Abstract
The activity of the epidermal growth factor receptor (EGFR) antibodies cetuximab and panitumumab in metastatic colorectal carcinoma (mCRC) is significantly limited by molecular mechanisms leading to intrinsic or acquired resistance. The S492R mutation of the EGFR, which is caused by either the 1476C>A or the 1474A>C substitution, interferes with binding to cetuximab but not to panitumumab, and has been detected in mCRC with acquired resistance to cetuximab. Since mechanisms of acquired and intrinsic resistance to EGFR monoclonal antibodies in CRC significantly overlap, we evaluated the frequency of the S492R mutation in a series of KRAS-exon 2 wild-type CRC patients. Genomic DNA was extracted from formalin fixed paraffin embedded (FFPE) tissues that were obtained from 505 systemic therapy-naïve CRC patients. A PCR/sequencing method for the detection of the S492R mutation was developed, by using as positive control a plasmid in which the 1474A>C mutation was generated by site directed mutagenesis. The lowest level of detection of this assay was approximately 10% mutant DNA in a background of wild-type DNA. PCR sequencing analysis revealed no S492R mutations in any of the analyzed 505 CRC specimens. Our findings suggest that the S492R mutation is not involved in primary resistance to cetuximab in CRC. Therefore, patients with mCRC should not be routinely screened for this mutation prior therapy with cetuximab.
Introduction
The epidermal growth factor receptor (EGFR) is expressed in a majority of colorectal carcinomas (CRC), and the EGFR antibodies cetuximab and panitumumab have been approved for treatment of metastatic CRC (mCRC).Citation1,Citation2 However, the activity of EGFR antibodies is significantly limited by molecular mechanisms leading to intrinsic or acquired resistance to these agents.Citation3,Citation4
Intrinsic resistance to EGFR monoclonal antibodies in CRC is due to constitutive activation of signaling pathways leading to EGFR-independent cell growth. Indeed, mutations in the KRAS or NRAS genes have been associated with resistance to both cetuximab and panitumumab in different studies. These drugs have been approved for patients that do not carry exon 2 KRAS mutations, although the use of panitumumab has been recently restricted only to KRAS/NRAS exons 2, 3, and 4 wild-type patients.Citation1,Citation5-Citation7 The role of BRAF mutations is more controversial, since some studies have demonstrated a prognostic rather than a predictive value of these molecular alterations.Citation5,Citation6,Citation8,Citation9 However, there is common agreement on the fact that BRAF mutant patients do not respond to currently available therapies and should undergo more intensive treatments. Molecular alterations in other signaling proteins, such as PI3K and PTEN, have also been hypothesized to play a role in regulating sensitivity to anti-EGFR agents.Citation3,Citation5,Citation10-Citation12
Some recent studies have shed light on the mechanisms of acquired resistance to EGFR monoclonal antibodies in CRC. Amplification of ErbB-2 and/or increased serum levels of the ErbB-2 ligand heregulin, as well as MET amplification, have been reported to be associated with acquired resistance to cetuximab and panitumumab in mCRC.Citation13-Citation15 Interestingly, KRAS mutations have also been detected, at the time of tumor progression, in tumors from KRAS-wild-type patients that initially responded to EGFR monoclonal antibodies.Citation16,Citation17 Conversely, ErbB-2 gene amplification has been detected in approximately 3% KRAS wild-type mCRC prior to exposure to EGFR monoclonal antibodies and is associated with reduced response to these agents.Citation14 These findings imply that mechanisms of acquired and intrinsic resistance might significantly overlap.
All the mechanisms of resistance to EGFR monoclonal antibodies in CRC described up to now affect sensitivity to both cetuximab and panitumumab with one exception. A recent study reported that cell lines with acquired resistance to cetuximab showed a mutation of the extracellular domain of the EGFR, 1476C>A, leading to a substitution of serine to arginine at amino acid 492 (S492R).Citation18 This mutation interferes with binding to cetuximab but not to panitumumab. Indeed, cell lines with the EGFR S492R mutation showed sensitivity to panitumumab but not to cetuximab. Importantly, a S492R mutation was detected in two patients with mCRC and acquired resistance to cetuximab. One patient carried the 1476C>A substitution, the other a 1474A>C mutation causing the same amino acid substitution. Mutations in this codon were not detected in a small cohort (n.156) of tumors from therapy-naïve subjects with mCRC. However, the frequency of this mutation has not been investigated up to now in an adequate cohort of patients.
Since mechanisms leading to acquired resistance might also be involved in the intrinsic resistance to EGFR monoclonal antibodies in mCRC, the S492R mutations might represent a potential mechanism of resistance to cetuximab in patients that have not been exposed yet to this drug. In this regard, it is important to define the frequency of the S492R mutation in untreated patients, in order to assess whether mCRC patients should be routinely screened for this molecular alteration. To this end, we evaluated the frequency of the S492R mutation in a relatively large series of KRAS-exon 2 wild-type mCRC patients.
Results
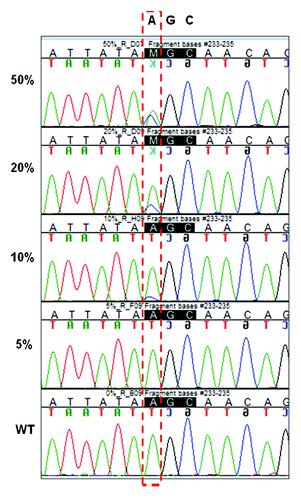
We first developed an assay to detect the S492R mutation. Since two different nucleotide substitutions have been described to cause this change in mCRC patients, we set up an assay based on PCR amplification of genomic DNA and direct sequencing of the PCR product by Sanger sequencing. In order to assess the sensitivity and specificity of the assay, we generated a positive control carrying the 1474A>C mutation by site directed mutagenesis, as described in the methods. Next, the sensitivity of the test was determined by assaying dilutions of mutant DNA in wild-type DNA in proportions of 100%, 80%, 50%, 40%, 20%, 10%, and 5%. The DNA mixtures were amplified by PCR and sequences were analyzed to identify the presence of the 1474A>C mutation. Samples were defined as mutant only when the mutation was evident in at least two different chromatograms obtained from two different PCR reactions. The lowest level of detection was approximately 10% mutant DNA in a background of wild-type DNA, as found in three independent experiments. Representative results are shown in .
Figure 1. Sensitivity of PCR sequencing: plasmid DNA carrying the c.1474A>C EGFR point mutation was mixed with plasmid wild-type DNA in dilutions of 50%, 20%, 10%, and 5%. Reverse sequencing chromatograms are shown.
We next analyzed 505 KRAS-exon 2 wild-type formalin fixed paraffin embedded (FFPE) samples from CRC patients. The characteristics of the patients from which the tumors were obtained are described in . In agreement with current indications for anti-EGFR therapies in CRC, 88% of the cases were in stage IV, with 66% of the patients having nodal involvement at the time of the diagnosis.
Table 1. Patients’ clinic-pathological characteristics
Tumor specimens were obtained before patients received any systemic therapy. Primary tumors were available in the majority of the cases (93%), metastases were analyzed for few patients. The tumor cell percentage of each specimen was assessed, and tumor macrodissection was performed for tumor samples containing a tumor cell percentage <50%. The final tumor cell content was ≥50% for all samples. All cases were wild-type for KRAS exon 2 mutations by PCR/sequencing analysis, that was performed as previously described.Citation19 For EGFR S492R mutational analysis, two independent PCR reactions and sequences in forward and reverse were obtained for each sample. PCR sequencing analysis revealed no S492R mutation in any of the analyzed specimens.
Discussion
The approval of EGFR monoclonal antibodies for mCRC patients without mutations in the exon 2 of KRAS, has led medical oncologist to classify CRC in two major subgroups on the basis of the presence or absence of these mutations. However, KRAS exon 2-wild-type tumors are an extremely heterogeneous population that includes carcinoma with different molecular alterations and, potentially, different possibility to respond to anti-EGFR agents. For example, a recent report suggested that patients with KRAS exon 3 and 4 mutations, as well as those carrying variants in NRAS exon 2, 3, and 4, do not benefit of panitumumab treatment.Citation7 Although conclusive data are not available yet, preliminary findings suggest that these molecular alterations might also predict resistance to cetuximab.Citation5 In this respect, assessment of mechanisms of intrinsic resistance is required in order to better select patients that might benefit of treatment with EGFR monoclonal antibodies.
Interestingly, some molecular alterations seem to be involved in both intrinsic and acquired resistance to anti-EGFR agents, such as recently hypothesized for ErbB-2 signaling activation, through either ErbB-2 gene amplification or increased secretion of heregulin, or KRAS mutations.Citation13,Citation14,Citation16 Here, we clearly demonstrate that this is not the case for the S492R mutation that is never detected in CRC prior to exposure to cetuximab. We must acknowledge that we used a relatively low sensitive technique to study the expression of this mutation, and therefore low levels of mutant alleles might not have been detected with our approach. However, deep sequencing of pre-treatment samples from tumors showing the S492R mutation at disease progression, failed to identify this molecular alteration before exposure to cetuximab, suggesting that this mutation might be acquired during treatment.Citation18
Interestingly, one of the cases in which the S492R mutation was initially discovered showed only 3% of the alleles carrying this variant.Citation18 Similarly, in KRAS wild-type patients showing an acquired KRAS mutation at progression after initial response to EGFR monoclonal antibodies, a low level of KRAS mutant alleles was shown in the majority of the cases (range 0.48–17.3%).Citation16 A recent report has indeed suggested that the presence of low levels of KRAS mutant alleles are associated with a reduced response rate and progression-free survival in mCRC patients treated with EGFR-based therapies.Citation20 However, a major bias of this latter study is that patients that showed resistance to anti-EGFR agents were also in more advanced lines of therapy as compared with sensitive patients, and this unbalance might have clearly affected the results of this study. Actually, the presence of resistance mutations in a small fraction of tumor cells is unlikely to cause primary resistance, although it may lead to a reduced progression free survival as demonstrated for treatment with EGFR tyrosine kinase inhibitors and the T790M resistance mutations in non-small-cell lung carcinoma (NSCLC).Citation21 In this respect, it is important to underline that the patient with only 3% of alleles carrying the S492R mutation had also a BRAF mutation that might have caused the resistance to cetuximab. Therefore, using techniques with high sensitivity to detect this mutation should be performed with extreme caution because it may lead to the exclusion from the treatment of patients potentially sensitive to anti-EGFR agents. Finally, the frequency of the S492R mutation in cetuximab-resistant CRC needs also to be assessed in larger cohorts of patients. In fact, a recent report failed to identify this mutation in post-treatment biopsies from 20 patients that received cetuximab-based therapy.Citation22
In conclusion, our findings suggest that the S492R mutation is not involved in primary resistance to cetuximab in CRC, and patients with mCRC should not be routinely screened for this mutation prior therapy with cetuximab.
Materials and Methods
Samples
FFPE tissues from 505 mCRC patients were obtained from different Italian Surgical Patholgy Departments. Written informed consent was obtained from all patients prior to testing. The tumor cell content of each sample was assessed by two pathologists (GB and FT). All patients were previously screened for KRAS exon 2 mutations by PCR/sequencing as previously described.Citation19
Site-directed mutagenesis
The EGFR region containing nucleotides 1474–1476 was amplified by PCR and the obtained amplicons were cloned into the pCRII-TOPO vector using TOPO TA Cloning Kit (Invitrogen). Plasmid DNA from positive clones was isolated using QIAprep spin Miniprep Kit® (Qiagen). The presence and correct orientation of the insert was confirmed by direct sequencing. To generate the S492R variant (c.1474A>C point mutation) into the plasmid DNA template, the QuikChange™ Lightning Multi Site-Directed Mutagenesis kit (Agilent Technologies) was used. The presence of the mutation was confirmed by direct sequencing.
EGFR sequencing
Genomic DNA was extracted from two 20 μm FFPE sections using the QIAamp® DNA FFPE Tissue kit (Qiagen) and the QIAcube apparatus (Qiagen). The genomic region of EGFR harboring the S492R mutation (c.1474A>C or c.1476C>A,) was amplified by PCR using the following primers:
EGFR_EX12_FOR: 5′-TGTAAAACGA CGGCCAGTGT GCTATGCAAA TACAATAAAC TGG-3′ and EGFR_EX12_REV: 5′-CAGGAAACAG CTATGACCGG ACCCATTAGA ACCAACTCC-3′
The nucleotides in bold correspond to M13 consensus sequences, and were used for cycle sequencing with M13 consensus primers.
Sequence analysis was performed on an ABI Prism 3500 Genetic Analyzer (LifeTechnologies). The collected data were evaluated with the Sequencer 4.8v Analysis Software (GeneCodes Corporation).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E, Ciardiello F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol 2009; 6:519 - 27; http://dx.doi.org/10.1038/nrclinonc.2009.111; PMID: 19636327
- Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006; 366:2 - 16; http://dx.doi.org/10.1016/j.gene.2005.10.018; PMID: 16377102
- De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol 2011; 12:594 - 603; http://dx.doi.org/10.1016/S1470-2045(10)70209-6; PMID: 21163703
- Ciardiello F, Normanno N. HER2 signaling and resistance to the anti-EGFR monoclonal antibody cetuximab: a further step toward personalized medicine for patients with colorectal cancer. Cancer Discov 2011; 1:472 - 4; http://dx.doi.org/10.1158/2159-8290.CD-11-0261; PMID: 22586650
- De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010; 11:753 - 62; http://dx.doi.org/10.1016/S1470-2045(10)70130-3; PMID: 20619739
- Peeters M, Oliner KS, Parker A, Siena S, Van Cutsem E, Huang J, Humblet Y, Van Laethem JL, André T, Wiezorek J, et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res 2013; 19:1902 - 12; http://dx.doi.org/10.1158/1078-0432.CCR-12-1913; PMID: 23325582
- Douillard J-Y, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013; 369:1023 - 34; http://dx.doi.org/10.1056/NEJMoa1305275; PMID: 24024839
- Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 2008; 26:5705 - 12; http://dx.doi.org/10.1200/JCO.2008.18.0786; PMID: 19001320
- Van Cutsem E, Köhne CH, Láng I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol 2011; 29:2011 - 9; http://dx.doi.org/10.1200/JCO.2010.33.5091; PMID: 21502544
- Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res 2009; 69:1851 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-08-2466; PMID: 19223544
- Souglakos J, Philips J, Wang R, Marwah S, Silver M, Tzardi M, Silver J, Ogino S, Hooshmand S, Kwak E, et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer 2009; 101:465 - 72; http://dx.doi.org/10.1038/sj.bjc.6605164; PMID: 19603024
- Loupakis F, Pollina L, Stasi I, Ruzzo A, Scartozzi M, Santini D, Masi G, Graziano F, Cremolini C, Rulli E, et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol 2009; 27:2622 - 9; http://dx.doi.org/10.1200/JCO.2008.20.2796; PMID: 19398573
- Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med 2011; 3:99ra86; http://dx.doi.org/10.1126/scitranslmed.3002442; PMID: 21900593
- Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Corà D, Di Nicolantonio F, Buscarino M, Petti C, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov 2011; 1:508 - 23; http://dx.doi.org/10.1158/2159-8290.CD-11-0109; PMID: 22586653
- Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov 2013; 3:658 - 73; http://dx.doi.org/10.1158/2159-8290.CD-12-0558; PMID: 23729478
- Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012; 486:532 - 6; PMID: 22722830
- Diaz LA Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012; 486:537 - 40; PMID: 22722843
- Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S, Tsai SP, et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat Med 2012; 18:221 - 3; http://dx.doi.org/10.1038/nm.2609; PMID: 22270724
- Carotenuto P, Roma C, Rachiglio AM, Tatangelo F, Pinto C, Ciardiello F, Nappi O, Iaffaioli RV, Botti G, Normanno N. Detection of KRAS mutations in colorectal carcinoma patients with an integrated PCR/sequencing and real-time PCR approach. Pharmacogenomics 2010; 11:1169 - 79; http://dx.doi.org/10.2217/pgs.10.86; PMID: 20712532
- Tougeron D, Lecomte T, Pagès JC, Villalva C, Collin C, Ferru A, Tourani JM, Silvain C, Levillain P, Karayan-Tapon L. Effect of low-frequency KRAS mutations on the response to anti-EGFR therapy in metastatic colorectal cancer. Ann Oncol 2013; 24:1267 - 73; http://dx.doi.org/10.1093/annonc/mds620; PMID: 23293113
- Rossi A, Pasquale R, Esposito C, Normanno N. Should epidermal growth factor receptor tyrosine kinase inhibitors be considered ideal drugs for the treatment of selected advanced non-small cell lung cancer patients?. Cancer Treat Rev 2013; 39:489 - 97; http://dx.doi.org/10.1016/j.ctrv.2012.09.001; PMID: 23022519
- Tougeron D, Cortes U, Ferru A, Villalva C, Silvain C, Tourani JM, Levillain P, Karayan-Tapon L. Epidermal growth factor receptor (EGFR) and KRAS mutations during chemotherapy plus anti-EGFR monoclonal antibody treatment in metastatic colorectal cancer. Cancer Chemother Pharmacol 2013; 72:397 - 403; http://dx.doi.org/10.1007/s00280-013-2211-0; PMID: 23765179