Abstract
ATG7 is a key autophagy-promoting gene that plays a critical role in the regulation of cell death and survival of various cell types. We report here that microRNAs (miRNAs), a class of endogenous 22–24 nucleotide noncoding RNA molecules able to affect stability and translation of mRNA, may represent a novel mechanism for regulating ATG7 expression and therefore autophagy. We demonstrated that ATG7 is a potential target for miR-17, and this miRNA could negatively regulate ATG7 expression, resulting in a modulation of the autophagic status in T98G glioblastoma cells. Treatment of these tumor cells with the miR-17 mimic decreased, and with the antagomir increased, the expression of ATG7 protein. Dual luciferase reporter assay confirmed that a specific miR-17 binding sequence in the 3′-UTR of ATG7 contributed to the modulation of the expression of the gene by miR-17. Interestingly, our results showed that anti-miR-17 administration activated autophagy through autophagosome formation, as resulted by LC3B and ATG7 protein expression increase, and by the analysis of GFP-LC3 positive autophagosome vesicles in living cells. Furthermore, the autophagy activation by anti-miR-17 resulted in a decrease of the threshold resistance at temozolomide doses in T98G cells, while miR-17 modulation in U373-MG glioblastoma cells resulted in a sensitization to low ionizing radiation doses. Our study of the role of miR-17 in regulating ATG7 expression and autophagy reveals a novel function for this miRNA sequence in a critical cellular event with significant impacts in cancer development, progression and treatment.
Keywords: :
Introduction
Autophagy (from the Greek words for “self” and “eating”), is an evolutionarily conserved program responsible for intracellular degradation of proteins and whole organelles such as mitochondria. Autophagy is activated, under normal growth condition, in response to several stresses (e.g., starvation, hypoxia) by providing recycled metabolic substrates necessary for survival. Beside this, autophagy is activated to maintain cellular homeostasis in numerous pathophysiological processes, such as pathogen infection, neurodegenerative disorders, aging, myopathies, and cancer.Citation1,Citation2 Briefly, the canonical autophagic pathway is stimulated by internal and external stimuli, converging on phosphatidylinositol 3-kinase, protein kinase B, and mTOR (PI3K/Akt/mTOR) axis and triggering the activity of 34 ATGs.Citation3-Citation5 The process involves the formation of double membrane vesicles, namely autophagosomes, which engulf portions of cytoplasm, mature along the endocytic pathway, acidify, and eventually fuse with lysosome, forming autolysosomes and leading to their degradation.Citation4,Citation6
In cancer cells, in the attempt to cope with stress conditions such as low nutrient and oxygen, autophagy is used both as pro-survival and pro-death mechanism. To date, it is still debated how autophagy switches between these two cellular fates; probably, the degradation of cytosol and organelles beyond a certain threshold, depending on nature and duration of the induced cellular stress as well as on the involved tumor type, lead to the autophagic-related cell death.Citation1 Several researches are now directed toward the dissection of the autophagic pathway in order to identify novel potential targets, effective in the regulation of the autophagy process, and to enhance the ratio of cell death induction, combining gene therapy to traditional protocols.Citation7-Citation9 In particular, microRNAs (miRNAs) are small non coding RNAs that have been estimated to regulate more than 60% of all mammal protein-coding genes.Citation10 They are mainly known as cytoplasmic post-transcriptional regulators of gene expression, inhibiting protein synthesis by base-pairing to target mRNA in their 3′-UTR and leading to mRNA decay or destabilization.Citation11 However, several evidences indicate that they act also on different mRNA regions and different cellular compartments, including nucleus, either enhancing or suppressing gene expression.Citation12-Citation15
Recently, an increasing number of miRNAs are being characterized to modulate the expression of ATG genes and their regulators at different autophagic stages: induction, vesicle nucleation, elongation, and completion.Citation16,Citation17 Nonetheless, despite recent advances in understanding miRNA regulation of the autophagy process, several gaps remain.
Following bioinformatics analysis, we identified miR-17-5p (hereafter miR-17) as a putative modulator of different ATGs. This miRNA is part of the miR-17-92 cluster, already known for its role in tumor development via MYC and E2F1 signals.Citation18-Citation20 The primary transcript of this miR was named “OncomiR-1”.Citation21 In particular, miR-17 was already demonstrated to act specifically at the G1/S-phase cell cycle boundary, targeting many genes involved in the transition between these phases.Citation22 However, the functional involvement of miR-17 in tumor development is still debated, reporting both oncogenicCitation23,Citation24 and tumor suppressive roles.Citation18,Citation25,Citation26
In addition, the induction of autophagic cell death (type II cell death) by pro-autophagic drugs is an alternative and emerging concept to trigger glioma cell death and to exploit caspase-independent programmed cell death pathways for the development of novel glioma therapies.Citation27 A variety of chemical or physical treatments, including rapamycin (mTOR inhibitor), radiation, arsenic trioxide, ceramide, temozolomide, dopamine, endostatin, the histone deacetylase (HDAC) inhibitors butyrate and suberoylanilide hydroxamic acid, neodymium oxide, the chemotherapeutic vitamin D analog EB1089, saponins, and resveratrol, have been reported to induce autophagy in vitro and in vivo in certain cancer cells.Citation28 Notably, temozolomide brings significant therapeutic benefits in glioblastoma treatment and its cytotoxic activity is exerted through pro-autophagic processes.Citation29
In this work, we demonstrated that the modulation of miR-17 expression in GBM cells modulated the autophagic level. Specifically, ATG7 gene was identified as a molecular target of miR-17. In addition, we demonstrated that autophagy activation, by means of miR-17 downregulation, leads to further sensitization of GBM cells to chemotherapeutic and ionizing radiation treatments, resulting in reduced long-term tumor cell viability.
Results
Computational identification of miRNAs targeting autophagy-related genes
According to bioinformatics prediction tools using miRWalk software,Citation30 miR-17, already known to be entangled in tumor developmentCitation18,Citation20,Citation21 and demonstrated to be overexpressed in GBM,Citation31 was predicted to target various ATGs (i.e., ATG2, ATG4, ATG5, ATG7, ATG10, ATG12, and ATG16), suggesting a multiple role in the modulation of the autophagy process. Next, in refining miR-17 and ATGs interactions, Miranda target tool predicted four putative interaction sites between the miR-17 seeding core and the 3′-UTR of ATG7 transcript, while fewer complementary sites were predicted for the other ATG genes.
Prediction sites were provided with ranking scores (mirSVR) and reported in : according to Betel et al.,Citation32 these scores were interpreted as an empirical probability of transcript downregulation. In particular, the miR-17 complementary site with nucleotide coordinates 1218–1243 on ATG7 3′-UTR, showed the highest ranking score, i.e., mirSVR = -0.772. Using Targetscan tool, this site also exhibited the highest probability of conserved targeting (PCT = 0.96), as described in Friedman et al.Citation10 for conserved miRNA families. Accordingly, this site was considered for next experiments as a bona fide target of miR-17 interaction on the ATG7 transcript and subjected to a validation assay.
Figure 1. Sequence alignment prediction between miR-17 and ATG7 and validation assays. (A) miRanda predicted miR-17 targets (squares) within the 3′-UTR region (with nucleotides coordinates) of human ATG7 sequence (NM_006395). miRanda likelihood of miR-17-mediated mRNA downregulation scores (mirSVRs) are reported. (B) Nucleotide alignments between miR-17 seed sequence and ATG7 wild-type and mutated sequences, respectively cloned into pMIRATG7WT and pMIRATG7MUT vectors (see Materials and Methods) (top); results of the luciferase reporter assay at 24 and 48 h post-transfection (p.t.) in T98G cells are expressed as Relative Luciferase Units (left); results were normalized to pMIR-REPORT vector (vehicle) transfections. (C) Real-time PCR quantitative expression levels of miR-17 and ATG7 in T98G cells, from a RIP-assay using EIF2C2/AGO2-co-immunoprecipitated RNA, described in Materials and Methods; controls are represented by mock (input) and IgG isotype RNA samples. Expression levels are normalized to miR-17 and ATG7 expression levels of input samples. RIP-assay is performed using Protein A (top) and G sepharose matrices (bottom). Results of three independent replicas are indicated with error bars. *P < 0.005, **P < 0.001.
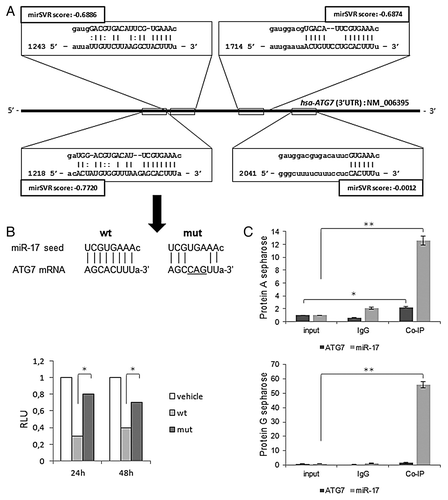
miR-17 directly inhibits ATG7 expression by targeting its 3′-UTR region
A luciferase reporter assay was used to test whether the negative effects of miR-17 on ATG7 were through directly targeting the above mentioned specific complementary sequence on the 3′-UTR region of the transcript. The schematic structure of ATG7 3′-UTR, the potential miR-17 binding site and its mutation which disrupts the “seed” region was reported in . T98G cells were then co-transfected using either a plasmid vector expressing wild-type ATG7 (pMIRATG7WT) or its mutated counterpart (pMIRATG7MUT) and miR-17 precursor moieties (PmiR-17, 2.5 µM). A 62.5% and 42.9% reduction of relative luciferase emission between pMIRATG7WT and pMIRATG7MUT transfected cells was scored after 24 and 48h p.t., respectively (P < 0.005). The association of miR-17 and ATG7 transcript in T98G cells was also tested by a RIP-Assay, followed by Real-time PCR detection. Cellular EIF2C2/AGO2-associated RNA from T98G cultured cells was isolated with RIP-Assay kit for microRNA coupled with a RIP-Certified Anti-EIF2C2/AGO2 monoclonal antibody. An equal amount of IgG2a isotype control was used as a negative control. As reported in , a significant enrichment of ATG7 and miR-17 expression, compared with input and isotype controls was reported within the anti-EIF2C2/AGO2 immunoprecipitates, using either Protein A or G sepharose beads. These data suggested that miR-17 directly targeted the 3′-UTR of ATG7 to mediate a repressive effect on the protein expression.
miR-17 regulates autophagy in T98G cells
In order to verify the computational prediction above reported, miR-17 expression was modulated in T98G human glioblastoma cells by in vitro administration of specific inhibitor (AmiR-17) or precursor (PmiR-17) miRNA molecules at 2.5 µM concentration and these assays were compared with untreated endogenous miR-17 levels. In general, the transfection of these molecules, did not significantly affect cell viability and morphology, even at different post-transfection (p.t.) times and concentrations (data not shown). At 48 h p.t. the levels of miR-17 expression were specifically evaluated by Real-time PCR. As reported in , a significant decrease of miR-17 expression was observed after AmiR-17 transfection (P < 0.005); differently, an increase in miR-17 expression was scored after PmiR-17 administration (P < 0.005). Following the above mentioned treatments, miR-17 variations significantly altered ATG7 protein expression compared with mock untreated cells (P < 0.001; ). Then, T98G cells, subjected to the above mentioned miR-17 modulations, were assayed for the autophagy process activation, by means of the expression of the well-established autophagosome marker, i.e., light chain microtubule associated protein 3 (LC3B).Citation33 As previously stated, during autophagy LC3B-I proteins are processed, conjugated with phosphatidilethanolammine and recruited to the autophagosomal membrane as LC3B-II isoforms, therefore the amount of LC3B-II correlates with the number of autophagosomes.Citation4 Here, LC3B-II expression was evaluated and it increased sensibly after AmiR-17 administration, while it decreased with PmiR-17 treatment (, darker bars). Given that LC3B-II isoform may be ectopically generated in an autophagy-independent manner, to discriminate among the different LC3B-II endogenous forms, BafilomycinA1 (BafA1, 10 nM) was directly added in cultured T98G cells, previously transfected with miR-17 inhibitors or mimics molecules. BafA1 promotes accumulation of autophagic vacuoles, preventing fusion between autophagosomes and lysosomes, thus inhibiting LC3B-II degradation by acidic organelles. Accordingly, different amount of LC3B-II protein levels, in presence or absence of lysosomal inhibitors represent the amount of LC3B-II that is delivered to lysosomes for degradation (i.e., autophagic flux).Citation33 As a result, in a similar trend of ATG7 protein expression, the in vitro incubation with BafA1 further increased LC3B-II levels in AmiR-17 transfected cells, whereas LC3B-II significantly decreased after PmiR-17 transfection coupled with BafA1 administration (P < 0.001). To further evaluate the modulation of the autophagy process by miR-17 in T98G cells, a well-established autophagy inducer rapamycin was administered at different concentrations (50–100 nM), after separate transfections of AmiR-17 or PmiR-17 molecules, at 2.5 µM concentration each. As we previously reported in T98G cells,Citation34 rapamycin administration was able to block mTOR kinase activity, decreasing the phosphorylation of the mTOR-substrate p70S6K, enabling Ulk1/2 complex activation and autophagosome initiation. As reported in , both rapamycin administration and AmiR-17 transfection alone promoted ATG7 and LC3B-II expression increase, compared with mock untreated samples. However, the most effective increase in ATG7 and LC3B-II proteins expression was scored after rapamycin administration (100 nM). To note, the combined treatment of AmiR-17 and rapamycin did not give any cumulative effects in terms of ATG7 and LC3B-II expression variations. However, as above described, miR-17 modulation significantly affected ATG7 and LC3B expression in T98G cells. To directly visualize in T98G living cells the activation of the autophagy process through the formation of autophagosomes, the Premo Autophagy Sensor (LC3B-FP) BacMam 2.0 system was employed. Briefly, LC3B-FP and LC3B(G120A)-FP viral vectors (each at MOI = 30) were transduced into T98G cells, enabling the expression of fluorescent LC3B protein, and consequently, monitoring of autophagosomes dynamics using an inverted fluorescent microscope in real-time analysis. The mutant chimera LC3B(G120A)-FP was employed as a negative control. In this construct, a G120A mutation prevents LC3B-I cleavage and its subsequent lipidation, thus showing a cytosolic and diffuse protein localization. Thus, transduced T98G cells were transfected with AmiR-17 or PmiR-17 molecules (2.5 µM) and treated with rapamycin (0–50–100 nM). After an incubation of additional 24h, DIC phase contrast-fluorescent photographs were recorded. According to the results reported in , it was evident that BacMam LC3B(G120A)-FP transduced cells showed a marked cytosolic and diffuse expression pattern; moreover, untreated or PmiR-17 transfected LC3-FP expressing cells, presented only few fluorescent vesicles; differently, T98G cells treated with miR-17 inhibitor (AmiR-17), presented an extensive punctate fluorescent distribution pattern, suggesting LC3B-FP protein accumulation in nascent autophagosomes. In addition, rapamycin administration increased LC3B-FP signals, further enforcing the punctuated pattern of LC3B-FP in AmiR-17 transfected cells. Statistics on vesicles area and numbers in T98G transfected cells, exposed to the above mentioned rapamycin doses and AmiR-17 or PmiR-17 transfection, were calculated using the AUTOCOUNTER ImageJ Javascript tool:Citation35 according to these analyses, AmiR-17 administration induced a significant increase in the autophagic-like vesicles areas, particularly within rapamycin-treated cells (P < 0.005), rather than a direct increase in their total number within the cells ().
Figure 2. miR-17 modulation of ATG7 and LC3B expression in T98G cells. (A) Relative expression of miR-17 (Real-time PCR) and ATG7 protein (immunoblotting), in T98G cells, transfected with miR-17 specific inhibitor (2.5 μM) and precursor molecules (2.5 μM) evaluated at 72h p.t. Data are normalized using β-Actin mRNA/protein levels. (B) LC3B protein expression after miR-17 inhibitory or precursor treatments (as above) in absence/presence of BafilomycinA1 (10 nM). LC3B-II expression levels were calculated normalizing LC3B-II to β-actin expression, according to the current autophagy guidelines.Citation33 Protein expression and densitometric analysis using ImageJ tool are evaluated at 72h p.t. Results of three independent experiments are indicated with error bars. *P < 0.005, **P < 0.001.
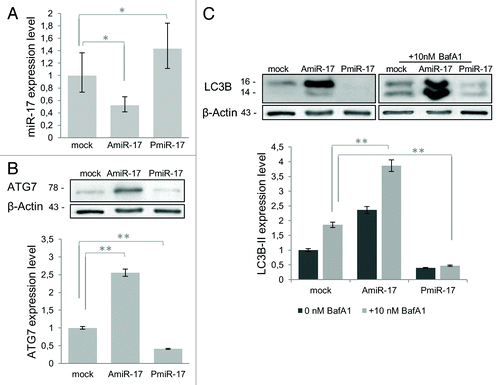
Figure 3. Combined effects of rapamycin and miR-17 inhibitor/precursor administration in T98G cells on LC3B and ATG7 protein expression. To evaluate the autophagy process activation by miR-17 modulation in T98G cells, rapamycin is administered at different concentrations (50−100 nM), after separate transfection of AmiR-17 or Pmir-17 molecules, at 2.5 µM concentration. LC3B-II and ATG7 protein expression, followed by densitometric analysis using ImageJ software, are evaluated after 72 h p.t. LC3B-II expression levels are calculated normalizing LC3B-II to β-Actin expression, according to the current autophagy guidelines.Citation33 Data are normalized using β-Actin protein levels and referred to mock untreated samples.
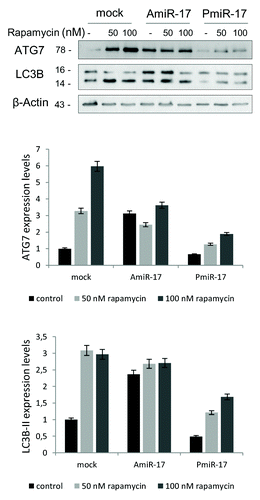
Figure 4. Autophagosomal LC3-FP expression in T98G cells after rapamycin and miR-17 inhibitor/precursor administration. T98G cells are transduced with BacMam LC3B-FP or BacMam LC3B(G120A)-FP viral particles (MOI = 30 each), and after 24 h treated with rapamycin (0−50−100 nM) or transfected with AmiR-17 or PmiR-17 (2.5 µM each). LC3B-FP or LC3B(G120A)-FP expression are analyzed after additional 24h. For DIC phase contrast-fluorescent photographs an inverted microscope (Eclipse TS100, Nikon) with 40× magnification is employed. (Scale bar: 10 μM).
Figure 5. Autophagosomal LC3-FP evaluation in T98G cells after Rapamycyn and miR-17 inhibitor/precursor administration. Ratio percentage between vesicles area and cells areas (Aves/Atot [%]) and number of total vesicles (Vestot) are calculated using the AUTOCOUNTER ImageJ Javascript tool as described.Citation35 Results are obtained analyzing different DIC phase contrast-fluorescent photographs for a total of 30 T98G cells for each of the mentioned mock, rapamycin, AmiR-17, and PmiR-17 treatments. *P < 0.005.
![Figure 5. Autophagosomal LC3-FP evaluation in T98G cells after Rapamycyn and miR-17 inhibitor/precursor administration. Ratio percentage between vesicles area and cells areas (Aves/Atot [%]) and number of total vesicles (Vestot) are calculated using the AUTOCOUNTER ImageJ Javascript tool as described.Citation35 Results are obtained analyzing different DIC phase contrast-fluorescent photographs for a total of 30 T98G cells for each of the mentioned mock, rapamycin, AmiR-17, and PmiR-17 treatments. *P < 0.005.](/cms/asset/02ec0a0f-bb2f-4887-a3d2-005ee09932ef/kcbt_a_10924597_f0005.gif)
miR-17 downregulation induces TMZ toxicity in T98G cells
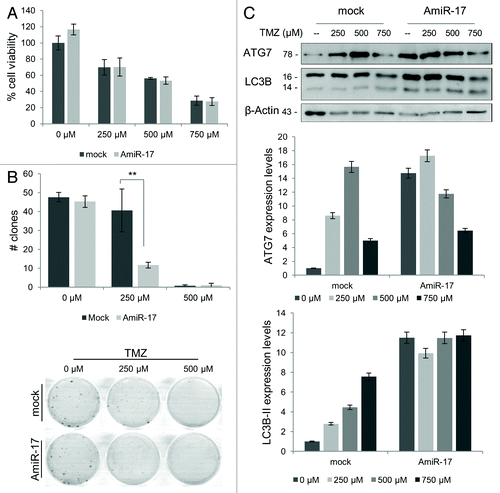
The chemotherapeutic approach for malignant gliomas, that is TMZ, at clinically achievable doses, is proved to exert its anticancer activity inducing autophagy.Citation36 However, T98G cells, differently to most of human glioblastoma cell lines, have been found resistant to the TMZ-induced toxicity.Citation36 Here, to increase the efficacy of TMZ in T98G cells, miR-17 was downregulated, using AmiR-17 (2.5 µM) interference, 24 h before different TMZ treatments (250–500–750 µM). Next, the ability of the combinatorial treatment to enhance TMZ-mediated cell death was examined. Short and long-term viability, assessed respectively by MTT and clonogenic assays, were evaluated. While short-term viability did not evidence clear TMZ and AmiR-17 pro-death combinatory effects (), long-term viability assays showed a marked decrease at the lowest TMZ concentration combined with AmiR-17 administration (P < 0.001). Differently, using a high TMZ concentration (500 µM), the re-plating efficacy was already almost null (). In Table S1, the long-term survival statistics were reported, along with TMZ and AmiR-17 administration doses. To verify the involvement of autophagy on the detected long-term viability decrease, the expression of LC3B-II autophagic marker as well as ATG7 was evaluated, in combination with TMZ administration. As shown in , ATG7 expression generally increased after TMZ administration, with, however, a maximum relative expression at 500 µM; when combined with AmiR-17 transfection, ATG7 increased independently of TMZ as well as with TMZ at 250 µM dosage. Differently, in mock not-transfected cells, LC3B-II expression increase was proportional to TMZ treatment, following a clear linear trend, while the additional AmiR-17 treatment independently re-enforced the LC3B-II increased expression in a dose-independent manner.
Figure 6. Combined effects of TMZ and miR-17 inhibitor (AmiR-17) treatment on short/long-term viability and ATG7/LC3B protein expression in T98G cells. T98G cells are transfected with AmiR-17 (2.5 μM) and, after 24 h, treated with TMZ (0−250−500 μM). (A) Short-term viability in T98G cells is evaluated using MTT assay after 48 h p.t. (B) For T98G long-term viability a clonogenic assay is performed after a two-weeks culturing condition. Statistics (number of clones and mean of each treatment) are reported in Table S1. (C) ATG7 and LC3B expression are evaluated by immunoblotting. For densitometric analysis, LC3B-II expression levels are calculated normalizing LC3B-II to β-actin expression, according to the current autophagy guidelines.Citation33 Results are normalized to mock untreated samples. **P < 0.001.
Anti-miR-17 treatment induces low-dose radio-sensitivity in U373-MG cells
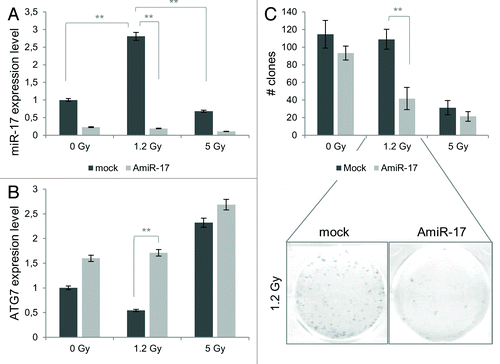
As recently reported, the autophagic process is involved and able to enhance radiation effects in human glioblastoma cell lines;Citation9 in particular, it has been demonstrated that U373-MG, a well-established human glioblastoma cell line, exhibited a low-dose resistant phenotype, while this behavior was not observed in T98G cells.Citation34 First, in order to shed light into a putative involvement of miR-17 in this differential radio-sensitivity, miR-17 levels were evaluated at 48 h p.t. after a designed IR scheme, comprising low (1.2 Gy) and high X-rays doses (5 Gy). As presented in , miR-17 showed a significant expression increase at the lowest radiation doses (1.2 Gy; P < 0.001). Concordantly, ATG7 expression decreased in correspondence of the lowest IR dose employed. According to these results, to promote a radiosensitive phenotype in U373-MG cells at low IR doses (1.2 Gy), AmiR-17 molecules (2.5 µM) were transfected before IR treatment to induce the activation of the autophagic process (). At the end of the combined experimental scheme, as previously described, miR-17 levels significantly decreased after AmiR-17 administration, independently of the IR treatment employed, compared with mock IR treated samples. Accordingly, a significant increase of ATG7 expression was scored at the different IR doses compared with their respective not-transfected mock samples: in particular, the 1.2 Gy/AmiR-17 combined treatment induced a significant upregulation of ATG7 expression (P < 0.001). Importantly, AmiR-17 transfection induced a significant surviving fraction (SF) decrease in U373-MG cells when irradiated with 1.2 Gy (P < 0.001), showing a massive reduction of re-plating efficacy (; Table S2).
Figure 7. Combined effects of IR and miR-17 inhibitor (AmiR-17) treatment on miR-17/ATG7 expression and on long-term viability in U373-MG cells. (A) U373-MG cells are transfected with AmiR-17 (2.5 μM) 48 h before IR administration (0−1.2–5 Gy). miR-17 and ATG7 protein expression are evaluated by means of real-time quantitative PCR and immunoblotting/densitometric analysis, respectively. Results are normalized to mock untreated samples. (B) For U373-MG long-term viability a clonogenic assay is performed after a 2-weeks culturing condition. Statistics (clones number and mean of each treatment) are reported in Table S2. *P < 0.005, **P < 0.001.
Discussion
In this study, it is originally reported that miR-17 can regulate ATG7 expression in glioblastoma-derived cell lines, activating the endogenous autophagic process. To date, an increasing number of miRNAs are being discovered as key regulators of many different cellular processes, in particular those related to autophagy death in cells of different tumor, like melanoma,Citation37 hepatocellular carcinoma,Citation38 myelogenous leukemia,Citation39 colon,Citation40 cervical,Citation41 and breast cancer;Citation42 however, to date, only few reports identified miRNA sequences (i.e., miR-30a, miR-21, and miR-10b) directly involved in the regulation of the autophagic process in glioma cells.Citation43-Citation45 Among oncogenic miRNAs, one of the best-characterized is miR-17-92, a polycistronic miRNA cluster, designated as OncomiR-1.Citation21 The precursor transcript contains six tandem stem-loop hairpin structures that ultimately yield six mature miRNAs: miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1 and miR-92–1.Citation46 In particular, human OncomiR-1 is located at 13q31.3, a region amplified in several hematopoietic malignancies and solid tumors, including diffuse B-cell lymphomas, follicular lymphomas, Burkitt lymphomas, and lung carcinoma.Citation47
Recent findings indicate that these miRNAs are integrated components of the molecular pathways that regulate tumor development and tumor maintenance.Citation48 In detail, miR-17 has been associated with the regulation of many genes involved in G1/S-phase cell cycle boundary.Citation22 Transcriptome analysis in human GBMs showed that miR-17, together with miR-20, is upregulated in high grade tumors, with a higher miR-17 expression correlating with increased patients’ survival.Citation31 In addition, a lower expression of E2F1, a miR-20 target, and cyclin D1, a target of both miR-20 and miR-17, was found associated with longer survival outcomes in other cancer patients.Citation25,Citation26
Within several human autophagic genes, our bioinformatics predictions identified ATG7 as a bona fide miR-17 target, having multiple potential miR-17 complementary sites in the 3′-UTR of its transcript. Importantly, ATG7 is one of the master regulators of the autophagy process, responsible for two major reactions involved in autophagosome formation and in vesicle progression.Citation4 Moreover, it was already established that Atg7−/− knockout mice die within one day from birth and show reduced pup size, due to an impaired autophagy pathway.Citation46 Through the use of mimic and antagomir molecules, we demonstrated that miR-17 was able to modulate autophagy varying endogenous ATG7 expression levels, and this effect was mediated via a miR-17 consensus sequence contained in the 3′-UTR of the gene. The role of miR-17 in the regulation of ATG7 expression was evidenced by our experiments showing that transfection of T98G glioblastoma cells with the miR-17 mimic resulted in a decrease of ATG7 protein expression and, in parallel, in a reduction of the activity of a reporter gene plasmid containing the consensus sequences for miR-17. By contrast, in the absence of the miR-17 consensus sequence, the inhibitory effect of miR-17 on the reporter activity was abolished. Furthermore, we showed that transfection of T98G cells with miR-17 antagomir molecules caused an increase in ATG7 profile, providing additional evidence for a possible role of miR-17 in controlling ATG7 expression. Consequently, we observed that miR-17-mediated decrease of ATG7 had an inhibitory effect on autophagy, showing lower level of LC3B-II expression and, conversely, miR-17 inhibition lead to the activation of the autophagy process, inducing higher level of the LC3B-II expression. This effect was further confirmed with the use of Bafilomycin A1 (BafA1), as suggested for proper autophagy assays interpretations.Citation33 In fact, BafA1, a proton pump inhibitor, raises the lysosomal pH, preventing fusion with autophagosomal compartment, inducing an LC3B-II increase in presence of an autophagic flux. Moreover, in line with these observations, miR-17 mediated autophagy regulation was comparable with the effects of rapamycin. This compound is a PI3K/Akt/mTOR pathway inhibitor, therefore a well-established autophagy inducer. We reported that the rapamycin-induced upregulation of the autophagic markers (LC3B-II and ATG7) was similar to the induction after anti-miR-17 administration; the molecular interfering effect of miR-17 might be directed to antagonize ATG7 E1-like activity, responsible for ATG12-ATG5 conjugation and LC3B activation, both processes necessary for autophagic vesicles elongation.Citation4 On the contrary, an additive effect of rapamycin and anti-miR-17 treatment on the overall autophagy status was not highlighted, probably due to other processes activated by the cells to counteract an excessive activation of the autophagy process itself. The miR-17-mediated activation of autophagy, through the formation of autophagosomes, was also scored in T98G living cells, transduced with the Premo Autophagy Sensor (LC3B-FP) BacMam 2.0 system and subsequently analyzed with the AUTOCOUNTER ImageJ Javascript tool.Citation35 As a result, T98G cells treated with miR-17 inhibitor scored an increase in the surface amount of punctate fluorescent vesicles, suggesting a LC3B-FP protein accumulation in nascent autophagosomes.Citation49
Next, autophagy induction due to miR-17 downregulation was assayed as an adjuvant strategy to current in vitro chemo- and radio-therapeutic schemes. In standard clinical protocols, brain tumor resection has to be followed by radiation therapy and chemotherapy with alkylating drugs, mainly TMZ and by a combination of them. Unfortunately the patients’ prognosis is still poor, since most of the patients had recidivist tumor forms and their tumor cells showed an intrinsic resistance to radio- or chemo-therapy death induction.Citation50 To this regard, very recently, Gwak et al.Citation46 demonstrated that miR-21, which plays an anti-apoptotic role in cancer, was a target molecule for radio-sensitization of glioma cells. The authors reported a radiation-induced modulation of miR-21 endogenous levels, and that its expression correlated with a radio-resistance phenotype in different glioma cell lines. In order to perform in vitro studies to modulate chemotherapy and IR resistance, two different human glioblastoma cell lines were assayed: T98G as TMZ resistantCitation37 and U373-MG as radio-resistant cells.Citation31 Importantly, many evidences reported that both treatments lead to autophagy activationCitation34,Citation36,Citation51,Citation52 and that, a further activation of the autophagy process, by means of drugs like rapamycin or cannabinoids, lead to an enhanced sensitization of glioblastoma cells to chemotherapy and IR treatments.Citation53 We observed that the in vitro combination of miR-17 inhibition and TMZ treatment was highly effective in a relatively long period of cell culture time, further decreasing the threshold resistance at low TMZ doses in T98G cells. Similarly to the above mentioned results comparing anti-miR-17 and rapamycin administration, TMZ and miR-17 inhibition did not display a cumulative effect on the expression of the investigated autophagy markers LC3B and ATG7. Importantly, it was proved that the autophagic process activated by miR-17 inhibition in U373-MG cells sensitized them to a low IR dose treatment scheme, sensibly decreasing their long-term viability. Within these radiation treatment schemes, differences in miR-17 expression were identified. Specifically, a significant increase of miR-17 expression was observed at 1.2 Gy (low dose), but not at 5 Gy (high dose), thus, suggesting that miR-17 expression might be a factor involved in the low dose radio-resistance phenotype in U373-MG cells. Altogether these results showed that the activation of the autophagy process by means of a downregulation of endogenous miR-17 levels might be therapeutically beneficial, enhancing the in vitro effect of TMZ and radiation treatments. In conclusion, miR-17 was confirmed as an effective key regulator in important cellular pathways, such as the autophagy process. In particular, it was here originally reported that miR-17 can regulate ATG7 expression in glioblastoma-derived cell lines, modulating the endogenous autophagic process. Subsequently, the molecular interference on miR-17 expression was also successfully applied in two experimental pre-clinical therapeutic strategies using TMZ and ionizing radiation. The identification of miR-17 as one of the actors in regulating autophagy and radio-resistance might provide novel therapeutic hints for future treatment of malignant glioblastoma. However, since miRNAs might control the expression of several targets, further studies will be needed to better clarify the molecular circuits interested by miR-17 modulation.
Materials and Methods
Computational identification of miRNA targeting autophagy genes
The miRWalk algorithm (www.umm.uni-heidelberg.de/apps/zmf/mirwalk/index.html) was used to predict miRNA sequences that might regulate the expression of human ATGs. miRWalk integrates different parameters of common miRNA-specific target prediction programs, like DIANAmT, miRanda, miRDB, PICTAR5. A minimum seed length of 7 nucleotides and a p Value of 0.05 between microRNA and target gene was considered as a significant prediction threshold. Sequence alignments between microRNA and ATGs were performed using miRanda (www.microRNA.org) algorithm and their significance were measured by mirSVR scores according to Betel et al.Citation32 TargetScan microRNA tool (www.targetscan.org) was used to further classify the probability of putative miR-17/ATG7 binding sites, using the PCT ranking score as recommended.Citation10
Cell cultures and chemicals
T98G and U373-MG established human malignant glioma cell lines (ECACC) were cultivated in D-MEM medium supplemented with 10% FBS, 100 units/ml penicillin, 0.1 mg/ml streptomycin and 1% l-glutamine (Invitrogen, Life Technologies), at 37 °C and 5% CO2 atmosphere. Cells were cultured two days before treatments. Bafilomycin A1 was used as a lysosomal inhibitor, added 6h before cell harvesting at 10 nM concentration TMZ and rapamycin were employed as chemotherapeutic agent and autophagy inducer, respectively. All these reagents, provided by Sigma, were dissolved in dimethyl sulfoxide (DMSO, Sigma) and diluted in culture medium to the appropriate concentration.
Plasmid constructs and luciferase assay in T98G cells
A portion of 969 bp fragment of human ATG7 3′-UTR sequence (GenBank Acc. NM_006395) was amplified from genomic DNA using primers ATG7FOR (CCTAGACTAG TCATGGGGAC ACAGCCGGCA C) and ATG7REV (CCTAGGAGCT CTGGCACTGG GTGAAAGGTC GG) (30 cycles at 94 °C × 15 sec; 62 °C × 20 sec; 72 °C × 60 sec) and then subcloned into the SpeI and SacI sites into pMIR-REPORT vector (Ambion, Life Technologies), thus originating pMIRATG7wt plasmid. A three-point mutation was then introduced into the seed region of miR-17/ATG7 putative interacting sequence (see ) using primers MIRMUTFOR (5′-CCCACCCAGC CTGGCGCCAG AACTGCACAC GTACACTATG TGG) and MIRMUTREV (5′-CCACATAGTG TACGTGTGCA GTTCTGGCGC CAGGCTGGGT GGG), according to a DpnI mediated site-directed mutagenesis protocol (Quick-Change Site Directed Mutagenesis, Stratagene), producing the pMIRATG7MUT plasmid. Constructs were all sequence verified. T98G cells were transfected with 1 μg of each plasmid, using a Lipofectamine 2000 protocol (Invitrogen, Life Technologies). Luciferase activity assay was performed in 96-well plates in presence of 1000 cells and 100 μl of MISSION LightSwitch Luciferase Assay Reagent (Sigma). After a room-temperature incubation of 30 min, 6-fold replicated samples were read for 4 sec using a luminometer (Perkin Elmer Microbetatrilux 1450 LSC) at 24 and 48 h p.t. Results expressed as Relative Luciferase Units (RLU) were normalized to pMIR-REPORT vector (vehicle) transfections.
ATG7 and miR-17 immunoprecipitation (RIP-Assay) in T98G cells
The RIP-Assay kit for microRNA (MBL) was employed following the manufacturer’s specifications. Briefly, fresh cellular extracts from T98G cells (7.5 × 106) were co-immunoprecipitated overnight at 4 °C with 20 μg of RIP-certified anti-EIF2C2/AGO2 mouse monoclonal antibody (MBL), one of the RISC protein components, previously conjugated with Sepharose Protein A or G beads (Amersham Biosciences, GE Healthcare). Rabbit IgG and input-alone (mock) were used as negative controls. ATG7 and miR-17 expression levels were evaluated after total RNA isolation from antibody-immobilized Protein A or G agarose beads-RNP complexes by Real-time PCR using custom-validated Taqman assays (Applied Biosystems, Life Technologies). Results, represented by 3 independent experiments, were normalized to mock samples.
miRNA-17 modulation and rapamycin-mediated autophagy induction
To modulate miR-17 expression, T98G glioblastoma cells were transfected using NEON Transfection System (Invitrogen, Life Technologies), according with the manufacturer’s instructions. 3 × 105 cells were transfected with specific miR-17 inhibitor (AmiR-17, 2.5 µM) or miR-17 precursor (PmiR-17, 2.5 µM) molecules, both provided by Applied Biosystem (Life Technologies) or with mock solution alone. After transfections, cells were seeded into 6-well plates and incubated for RNA and protein analysis.
For rapamycin-mediated autophagy induction in T98G cells, the compound was employed as previously described.Citation54 Specifically, rapamycin was added at different concentrations (50–100 nM), 48 h after transfection of AmiR-17 or PmiR-17 molecules. Transfected cells were incubated for 24 h in 6-well (for protein and RNA analysis) or in 96-well flat-bottom plates (for LC3B-FP expression analysis).
LC3B-FP expression analysis and autophagosome quantitative analysis
For autophagy detection, T98G cells, seeded at 1 × 103 into 96-well plates, were transduced with BacMam LC3B-FP or BacMam LC3B(G120A)-FP viral particles (MOI = 30), according with the Premo Autophagy Sensor Kit (Invitrogen, Life Technologies). After 24 h, AmiR-17 or PmiR-17 were transfected (both at 2.5 µM) and combined with rapamycin (50–100 nM) administration. After an additional 24 h, LC3B-FP and LC3B(G120A)-FP signals were monitored using an inverted fluorescence microscope (Eclipse Nikon TS100). DIC phase contrast-fluorescent photographs (40× magnification) were collected and analyzed.
To quantify the LC3B-FP puncta per cell in an unsupervised manner, the AUTOCOUNTER specific software was used.Citation35 Briefly, the program analyzed DIC phase contrast-fluorescent photographs with operator-defined cells contours; then, the number of intracellular fluorescent spots (Vestot) and their areas percentage within the cell (Aves/Acell) were calculated by an operator-independent algorithm. Results were obtained analyzing a total of 30 T98G cells for each of the mentioned mock, rapamycin, AmiR-17/PmiR-17 treatments.
TMZ and IR treatments
TMZ was added at different concentrations to T98G cells, 24h after transfection with AmiR-17 or PmiR-17 molecules. For protein and RNA analysis, TMZ was used at 250–500–750 µM concentrations, as well as for short-term viability; for long-term viability, only 250–500 µM concentrations were employed. For IR treatment, 3 × 105 U373-MG glioblastoma cells seeded into 25 cm2 culture flasks were irradiated 48 h after AmiR-17 (2.5 µM) transfection and then placed in a water phantom for dose homogeneity. Cells were irradiated with a 6-MV X-ray linear accelerator (Varian Clinac 600) at a dose-rate of 244.5 cGy/min, with 1.2 and 5 Gy.
Viability and clonogenic survival assays
To evaluate the short-term viability, the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega) was employed. After transfection, T98G cells were plated into 96-well plates at a density of 1.5 × 103 cells per well. After 48 h of incubation with TMZ (250–500–750 µM), cells were tested for viability. CellTiter 96 Aqueous One Solution Reagent was added to the culture volume and incubated at 37 °C for 3 h; then, absorbance was measured with a microplate reader (Titertek Multiskan) at 490 nm wavelength and results were expressed as percentage of cell viability. Three replicas were performed for each sample.
To evaluate plating efficiency, a clonogenic survival assay was performed as described by Palumbo et al.Citation34 Briefly, 48h after TMZ (0–250–500 µM) or IR treatments (0–1.2–5 Gy), cells were trypsinized and seeded into 6-well plates (2 × 102 cells per well), incubated for two weeks at 37 °C and then fixed with ethanol. Cells were stained with 0.5% Crystal Violet (Sigma), and colonies that contained more than 50 cells were automatically counted using a clono-counter software;Citation34 the number of clones/well was calculated and normalized to the corresponding control samples. Each experiment was performed in triplicate.
Immunoblotting analysis
To evaluate the expression of autophagy markers ATG7 and LC3B, cells were collected and lysated in ice-cold RIPA buffer (150 mM NaCl, 50 mM TRIS-HCl pH 8.0, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% sodium dodecylsulphate), supplemented with Complete Mini protease inhibitor cocktail 7X (Roche). Then, cell extracts were added with Laemmli sample buffer (2% SDS, 6% glycerol, 150 mM β- mercaptoethanol, 0.02% bromophenol blue and 62.5 mM TRIS-HCl pH 6.8), denatured for 5′ at 95 °C and loaded on 12% SDS-PAGE gels. After electrophoresis, proteins were transferred onto nitro-cellulose membrane Hybond-C Extra (GE Healthcare), using the semi-dry blotter TE70 PWR apparatus (GE Healthcare). Membranes were blocked 1 h with 8% non-fat milk in TBS (138 mM NaCl, 20 mM TrisOH pH 7.6) containing 0.1% Tween 20 and incubated over-night at 4 °C with primary antibodies. Anti-β-Actin (for internal and loading control), anti-ATG7 and anti-LC3B primary antibodies were employed (Cell Signaling Technology, 1/2000 dilution). Species-specific peroxidase-labeled ECL secondary antibodies (Cell Signaling Technology, 1/4000 dilution) were used. Protein signals were revealed using the ECL Advance Western Blotting Detection Kit (GE Healthcare). Protein expression was quantified by densitometric analysis with ImageJ software (http://rsbweb.nih.gov/ij/) according to the guidelines.
Real-time PCR expression analysis
Total RNA from T98G and U373-MG cells was extracted 48h after transfection using the Trizol reagent (Invitrogen, Life Technologies), according to the manufacturer’s instructions. After RNA quantitation (Quant-it RNA Assay, Invitrogen, Life Technologies), cDNA synthesis was performed using the High Capacity cDNA Archive kit (Applied Biosystems, Life Technologies). The quantitative relative expression of ATG7 was performed using a custom-specific Taqman Assay (Applied Biosystems, Life Technologies). Differently, miR-17 was retro-transcribed using a sequence-specific hairpin-primer and amplified with a TaqMan MicroRNA Assays (Applied Biosystems, Life Technologies). Real-time PCRs were performed using 250 ng of each cDNAs and using a DNA Engine Opticon 2 (MJ Research) platform. Samples were subjected to 40 amplification cycles of 95 °C for 15 min and 60 °C for 1 min.
Statistical analysis
Statistical analyses were performed using ANOVA-one way tests. Results represent the mean of at least 3 independent experiments. All P values lower than 0.005 were considered statistically significant.
| Abbreviations: | ||
| ATGs | = | autophagy-related genes |
| 3′-UTR | = | 3′ untranslated region |
| GBM | = | human glioblastoma |
| TMZ | = | temozolomide |
| IR | = | ionizing radiation |
| MOI | = | multiplicity of infection |
Additional material
Download Zip (194.2 KB)Acknowledgments
The present study was funded by PRIN-MIUR 2008.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 1998; 273:3963 - 6; http://dx.doi.org/10.1074/jbc.273.7.3963; PMID: 9461583
- Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem 2011; 80:125 - 56; http://dx.doi.org/10.1146/annurev-biochem-052709-094552; PMID: 21548784
- Pópulo H, Lopes JM, Soares P. The mTOR signalling pathway in human Cancer. Int J Mol Sci 2012; 13:1886 - 918; http://dx.doi.org/10.3390/ijms13021886; PMID: 22408430
- Mijaljica D, Prescott M, Devenish RJ. The intriguing life of autophagosomes. Int J Mol Sci 2012; 13:3618 - 35; http://dx.doi.org/10.3390/ijms13033618; PMID: 22489171
- Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 2010; 6:322 - 9; http://dx.doi.org/10.4161/auto.6.3.11625; PMID: 20224296
- Rosenfeldt MT, Ryan KM. The role of autophagy in tumour development and cancer therapy. Expert Rev Mol Med 2009; 11:e36; http://dx.doi.org/10.1017/S1462399409001306; PMID: 19951459
- Palumbo S, Comincini S. Autophagy and ionizing radiation in tumors: the “survive or not survive” dilemma. J Cell Physiol 2013; 228:1 - 8; http://dx.doi.org/10.1002/jcp.24118; PMID: 22585676
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009; 19:92 - 105; http://dx.doi.org/10.1101/gr.082701.108; PMID: 18955434
- Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, et al. A uniform system for microRNA annotation. RNA 2003; 9:277 - 9; http://dx.doi.org/10.1261/rna.2183803; PMID: 12592000
- Rossi JJ. Transcriptional activation by small RNA duplexes. Nat Chem Biol 2007; 3:136 - 7; http://dx.doi.org/10.1038/nchembio0307-136; PMID: 17301798
- Lee I, Ajay SS, Yook JI, Kim HS, Hong SH, Kim NH, et al. New class of microRNA targets containing simultaneous 5′-UTR and 3′-UTR interaction sites. Genome Res 2009; 19:1175 - 83; http://dx.doi.org/10.1101/gr.089367.108; PMID: 19336450
- Ajay SS, Athey BD, Lee I. Unified translation repression mechanism for microRNAs and upstream AUGs. BMC Genomics 2010; 11:155; http://dx.doi.org/10.1186/1471-2164-11-155; PMID: 20205738
- Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 2010; 79:351 - 79; http://dx.doi.org/10.1146/annurev-biochem-060308-103103; PMID: 20533884
- Fu LL, Wen X, Bao JK, Liu B. MicroRNA-modulated autophagic signaling networks in cancer. Int J Biochem Cell Biol 2012; 44:733 - 6; http://dx.doi.org/10.1016/j.biocel.2012.02.004; PMID: 22342941
- Xu J, Wang Y, Tan X, Jing H. MicroRNAs in autophagy and their emerging roles in crosstalk with apoptosis. Autophagy 2012; 8:873 - 82; http://dx.doi.org/10.4161/auto.19629; PMID: 22082964
- Aguda BD, Kim Y, Piper-Hunter MG, Friedman A, Marsh CB. MicroRNA regulation of a cancer network: consequences of the feedback loops involving miR-17-92, E2F, and Myc. Proc Natl Acad Sci U S A 2008; 105:19678 - 83; http://dx.doi.org/10.1073/pnas.0811166106; PMID: 19066217
- Grillari J, Hackl M, Grillari-Voglauer R. miR-17-92 cluster: ups and downs in cancer and aging. Biogerontology 2010; 11:501 - 6; http://dx.doi.org/10.1007/s10522-010-9272-9; PMID: 20437201
- Li Y, Li Y, Zhang H, Chen Y. MicroRNA-mediated positive feedback loop and optimized bistable switch in a cancer network Involving miR-17-92. PLoS One 2011; 6:e26302; http://dx.doi.org/10.1371/journal.pone.0026302; PMID: 22022595
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microRNA polycistron as a potential human oncogene. Nature 2005; 435:828 - 33; http://dx.doi.org/10.1038/nature03552; PMID: 15944707
- Cloonan N, Brown MK, Steptoe AL, Wani S, Chan WL, Forrest AR, et al. The miR-17-5p microRNA is a key regulator of the G1/S phase cell cycle transition. Genome Biol 2008; 9:R127; http://dx.doi.org/10.1186/gb-2008-9-8-r127; PMID: 18700987
- Fontana L, Fiori ME, Albini S, Cifaldi L, Giovinazzi S, Forloni M, et al. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS One 2008; 3:e2236; http://dx.doi.org/10.1371/journal.pone.0002236; PMID: 18493594
- Luo H, Zou J, Dong Z, Zeng Q, Wu D, Liu L. Up-regulated miR-17 promotes cell proliferation, tumour growth and cell cycle progression by targeting the RND3 tumour suppressor gene in colorectal carcinoma. Biochem J 2012; 442:311 - 21; http://dx.doi.org/10.1042/BJ20111517; PMID: 22132820
- Hossain A, Kuo MT, Saunders GF. Mir-17-5p regulates breast cancer cell proliferation by inhibiting translation of AIB1 mRNA. Mol Cell Biol 2006; 26:8191 - 201; http://dx.doi.org/10.1128/MCB.00242-06; PMID: 16940181
- Yu Z, Wang C, Wang M, Li Z, Casimiro MC, Liu M, et al. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J Cell Biol 2008; 182:509 - 17; http://dx.doi.org/10.1083/jcb.200801079; PMID: 18695042
- Lefranc F, Facchini V, Kiss R. Proautophagic drugs: a novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist 2007; 12:1395 - 403; http://dx.doi.org/10.1634/theoncologist.12-12-1395; PMID: 18165616
- Kögel D, Fulda S, Mittelbronn M. Therapeutic exploitation of apoptosis and autophagy for glioblastoma. Anticancer Agents Med Chem 2010; 10:438 - 49; http://dx.doi.org/10.2174/1871520611009060438; PMID: 20879985
- Lefranc F, Rynkowski M, DeWitte O, Kiss R. Present and potential future adjuvant issues in high-grade astrocytic glioma treatment. Adv Tech Stand Neurosurg 2009; 34:3 - 35; http://dx.doi.org/10.1007/978-3-211-78741-0_1; PMID: 19368079
- Dweep H, Sticht C, Pandey P, Gretz N. miRWalk--database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J Biomed Inform 2011; 44:839 - 47; http://dx.doi.org/10.1016/j.jbi.2011.05.002; PMID: 21605702
- Srinivasan S, Patric IR, Somasundaram K. A ten-microRNA expression signature predicts survival in glioblastoma. PLoS One 2011; 6:e17438; http://dx.doi.org/10.1371/journal.pone.0017438; PMID: 21483847
- Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol 2010; 182:509 - 17; PMID: 20799968
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Palumbo S, Pirtoli L, Tini P, Cevenini G, Calderaro F, Toscano M, et al. Different involvement of autophagy in human malignant glioma cell lines undergoing irradiation and temozolomide combined treatments. J Cell Biochem 2012; 113:2308 - 18; http://dx.doi.org/10.1002/jcb.24102; PMID: 22345070
- Fassina L, Magenes G, Inzaghi A, Palumbo S, Allavena G, Miracco C, et al. AUTOCOUNTER, an ImageJ JavaScript to analyze LC3B-GFP expression dynamics in autophagy-induced astrocytoma cells. Eur J Histochem 2012; 56:e44; http://dx.doi.org/10.4081/ejh.2012.e44; PMID: 23361240
- Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 2004; 11:448 - 57; http://dx.doi.org/10.1038/sj.cdd.4401359; PMID: 14713959
- Chen Y, Liersch R, Detmar M. The miR-290-295 cluster suppresses autophagic cell death of melanoma cells. Sci Rep 2012; 2:808; http://dx.doi.org/10.1038/srep00808; PMID: 23150779
- Chang Y, Yan W, He X, Zhang L, Li C, Huang H, et al. miR-375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology 2012; 143:177 - 87, e8; http://dx.doi.org/10.1053/j.gastro.2012.04.009; PMID: 22504094
- Kovaleva V, Mora R, Park YJ, Plass C, Chiramel AI, Bartenschlager R, et al. miRNA-130a targets ATG2B and DICER1 to inhibit autophagy and trigger killing of chronic lymphocytic leukemia cells. Cancer Res 2012; 72:1763 - 72; http://dx.doi.org/10.1158/0008-5472.CAN-11-3671; PMID: 22350415
- Zhai H, Song B, Xu X, Zhu W, Ju J. Inhibition of autophagy and tumor growth in colon cancer by miR-502. Oncogene 2013; 32:1570 - 9; http://dx.doi.org/10.1038/onc.2012.167; PMID: 22580605
- Zou Z, Wu L, Ding H, Wang Y, Zhang Y, Chen X, et al. MicroRNA-30a sensitizes tumor cells to cis-platinum via suppressing beclin 1-mediated autophagy. J Biol Chem 2012; 287:4148 - 56; http://dx.doi.org/10.1074/jbc.M111.307405; PMID: 22157765
- Frankel LB, Wen J, Lees M, Høyer-Hansen M, Farkas T, Krogh A, et al. microRNA-101 is a potent inhibitor of autophagy. EMBO J 2011; 30:4628 - 41; http://dx.doi.org/10.1038/emboj.2011.331; PMID: 21915098
- Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X, et al. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy 2009; 5:816 - 23; PMID: 19535919
- Gabriely G, Yi M, Narayan RS, Niers JM, Wurdinger T, Imitola J, et al. Human glioma growth is controlled by microRNA-10b. Cancer Res 2011; 71:3563 - 72; http://dx.doi.org/10.1158/0008-5472.CAN-10-3568; PMID: 21471404
- Gwak HS, Kim TH, Jo GH, Kim YJ, Kwak HJ, Kim JH, et al. Silencing of microRNA-21 confers radio-sensitivity through inhibition of the PI3K/AKT pathway and enhancing autophagy in malignant glioma cell lines. PLoS One 2012; 7:e47449; http://dx.doi.org/10.1371/journal.pone.0047449; PMID: 23077620
- Tanzer A, Stadler PF. Molecular evolution of a microRNA cluster. J Mol Biol 2004; 339:327 - 35; http://dx.doi.org/10.1016/j.jmb.2004.03.065; PMID: 15136036
- Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, et al. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res 2004; 64:3087 - 95; http://dx.doi.org/10.1158/0008-5472.CAN-03-3773; PMID: 15126345
- Olive V, Jiang I, He L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. Int J Biochem Cell Biol 2010; 42:1348 - 54; http://dx.doi.org/10.1016/j.biocel.2010.03.004; PMID: 20227518
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425 - 34; http://dx.doi.org/10.1083/jcb.200412022; PMID: 15866887
- Perry J, Okamoto M, Guiou M, Shirai K, Errett A, Chakravarti A. Novel therapies in glioblastoma. Neurol Res Int 2012; 2012:428565; http://dx.doi.org/10.1155/2012/428565; PMID: 22530121
- Torres S, Lorente M, Rodríguez-Fornés F, Hernández-Tiedra S, Salazar M, García-Taboada E, et al. A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol Cancer Ther 2011; 10:90 - 103; http://dx.doi.org/10.1158/1535-7163.MCT-10-0688; PMID: 21220494
- Zhuang D, Liu Y, Mao Y, Gao L, Zhang H, Luan S, et al. TMZ-induced PrPc/par-4 interaction promotes the survival of human glioma cells. Int J Cancer 2012; 130:309 - 18; http://dx.doi.org/10.1002/ijc.25985; PMID: 21328340
- Zhuang W, Qin Z, Liang Z. The role of autophagy in sensitizing malignant glioma cells to radiation therapy. Acta Biochim Biophys Sin (Shanghai) 2009; 41:341 - 51; http://dx.doi.org/10.1093/abbs/gmp028; PMID: 19430698
- Barbieri G, Palumbo S, Gabrusiewicz K, Azzalin A, Marchesi N, Spedito A, et al. Silencing of cellular prion protein (PrPC) expression by DNA-antisense oligonucleotides induces autophagy-dependent cell death in glioma cells. Autophagy 2011; 7:840 - 53; http://dx.doi.org/10.4161/auto.7.8.15615; PMID: 21478678