD1 and ERK is not a simple unidirectional linear one, at least in lung adenocarcinoma with KRAS mutations.
Key molecules in the cell cycle could serve as targets for anticancer agents to halt uncontrolled proliferation of tumor cells and to initiate apoptosis.Citation35 Our study demonstrated that apoptotic induction with increased Bax, cleaved PARP, and cleaved caspase3 occurred after CINK4 treatment (72 h, 10 μM) in H23, H358, and PC14, but not in A549 and SKLU-1. However, we found significantly increased cleaved PARP in A549 cells treated with CINK4 (72 h, 20 μM) (data not shown). CINK4 inhibits CDK4 in similar fashion of p16, an endogenous inhibitor of CDK4.Citation30 In present study, when treated with same concentration of CINK4, tumor cells had dissimilar apoptotic induction by CINK4. A previous study reported that p16INK4 mediated apoptosis was induced when p16-deficient lung cancer cells was infected with an adenovirus vector (Adv/p16).Citation36 Our result suggested that apoptosis be positively correlated with expression level of endogenous p16INK4. And our results are consistent with a previous report that low-concentration CINK4 also induced senescence, which was “bypassed” when cells were treated with either a higher dose or prolonged exposure to CINK4, both of which caused apoptotic induction.Citation30 Katsuda et al. reported that Rb was cleaved after activation of caspase-3; when Rb expression was significantly diminished, apoptosis began.Citation36 Our experiment showed that higher doses of CINK4 remarkably reduced Rb in A549 and H23 cells, suggesting that apoptosis induced by CINK4 treatment is also associated with Rb activation.
Apoptosis in cancer has been of particular interest from a therapeutic standpoint, since therapies should ultimately lead to the eradication of cancer cells through apoptosis.Citation37 We were surprised to observe the synergistic anti-proliferative activity of paclitaxel simultaneously combined with CINK4, especially more than 10 µM concentration (>IC50). This result be associated with potent apoptosis induced by higher concentration of CINK4. Apoptotic induction is a key mechanism for this combination in KRAS mutated adenocarcinoma with active Rb. In addition, the synergistic anti-proliferative activity also occurred by this combination in KRAS wild type PC14 cells (Fig. S1). It’s suggested that the combination therapy is generally for tumor cells regardless of KRAS.
Alterations in the expression of genes that control the cell cycle may be of critical importance in determining drug sensitivity to anti-cancer agents. Cyclin D1 has been proposed as a chemoprevention target and a surrogate marker of chemopreventive response in lung cancer.Citation38,Citation39 Targeting of cyclin D1 suppresses tumor growth via cell cycle arrest and promotes tumor apoptosis in NSCLC.Citation40-Citation42 Inhibition of cyclin D1 leads to cellular apoptosis through stimulation of the NFκB pathway with nucleolar translocation of RelA.Citation43,Citation44 Basseres et al. showed that deletion of RelA reduced the number of KRAS-mutant induced lung tumors with a higher number of apoptotic cells.Citation45 Therefore, our results suggest cyclin D1 plays a key role in apoptotic induction by CINK4 treatment in KRAS-mutated lung cancer.
The downregulation of Bcl-2 is thought to be one of the important modes of apoptosis induced by treatment with taxanes.Citation46 We observed that paclitaxel induces apoptosis, even in the absence of the reduction in Bcl-2 expression, suggesting that it may be attributed to the lower concentration of paclitaxel.Citation47 Our experiment demonstrated that CINK4 alone did not reduce Bcl-2 expression, but when combined with paclitaxel, Bcl-2 expression was significantly reduced. Paclitaxel alone did not reduce cyclin D1, but greatly reduced it when combined with CINK4. Our results suggest both Bcl-2 and cyclin D1 are important molecules for synergistic anti-proliferative activity when paclitaxel is combined with CINK4. We propose that downregulation of cyclin D1 by CINK4 may enhance sensitivity to paclitaxel, subsequently leading to increased apoptosis in KRAS-mutated adenocarcinoma cells.
In summary, our preclinical study demonstrated that genetic knockdown or pharmacological inhibition of CDK4 reduced the population of phosphorylated Rb and induced G1 arrest. We also found the CDK4/6 inhibitor CINK4 promotes apoptotic induction through the downregulation of Rb and cyclin D1. In addition, cyclin D1 and Bcl-2 play critical roles in synergistic anti-tumor efficacy with significant apoptotic induction by combined treatment with CINK4 and paclitaxel in adenocarcinoma cells harboring KRAS mutations. We suggest CDK4 inhibition combined with taxane therapy may be a clinically useful strategy for KRAS mutant lung cancers.
Material and Methods
Drug preparation
The CDK4/6 inhibitor (CINK4) was purchased from EMD Biosciences and paclitaxel was kindly provided by Hanmi Pharmaceuticals. For in vitro experiments, CINK4 and paclitaxel were dissolved in 100% DMSO (Sigma) and diluted with culture medium to the desired concentration with a final DMSO concentration of 0.1%. DMSO was added to untreated cultures at 0.1% (v/v) as a solvent control.
Cancer cell lines
NSCLC cell lines A549 (KRASG12S), H358 (KRASG12C), SKLU-1 (KRASG12S), H23 (KRASG12C), and PC14 (KRASwild) were obtained from the Korea Cell Line Bank. H358 and H23 cells carry a wild-type CDKN2A (p16) gene, but A549, SKLU-1, and PC14 carry homozygous deletions of the CDKN2A gene. The somatic mutation data for KRAS and CDKN2A were obtained from the COSMIC database (http://www.sanger.ac.uk/cosmic).Citation16,Citation48 A549 and SKLU-1 were propagated in Dulbecco’s modified Eagle medium (DMEM, Gibco BRL). H358, H23, and PC14 cells were maintained in RPMI 1640 medium (WelGENE). Culture media were supplemented with 10% fetal bovine serum (FBS; Gibco BRL), 1% penicillin-streptomycin (Gibco BRL) and 10 mmol/L HEPES (Amresco). Cells were cultivated in 100-mm plastic dishes (TPP) at 37 °C in 5% CO2 and 95% air and harvested with trypsin/EDTA at logarithmic growth.
siRNA transfection and drug sensitivity assay
The day before siRNA transfection, 200 000 and 6000 cells were plated in 6- and 96-well plates, respectively and incubated in antibiotic-free medium with 10% FBS. After 24 h, the medium was exchanged with serum- and antibiotic-free medium. Lipofectamine 2000 (Invitrogen) was used to transfect CDK4 siRNA or universal negative control siRNA (Sigma-Aldrich) into cells. Both on-target siRNA and negative control siRNA were used at the same concentration in all experiments. The efficiency of inhibition was determined by reverse-transcription polymerase chain reaction (RT-PCR) and western blotting after 48 h transfection. Twenty-four hours after siRNA transfection, cytotoxic activity was measured during 48 h incubation with paclitaxel. Cytotoxicity was normalized to control siRNA-transfected, drug-treated cultures.
RT-PCR
Total RNA was isolated using Trizol reagent (Invitrogen). cDNA was obtained with the Maxime RT premix kit (Intron). The following sense and antisense primers were used to amplify CDK4 (5′-CATCGTTCAC CGAGATCTGA-3′ and 5′-CCAACACTCC ACATGTCCAC-3′, 198-bp product) and GAPDH (5′-ACCCACTCCT CCACCTTTGA-3′ and 5′-TCCAGGGGTC TTACTCCTTG-3′, 147-bp product). Reaction conditions included a pre-denaturation step at 95 °C for 10 min followed by 35 cycles of 95 °C for 30 sec, 58 °C for 30 sec, 72 °C for 1 min, and extension for 5 min at 72 °C. PCR products were separated on 1.5% agarose gels.
Cell proliferation assay
Cells were seeded at 2000 per well in 96-well sterile plastic dishes (Nonclon), allowed to attach for 24 h and then treated with single-drug dilutions. The cells were treated with a series of final concentrations ranging from 0.1 μM to 40 μM for CINK4 and from 0.1 nM to 300 nM for paclitaxel. Inhibition of cell growth was measured with the sulforhodamine B (SRB) assay.Citation49 Absorbance was measured at 560 nm using an enzyme-linked immunosorbent assay (ELISA) reader (Spectra Max 250; Molecular Devices). The drug concentrations required to inhibit cell growth by 50% (IC50) were determined from dose-response curves created with Sigma Plot software (version 8.0). The results were presented as the mean ± standard deviation (SD) of at least 3 independent experiments.
Cell cycle analysis
Cells were seeded at 100 000 in 6-well plates (Nonclon) and incubated for 24 h. Then, each cell line was treated with CINK4 alone at 3, 5 and 10 μM for 48 h. After the treatments were completed, cells were trypsinized, washed twice with PBS and harvested by centrifugation. Briefly, cells were fixed with ice-cold 70% ethanol for at least 1 h, centrifuged, washed twice in cold PBS, resuspended in 1 mL PBS and stained with propidium iodide (PI) solution (0.05 mg/mL PI, 10 mg/mL RNase A) for 20 min at 37 °C in the dark. Fluorescence intensity was measured by flow cytometry (BD FACS Calibur); at least 10 000 cells were counted and DNA content analysis was performed with CellQuest software (Becton Dickinson). All experiments were performed in triplicate.
Apoptosis analysis
Cells were seeded in 6-well plates (Nonclon) at a density of 100,000 cells per well and incubated for 24 h prior to drug addition. After treatment, adherent and non-adherent cells were harvested and the FITC Annexin V Apoptosis Detection Kit I (BD PharMingen) was used according to the manufacturer’s protocol. Briefly, cells were washed with cold PBS, resuspended in 1 × binding buffer (0.1 M HEPES/NaOH, pH 7.4, 1.4 M NaCl, and 25 mM CaCl2), stained with annexin V-FITC and PI, incubated for 15 min at room temperature in the dark and analyzed by flow cytometry (BD FACS Calibur). Cells unstained with PI and annexin V were considered normal (i.e., no apoptosis). Cells stained with annexin V alone or both annexin V and PI were considered to have undergone early and late apoptosis, respectively; these cells were included in the total count of apoptotic cells. The PI-positive and annexin V-negative cells were counted as necrotic cells. All experiments were performed in triplicate.
Western blot analysis
Cells were seeded at 500 000 in 100-mm plastic dishes and incubated for 24 h prior to addition of drug. After drug treatment, cells were harvested with trypsin/EDTA, washed twice with PBS and lysed with RIPA cell lysis buffer (Gibco BRL) containing a protease inhibitor cocktail (Amresco). Thirty micrograms of protein from each cell treatment was used in Bio-Rad detergent-compatible protein assays (Bio-Rad); proteins were resolved on 8–12% polyacrylamide gels by standard sodium dodecyl sulfate PAGE (SDS-PAGE) and transferred onto PVDF membranes (0.45 μm; Millipore). Membranes were blocked with 5% skim milk (Becton Dickinson). Blots were probed with the following antibodies. Anti-rabbit antibodies were used against Rb, phospho-Rb (Ser807/811), p16INK4A, phospho-ERK1/2 (Thr202/Thr204), cleaved caspase3 and cleaved PARP (Cell Signaling). Anti-mouse antibodies were used for CDK4, cyclin D1, Bax (Santa Cruz); CDK6 (Cell Signaling); actin and GAPDH (Abcam). Anti-goat antibodies were used against Bcl-2 (Santa Cruz). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Santa Cruz. Blots were developed with an enhanced chemiluminescence reagent (Amersham) and detected with an LAS 3000 Image analyzer system (Fujifilm).
Combined drug efficacy
We chose the drug concentrations and treatment times based on the results of single drug treatment; 4 concentrations of paclitaxel (1, 3, 5, and 10 nM) were combined with 4 concentrations of CINK4 (1, 3, 5, and 10 µM) in 3 cell lines over 72 h. The control wells consisted of cells incubated with culture medium. Interactions between paclitaxel and CINK4 were calculated by the combination index (CI). The calculated CI was then divided into 3 categories: CI < 1, CI = 1, or CI > 1, indicating synergistic, additive and antagonistic effects.Citation50
Statistical analysis
Data are expressed as the mean ± SD. Statistically significant differences between groups were determined by the Student’s t-test. In each case, P values < 0.05 were considered statistically significant.
| Abbreviations: | ||
| CDK | = | cyclin-dependent kinase |
| siRNA | = | short interfering RNA |
| Rb | = | retinoblastoma |
| NSCLC | = | non-small cell lung cancer |
| EGFR TKI | = | growth factor receptor tyrosine kinase inhibitor |
| CI | = | combination index |
| RNAi | = | RNA interference |
Additional material
Download Zip (125 KB)Acknowledgments
This study was supported by a BK21 project for biomedical science and a grant for disease-oriented translational research from the Ministry of Education and Human Resources Development in Korea.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Porta M, Crous-Bou M, Wark PA, Vineis P, Real FX, Malats N, et al. Cigarette smoking and K-ras mutations in pancreas, lung and colorectal adenocarcinomas: etiopathogenic similarities, differences and paradoxes. Mutat Res 2009; 682:83 - 93; http://dx.doi.org/10.1016/j.mrrev.2009.07.003; PMID: 19651236
- Rodenhuis S, Slebos RJ, Boot AJ, Evers SG, Mooi WJ, Wagenaar SS, et al. Incidence and possible clinical significance of K-ras oncogene activation in adenocarcinoma of the human lung. Cancer Res 1988; 48:5738 - 41; PMID: 3048648
- Zhang X, Chang A. Molecular predictors of EGFR-TKI sensitivity in advanced non-small cell lung cancer. Int J Med Sci 2008; 5:209 - 17; http://dx.doi.org/10.7150/ijms.5.209; PMID: 18645621
- Keohavong P, DeMichele MA, Melacrinos AC, Landreneau RJ, Weyant RJ, Siegfried JM. Detection of K-ras mutations in lung carcinomas: relationship to prognosis. Clin Cancer Res 1996; 2:411 - 8; PMID: 9816185
- Mascaux C, Iannino N, Martin B, Paesmans M, Berghmans T, Dusart M, et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer 2005; 92:131 - 9; http://dx.doi.org/10.1038/sj.bjc.6602258; PMID: 15597105
- Ladanyi M, Pao W. Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Modern pathology: an official journal of the United States and Canadian Academy of Pathology. Inc 2008; 21:Suppl 2 S16 - 22
- Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, Liu DD, et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res 2007; 13:2890 - 6; http://dx.doi.org/10.1158/1078-0432.CCR-06-3043; PMID: 17504988
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 2007; 1773:1263 - 84; http://dx.doi.org/10.1016/j.bbamcr.2006.10.001; PMID: 17126425
- Zhang XH, Shin JY, Kim JO, Oh JE, Yoon SA, Jung CK, et al. Synergistic antitumor efficacy of sequentially combined paclitaxel with sorafenib in vitro and in vivo NSCLC models harboring KRAS or BRAF mutations. Cancer Lett 2012; 322:213 - 22; http://dx.doi.org/10.1016/j.canlet.2012.03.015; PMID: 22433711
- Trejo CL, Juan J, Vicent S, Sweet-Cordero A, McMahon M. MEK1/2 inhibition elicits regression of autochthonous lung tumors induced by KRASG12D or BRAFV600E. Cancer Res 2012; 72:3048 - 59; http://dx.doi.org/10.1158/0008-5472.CAN-11-3649; PMID: 22511580
- van Krieken JH, Jung A, Kirchner T, Carneiro F, Seruca R, Bosman FT, et al. KRAS mutation testing for predicting response to anti-EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Arch 2008; 453:417 - 31; http://dx.doi.org/10.1007/s00428-008-0665-y; PMID: 18802721
- Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J 2000; 351:289 - 305; http://dx.doi.org/10.1042/0264-6021:3510289; PMID: 11023813
- Yamamoto T, Ebisuya M, Ashida F, Okamoto K, Yonehara S, Nishida E. Continuous ERK activation downregulates antiproliferative genes throughout G1 phase to allow cell-cycle progression. Curr Biol 2006; 16:1171 - 82; http://dx.doi.org/10.1016/j.cub.2006.04.044; PMID: 16782007
- Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc Natl Acad Sci U S A 1998; 95:1091 - 6; http://dx.doi.org/10.1073/pnas.95.3.1091; PMID: 9448290
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993; 366:704 - 7; http://dx.doi.org/10.1038/366704a0; PMID: 8259215
- Hamada K, Kohno T, Kawanishi M, Ohwada S, Yokota J. Association of CDKN2A(p16)/CDKN2B(p15) alterations and homozygous chromosome arm 9p deletions in human lung carcinoma. Genes Chromosomes Cancer 1998; 22:232 - 40; http://dx.doi.org/10.1002/(SICI)1098-2264(199807)22:3<232::AID-GCC9>3.0.CO;2-X; PMID: 9624535
- Shapiro GI, Park JE, Edwards CD, Mao L, Merlo A, Sidransky D, et al. Multiple mechanisms of p16INK4A inactivation in non-small cell lung cancer cell lines. Cancer Res 1995; 55:6200 - 9; PMID: 8521414
- Serrano M, Gómez-Lahoz E, DePinho RA, Beach D, Bar-Sagi D. Inhibition of ras-induced proliferation and cellular transformation by p16INK4. Science 1995; 267:249 - 52; http://dx.doi.org/10.1126/science.7809631; PMID: 7809631
- Guan RJ, Fu Y, Holt PR, Pardee AB. Association of K-ras mutations with p16 methylation in human colon cancer. Gastroenterology 1999; 116:1063 - 71; http://dx.doi.org/10.1016/S0016-5085(99)70009-0; PMID: 10220498
- Lazarov M, Kubo Y, Cai T, Dajee M, Tarutani M, Lin Q, et al. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat Med 2002; 8:1105 - 14; http://dx.doi.org/10.1038/nm779; PMID: 12357246
- Puyol M, Martín A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010; 18:63 - 73; http://dx.doi.org/10.1016/j.ccr.2010.05.025; PMID: 20609353
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 2004; 3:1427 - 38; PMID: 15542782
- Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, et al. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer 2011; 104:1862 - 8; http://dx.doi.org/10.1038/bjc.2011.177; PMID: 21610706
- Konecny GE, Winterhoff B, Kolarova T, Qi J, Manivong K, Dering J, et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin Cancer Res 2011; 17:1591 - 602; http://dx.doi.org/10.1158/1078-0432.CCR-10-2307; PMID: 21278246
- Chang AY, Kim K, Glick J, Anderson T, Karp D, Johnson D. Phase II study of taxol, merbarone, and piroxantrone in stage IV non-small-cell lung cancer: The Eastern Cooperative Oncology Group Results. J Natl Cancer Inst 1993; 85:388 - 94; http://dx.doi.org/10.1093/jnci/85.5.388; PMID: 8094467
- Murphy WK, Fossella FV, Winn RJ, Shin DM, Hynes HE, Gross HM, et al. Phase II study of taxol in patients with untreated advanced non-small-cell lung cancer. J Natl Cancer Inst 1993; 85:384 - 8; http://dx.doi.org/10.1093/jnci/85.5.384; PMID: 8094466
- Rolle K, Nowak S, Wyszko E, Nowak M, Zukiel R, Piestrzeniewicz R, et al. Promising human brain tumors therapy with interference RNA intervention (iRNAi). Cancer Biol Ther 2010; 9:396 - 406; PMID: 20118657
- Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol 1994; 14:2077 - 86; PMID: 8114739
- Malumbres M, Sotillo R, Santamaría D, Galán J, Cerezo A, Ortega S, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 2004; 118:493 - 504; http://dx.doi.org/10.1016/j.cell.2004.08.002; PMID: 15315761
- Soni R, O’Reilly T, Furet P, Muller L, Stephan C, Zumstein-Mecker S, et al. Selective in vivo and in vitro effects of a small molecule inhibitor of cyclin-dependent kinase 4. J Natl Cancer Inst 2001; 93:436 - 46; http://dx.doi.org/10.1093/jnci/93.6.436; PMID: 11259469
- Clarke PR, Allan LA. Cell-cycle control in the face of damage--a matter of life or death. Trends Cell Biol 2009; 19:89 - 98; http://dx.doi.org/10.1016/j.tcb.2008.12.003; PMID: 19168356
- Klein EA, Assoian RK. Transcriptional regulation of the cyclin D1 gene at a glance. J Cell Sci 2008; 121:3853 - 7; http://dx.doi.org/10.1242/jcs.039131; PMID: 19020303
- Weber JD, Raben DM, Phillips PJ, Baldassare JJ. Sustained activation of extracellular-signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem J 1997; 326:61 - 8; PMID: 9337851
- Villanueva J, Yung Y, Walker JL, Assoian RK. ERK activity and G1 phase progression: identifying dispensable versus essential activities and primary versus secondary targets. Mol Biol Cell 2007; 18:1457 - 63; http://dx.doi.org/10.1091/mbc.E06-10-0908; PMID: 17314399
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature 2004; 432:316 - 23; http://dx.doi.org/10.1038/nature03097; PMID: 15549093
- Katsuda K, Kataoka M, Uno F, Murakami T, Kondo T, Roth JA, et al. Activation of caspase-3 and cleavage of Rb are associated with p16-mediated apoptosis in human non-small cell lung cancer cells. Oncogene 2002; 21:2108 - 13; http://dx.doi.org/10.1038/sj.onc.1205272; PMID: 11960384
- Sanchez RI, Mesia-Vela S, Kauffman FC. Challenges of cancer drug design: a drug metabolism perspective. Curr Cancer Drug Targets 2001; 1:1 - 32; http://dx.doi.org/10.2174/1568009013334296; PMID: 12188889
- Petty WJ, Dragnev KH, Dmitrovsky E. Cyclin D1 as a target for chemoprevention. Lung Cancer 2003; 41:Suppl 1 S155 - 61; http://dx.doi.org/10.1016/S0169-5002(03)00159-4; PMID: 12867074
- Pitha-Rowe I, Petty WJ, Feng Q, Koza-Taylor PH, Dimattia DA, Pinder L, et al. Microarray analyses uncover UBE1L as a candidate target gene for lung cancer chemoprevention. Cancer Res 2004; 64:8109 - 15; http://dx.doi.org/10.1158/0008-5472.CAN-03-3938; PMID: 15520223
- Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol 1994; 14:2066 - 76; PMID: 8114738
- Driscoll B, Wu L, Buckley S, Hall FL, Anderson KD, Warburton D. Cyclin D1 antisense RNA destabilizes pRb and retards lung cancer cell growth. Am J Physiol 1997; 273:L941 - 9; PMID: 9374720
- Saini SS, Klein MA. Targeting cyclin D1 in non-small cell lung cancer and mesothelioma cells by antisense oligonucleotides. Anticancer Res 2011; 31:3683 - 90; PMID: 22110187
- Sauter ER, Nesbit M, Litwin S, Klein-Szanto AJ, Cheffetz S, Herlyn M. Antisense cyclin D1 induces apoptosis and tumor shrinkage in human squamous carcinomas. Cancer Res 1999; 59:4876 - 81; PMID: 10519399
- Thoms HC, Dunlop MG, Stark LA. p38-mediated inactivation of cyclin D1/cyclin-dependent kinase 4 stimulates nucleolar translocation of RelA and apoptosis in colorectal cancer cells. Cancer Res 2007; 67:1660 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-06-1038; PMID: 17308107
- Bassères DS, Ebbs A, Levantini E, Baldwin AS. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res 2010; 70:3537 - 46; http://dx.doi.org/10.1158/0008-5472.CAN-09-4290; PMID: 20406971
- Ganansia-Leymarie V, Bischoff P, Bergerat JP, Holl V. Signal transduction pathways of taxanes-induced apoptosis. Curr Med Chem Anticancer Agents 2003; 3:291 - 306; http://dx.doi.org/10.2174/1568011033482422; PMID: 12769774
- Giannakakou P, Robey R, Fojo T, Blagosklonny MV. Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel-induced cytotoxicity. Oncogene 2001; 20:3806 - 13; http://dx.doi.org/10.1038/sj.onc.1204487; PMID: 11439344
- Okamoto A, Hussain SP, Hagiwara K, Spillare EA, Rusin MR, Demetrick DJ, et al. Mutations in the p16INK4/MTS1/CDKN2, p15INK4B/MTS2, and p18 genes in primary and metastatic lung cancer. Cancer Res 1995; 55:1448 - 51; PMID: 7882351
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990; 82:1107 - 12; http://dx.doi.org/10.1093/jnci/82.13.1107; PMID: 2359136
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22:27 - 55; http://dx.doi.org/10.1016/0065-2571(84)90007-4; PMID: 6382953