
Abstract
K-RAS mutated (K-RASmut) non-small cell lung cancer (NSCLC) cells are resistant to EGFR targeting strategies. We investigated the impact of K-RAS activity irrespective of mutational status in the EGFR-independent increase in clonogenic cell survival. An analysis of the K-RAS activity status revealed a constitutively high K-RAS activity in K-RASmut NSCLC cells and also in head and neck squamous cell carcinoma (HNSCC) cells overexpressing wild-type K-RAS (K-RASwt). Similar to K-RAS-mutated cells, increased K-RAS activity in HNSCC cells overexpressing K-RASwt was associated with the stimulated production of the EGFR ligand amphiregulin and resistance to EGFR tyrosine kinase (EGFR-TK) inhibitors such as erlotinib. Expression of mutated K-RAS stimulated Akt phosphorylation and increased plating efficiency. Conversely, knockdown of K-RAS in K-RASmut NSCLC cells and in HNSCC cells presenting overexpression of K-RASwt resulted in sensitization to the anti-clonogenic activity of erlotinib. K-RAS activity results in EGFR-dependent and EGFR-independent Akt activity. The short-term treatment (2 h) of cells with EGFR-TK or PI3K inhibitors (erlotinib and PI-103) resulted in the repression of Akt activation, whereas long-term treatment (24 h) with inhibitors led to the reactivation of Akt and improved clonogenicity. The Akt re-activation was MAPK-ERK2-dependent and associated with a lack of complete response to anti-clonogenic activity of PI-103. A complete response was observed when PI-103 was combined with MEK inhibitor PD98059. Together, clonogenicity inhibition in tumor cells presenting constitutive K-RAS activity independent of K-RAS mutational status can be achieved by targeting of EGFR downstream pathways, i.e., PI3K alone or the combination of PI3K and MAPK inhibitors.
Introduction
Epidermal growth factor receptor (EGFR), a member of the erbB receptor family, is frequently overexpressed or activated in many cancers and is implicated in tumor development. Ligand binding induces EGFR homo-/heterodimerization and activates the tyrosine kinase (TK) domain and the autophosphorylation of intracellular tyrosine residues.Citation1 Phosphorylation of these residues due to specific adaptor protein binding leads to the activation of specific downstream pathways, i.e., the Ras/mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K)/Akt, and signal transducers and activators of transcription pathways.Citation2 These pathways in turn regulate proliferation and are part of the regulatory mechanisms controlling the survival and metastatic potential of tumor cells. Therefore, EGFR targeting has been intensely pursued as a cancer treatment strategy. To this end, two classes of EGFR inhibitors, i.e., anti-EGFR monoclonal antibodies, such as cetuximab and panitumumab, and small-molecule EGFR-TK inhibitors, such as erlotinib and gefitinib, are routinely used clinically. However, the reported response rates to these drugs are low, primarily due to both intrinsic and acquired resistance.Citation3-Citation6 The above-mentioned anti-EGFR antibodies compete with ligands for receptor binding, whereas small-molecule inhibitors inhibit the TK activity of the receptor by binding to and blocking the ATP-binding pocket.
Activating EGFR-TK mutations, particularly deletions in exon 19 and a point mutation in exon 21 (L858R), have been identified in non-small cell lung cancer (NSCLC) as being associated with the response to EGFR-TK inhibitors.Citation7,Citation8 Similarly, acquired resistance to these inhibitors has also been reported to be in part due to inhibitor-induced point mutations in the TK domain (T790M) after a median of 10 to 16 mo of treatment.Citation4,Citation9 In contrast, mutations in the components of the EGFR cascade, such as mutations in codons 12 and 13 of K-RAS, which are present in 20–30% of NSCLCs, are associated with the resistance of NSCLC to the EGFR antibody cetuximabCitation6 and the EGFR-TK inhibitors gefitinib and erlotinib.Citation10 Similar to K-RAS mutations, mutations in the PIK3CA gene,Citation11 leads to the enhanced activation of the PI3K/Akt pathway.Citation10
However, the response of head and neck squamous cell carcinomas (HNSCCs) to EGFR targeting strategies is quite heterogeneous, and the extent to which the markers identified as predictors for NSCLC responses to EGFR inhibitors are relevant for HNSCC remains unclear. The mutations in EGFR described for NSCLC, such as deletions in exon 19 and a point mutation in exon 21 (L858R), are rare or have not been observed in HNSCC.Citation12,Citation13 However, the expression of EGFR variant III (EGFRvIII) has been demonstrated in approximately 40% of HNSCCs.Citation14 The EGFRvIII mutation was first identified in glioblastomas and results in constitutively active MAPK and PI3K/Akt cascades.Citation15 Tinhofer et al.Citation16 have reported that the expression of EGFRvIII together with the enhanced expression of amphiregulin (AREG) can identify HNSCC patients who are less likely to benefit from combination treatment with the anti-EGFR antibody cetuximab and docetaxel. Although mutations in K-RAS occur in HNSCC at a rather low frequency, amplification of the wild-type K-RAS gene (K-RASwt) has been demonstrated to promote the growth of HNSCC cells.Citation17 Moreover, and similar to NSCLC, a mutation in the PIK3CA gene increases PI3K activity in HNSCC cells, which leads to growth factor-independent colony formation.Citation18
It is known that a K-RAS mutation leads to constitutive K-RAS activity that is associated with the stimulated autocrine production of the EGFR ligand AREGCitation19 and resistance to EGFR-TK inhibitors in NSCLC. However, it is not known whether K-RASwt overexpression has a similar impact on K-RAS activity and resistance to EGFR-TK inhibitors. Because K-RAS mutations lead to the activation of the PI3K/Akt and MAPK/ERK pathways, the specific role of each pathway in clonogenicity needs to be investigated in both K-RASmut and K-RASwt overexpressing cells.
In the present study, we found that clonogenic activity in cells presenting either a K-RAS mutation or K-RASwt overexpression results from the activation of the EGFR-independent PI3K-Akt pathway. In contrast to a short-term inhibition (2 h), long-term inhibition (24 h) of PI3K by the specific PI3K inhibitor PI-103 leads to the K-RAS-mediated and ERK2-dependent reactivation of Akt and thus to a limited response to applied EGFR and PI3K inhibitors in terms of clonogenic cell survival.
Results
K-RAS-GTP level is correlated with increased proliferation and clonogenic activity
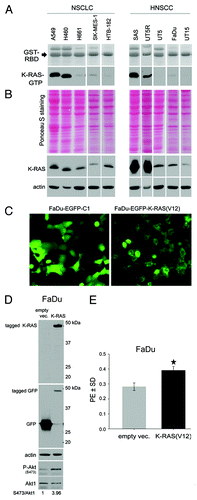
K-RAS mutation results in constitutive K-RAS activity, as demonstrated by a pull-down assay using the GST-tagged Raf1-Ras-binding domain (Raf1-RBD) protein (). Interestingly, although SAS and UT5R cells are K-RASwt, the level of K-RAS activity was comparable to that in the K-RASmut A549, and H460 cells (). Analyzing the expression level of K-RAS indicated that SAS and UT5R cells present overexpression of K-RAS protein (). A determination of the population doubling time (DT) of the cell lines indicated that the K-RASmut NSCLC cell lines A549 (20.98 ± 0.17 h) and H460 (22.34 ± 0.36 h) present a significantly shorter DT than the K-RASwt cell lines H661 (37.20 ± 1.91 h), SK-MES-1 (39.26 ± 2.17 h), and HTB-182 (37.65 ± 3.10 h) (P < 0.001). Similarly, for the HNSCC cell lines, the DTs of the SAS (24.01 ± 1.96 h) and UT5R (27.61 ± 2.34 h) cells were significantly shorter than that of either the UT5 (39.68 ± 8.55 h) or UT15 (48.08 ± 3.04 h) cells (P < 0.001) (Fig. S1A). The DT for FaDu cells (29.46 ± 1.90 h) was significantly longer than that of the SAS cells (P < 0.001) but was not significantly longer than that of the UT5R cells (P = 0.087) (Fig. S1A). Cells with a short DT (A549, H460, SAS, and UT5R) presented a significant increase in clonogenic activity, as shown by plating efficiency (PE) (Fig. S1B). K-RAS sequencing was performed to analyze whether the increased clonogenic activity in the NSCLC (A549 and H460) and HNSCC cells (SAS and UT5R) was due to a potential mutation in the K-RAS gene. The data for the mutational status of K-RAS, EGFR, PI3K, and TP53 (summarized in Table S1) indicate that the K-RAS gene was mutated only in the A549 (G12S) and H460 (Q61H) cells and not in the HNSCC SAS and UT5R cells presenting a short DT and high PE. On the basis of these results, it can be assumed that the level of K-RAS activity rather than its mutational status correlates with clonogenic activity (Fig. S1B). As an additional proof for the role of K-RAS in clonogenic activity, the HNSCC FaDu cells were transiently transfected with a plasmid expressing mutated K-RAS(V12); compared with the empty vector-transfected cells, K-RAS(V12) overexpression () resulted in a significant increase in clonogenicity ().
Figure 1. Impact of K-RAS activity on tumor cell clonogenicity. (A) The basal level of K-RAS-GTP was determined as described.Citation39 (B) Total cell lysates were subjected to sodium dodecyl sulfate-PAGE (SDS-PAGE). Following Ponceau staining, the expression level of K-RAS was analyzed by western blotting. Actin was detected as a loading control. (C) FaDu cells were transiently transfected with pEGFP-C1 empty vector or pEGFP/K-RAS(V12); 48 h after transfection, green fluorescent protein (GFP) expression was analyzed by fluorescent microscopy. (D) After microscopy analysis, the cells were lysed, and western blotting was performed. Following detection of K-RAS and P-Akt (S473), the blots were stripped and incubated with antibodies against GFP and Akt1. Actin was used as a loading control. The densitometric values represent the ratios of P-Akt (S473) to Akt1 normalized to 1 in the control vector-transfected cells. (E) FaDu cells were transiently transfected with empty vector or vector expressing K-RAS(V12); 48 h after transfection, the cells were plated for a clonogenic assay. Homozygous K-RAS(G12V) significantly increased PE. The data present the mean ± SD of 12 parallel experiments (*P < 0.05).
K-RAS activity limits the response to the EGFR-TK inhibitor erlotinib and is associated with the autocrine production of EGFR ligand
To investigate the possible role of K-RAS activity in the response pattern of tumor cells to EGFR-TK inhibitors, the effect of erlotinib on the clonogenic activity of NSCLC and HNSCC lines presenting different K-RAS activity levels was investigated. Erlotinib at 1 and 2.5 µM had no effect on the clonogenic activity of the K-RASmut NSCLC cell lines A549 and H460. In contrast, erlotinib strongly inhibited the colony formation of the H661 and SK-MES-1 cells (P < 0.001). The HTB-182 cells, with a very low expression of EGFR (Fig. S2), did not response to erlotinib (), and erlotinib (1 µM) had no effect on clonogenic activity in the HNSCC cells SAS and UT5R, which present high wild-type K-RAS activity, even at the higher concentration of 2.5 µM. In contrast, the clonogenic activity of HNSCC cells presenting low levels of K-RAS activity (UT5, UT15, and FaDu) was completely blocked ().
Figure 2. K-RAS activity is associated with erlotinib resistance and accompanied with increased autocrine production of AREG. (A and B) The effect of erlotinib on clonogenic activity was determined using a clonogenic assay. The data points shown represent the mean PE ± SD of at least 12 data from two independent experiments. The inhibition of clonogenic activity by erlotinib is dependent on the cell line (*P < 0.05; **P < 0.01; ***P < 0.001). (C) Cells were incubated in serum-free medium for 48 h, and the concentration of AREG was measured by ELISA. The data present the mean ± SD of 12 data from 4 independent cultures of SAS cells, 4 data from 2 independent cultures of UT5R, and 11 data from 4 independent cultures of UT5 cells (***P < 0.001).
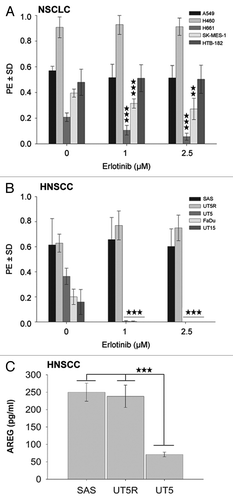
Previously, we showed that K-RAS mutation is associated with an enhanced autocrine production of the EGFR ligand AREG.Citation19,Citation20 As the K-RASmut cells were found to be resistant to erlotinib, we further investigated whether the erlotinib-resistant and K-RASwt-overexpressing SAS and UT5R cells also produce increased levels of AREG. The data shown in indicate that the erlotinib-resistant SAS and UT5R cells indeed exhibit an elevated production of AREG that was significantly higher than that of the erlotinib-sensitive UT5 cells (P < 0.001).
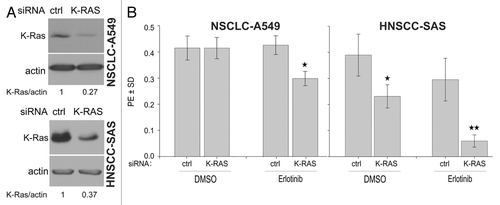
Based on the possible role of K-RAS activity in the response to erlotinib, the influence of this activity on erlotinib resistance in K-RASmut A549 and K-RASwt-overexpressing SAS cells was investigated using siRNA-dependent K-RAS protein repression. As demonstrated in , a marked reduction in the level of K-RAS protein led to a significant increase in the sensitivity of A549 and SAS cells to erlotinib ().
Figure 3. K-RAS knockdown sensitizes cells to erlotinib. (A) A549 and SAS cells were transfected with control (ctrl)-siRNA or K-RAS-siRNA. Two days after transfection, the efficiency of K-RAS-siRNA was analyzed by western blotting. (B) The cells were plated in 6-well plates for a clonogenic assay two days after transfection with the indicated siRNAs and then treated with erlotinib (1 µM) after 24 h. The histograms represent the mean PE ± SD of 12 parallel data in A549 cells and 18 data from two independent experiments in SAS cells (*P < 0.05).
Constitutive K-RAS activity regulates clonogenic cell survival through the PI3K/Akt pathway but not MAPK/ERK signaling
Transfection of mutated K-RAS in FaDu cells led to the enhanced phosphorylation of Akt at S473 (). Similarly, as indicated by the data presented in Figure S3, a 24 h treatment of the erlotinib-resistant K-RASmut A549 and K-RASwt-overexpressing SAS cells with erlotinib did not block Akt phosphorylation. In contrast, Akt phosphorylation was markedly inhibited by erlotinib in the erlotinib-responsive H661 and FaDu cells. Because erlotinib inhibited P-ERK1/2 in all cell lines tested (Fig. S3), we speculated that the clonogenic activity of the cell lines used in this study was not primarily dependent on the activation of the MAPK pathway. This hypothesis was tested using the specific MEK inhibitor PD98059. Cells pre-treated with 20 µM of PD98059 for 24 h presented markedly reduced ERK1/2 phosphorylation. A strong inhibition of ERK1/2 phosphorylation by approximately 80% was observed in FaDu cells, whereas the weakest effect (approximately 40% inhibition) was found in H661 cells (). Although PD98059 inhibited P-ERK1/2 in all the cell lines tested, MEK targeting did not efficiently block clonogenic activity (): a slight effect was only observed in the H661, UT5, and SAS cells (P < 0.05) (). Most interestingly, the clonogenic activity of FaDu cells (in which erlotinib and PD98059 blocked ERK1/2 phosphorylation) was blocked by erlotinib but not PD98059. This set of data indicates that the MAPK pathway is not the major regulator of clonogenic activity in the NSCLC and HNSCC cells used in this study.
Figure 4. The clonogenic activity of tumor cells depends mainly on the activation of PI3K-Akt but not on the MAPK-ERK1/2 pathway. (A) Cells were treated or not with the MEK inhibitor PD98059 (20 µM) for 24 h, and the level of P-ERK1/2 and ERK1/2 was analyzed by western blotting. (B) Cells were plated in 6-well plates for a clonogenic assay and were treated with 20 µM of PD98059 after 24 h. (C) Cells were treated or not with the indicated concentrations of PI3K inhibitor PI-103 for 24 h. The phosphorylation levels of Akt were analyzed by western blotting using isolated protein samples; the blots were re-probed with an anti-Akt1 antibody. (D) Effect of PI-103 on PE was determined by a clonogenic assay. The data points represent the mean PE ± SD of at least 12 data from two independent experiments. The statistical analysis indicated a differential effect of PD98059 (B) and PI-103 (D) on the clonogenic activity of the tested cell lines (*P < 0.05; **P < 0.01; ***P < 0.001). The densitometric values in (A and C) represent the ratios of P-Akt/Akt1 and P-ERK1/2 to ERK1/2 normalized to 1 in the DMSO-treated controls.
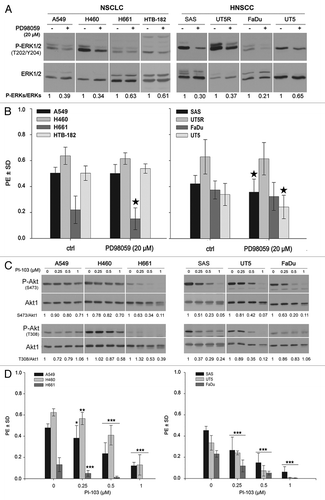
The kinase inhibitor PI-103, with a high specificity for PI3K, was used to investigate the specific role of the PI3K pathway in clonogenicity. The effect of PI-103 on Akt phosphorylation was tested after a 24 h treatment. Although, a dose-dependent inhibition of P-Akt (S473) was observed in all cell lines tested, the inhibition of S473 phosphorylation in K-RASmut A549 and H460 (30% inhibition) was not as efficient as in the H661, SAS, UT5, and FaDu cells (90–95% inhibition). Similar to the effect on S473 phosphorylation, a 24 h treatment with PI-103 only resulted in a slight inhibition of Akt phosphorylation at T308 in K-RASmut A549 and H460 cells, whereas a strong inhibition of Akt phosphorylation was observed in the H661, SAS, UT5, and FaDu cells (). As shown in , PI-103 also inhibited the clonogenic activity of all cell lines in a concentration-dependent manner (). Although PI-103 at the highest concentration (1 µM) blocked the clonogenicity of H661, the clonogenic activity of K-RASmut A549 and H460 cells was only reduced by 75% in A549 and 79% in H460, a difference that was even more pronounced when the cells were treated with lower concentrations of PI-103. A similar difference was observed in the HNSCC cells. PI-103 (1 µM) completely blocked the clonogenic activity of UT5 and FaDu cells, whereas clonogenic activity of SAS cells was reduced by 86%.
The ERK2-dependent reactivation of Akt following PI3K inhibition eliminates the anti-clonogenic effect of inhibitors
As described above, the PI3K inhibitor PI-103 exerted a limited effect on the clonogenic activity of K-RASmt and K-RASwt-overexpressing cells. Similarly, as shown in , erlotinib treatment did not affect the clonogenic activity of these cells. The molecular biology data presented in Figure S3 and indicate a lack of effect of erlotinib on Akt phosphorylation in erlotinib-resistant cells. Since PI-103 only slightly reduced Akt phosphorylation in K-RASmut cells, we hypothesized that long-term inhibition of PI3K activity following treatment with either erlotinib or direct inhibition of PI3K by PI-103 may lead to the reactivation of Akt, which interferes with the anticlonogenic effect of the inhibitors.
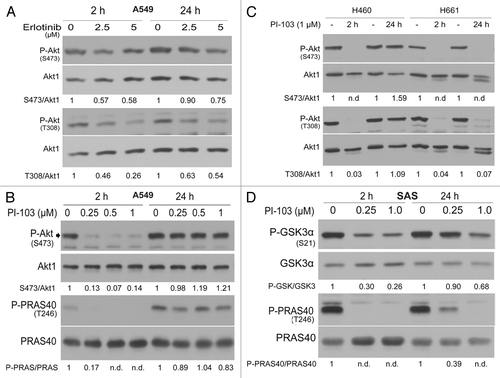
To confirm this hypothesis, the effect of erlotinib on Akt phosphorylation after 2 and 24 h of treatment was analyzed. The western blot data and relative densitometric analysis shown in indicate that the inhibition of Akt by erlotinib in A549 cells was more effective after 2 h than after 24 h of treatment. To verify whether the reactivation of Akt is dependent on PI3K activity, the cells were treated with the PI3K inhibitor PI-103, which completely blocked the phosphorylation of Akt at S473 and T308 and its substrate PRAS40 (T246) after a 2 h treatment (). In contrast, PI-103 treatment for 24 h only exerted a slight effect in the K-RASmut cells (). However, PI-103 completely blocked Akt phosphorylation at S473 and T308 in K-RASwt-H661 cells after 2 or 24 h (). In SAS cells overexpressing K-RASwt, a 2 h treatment of PI-103 reduced the phosphorylation of the Akt substrate GSKα at S21 by approximately 70% at 0.25 µM and 74% at 1 µM (). Interestingly, a 24 h pretreatment led to the restimulation of P-GSKα-S21, which reached approximately 90% and 68% of the control after treatment at 0.25 µM and 1 µM PI-103, respectively (). The analysis of the phosphorylation of the Akt substrate PRAS40 revealed that a 2 h treatment at both concentrations of PI-103 completely blocked PRAS40 phosphorylation, whereas treatment of the cells with 0.25 µM PI-103 for 24 h reduced the Akt activity only by approximately 60%, as tested by the phosphorylation of PRAS40.
Figure 5. Long-term inhibition of EGFR and PI3K results in the reactivation of Akt. (A) A549 cells were lysed at 2 h and 24 h after treatment with or without the indicated concentrations of erlotinib. (B–D) Cells were treated with the indicated concentrations of PI-103; at 2 and 24 h after treatment, protein samples were isolated and subjected to SDS-PAGE. The levels of P-Akt (S473 and T308), P-GSK3α/β (S21/S9), and P-PRAS40 (T246) were analyzed by western blotting. The blots were stripped and incubated with antibodies against Akt1, GSK3α/β, and PRAS40. The densitometric values represent the ratios of P-Akt (S473 and T308)/Akt1 (A and B), P-PARA40/PRAS40 (B–D), and P-GSK3α/GSK3α (D) normalized to 1 in the corresponding controls. n.d., non-detectable.
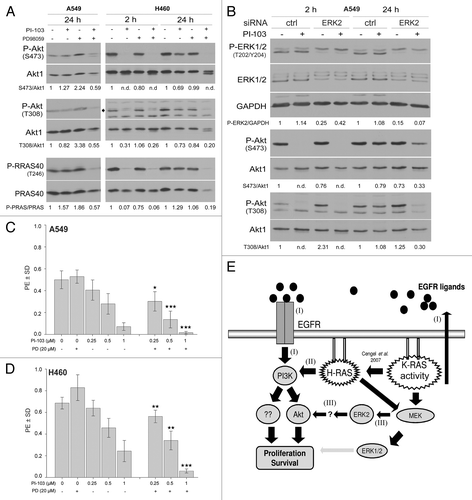
Based on the reported cross-talk between the PI3K-Akt and MAPK-ERK1/2 pathways,Citation21 we investigated whether the activation of PI3K-Akt after treatment with PI-103 is MAPK-ERK1/2 dependent. Using the specific MEK inhibitor PD98059 we were able to demonstrate that Akt phosphorylation after a 24 h treatment with PI-103 is dependent on the MAPK pathway (). An siRNA approach was then used to verify these results and assess the specific role of ERK2 on Akt activation. As shown in , the downregulation of ERK2 blocked the PI-103 dependent reactivation of Akt after 24 h of treatment. To correlate these results to a cellular endpoint, the influence of activated Akt on clonogenic survival was tested. In the K-RASmut NSCLC cell lines A549 and H460, PD98059 alone did not affect clonogenic activity, though the combination of PD98059 with PI-103 led to a significant synergistic effect when compared with PI-103 alone ().
Figure 6. The ERK2-dependent reactivation of Akt in K-RASmut cells following long-term treatment with PI-103 improves clonogenic survival. (A) A549 and H460 cells were treated with PI-103 (1 µM) for the indicated times, and protein samples were isolated and subjected to SDS-PAGE. The levels of P-Akt (S473 and T308) and P-PRAS40 (T246) were detected by western blotting; the blots were stripped, and total proteins were detected. (B) Cells transfected with control-siRNA (ctrl) or ERK2-siRNA were treated with DMSO or PI-103 at 3 d after transfection; 24 h after treatment, protein samples were isolated and subjected to SDS-PAGE. The levels of ERK1/2, PDK1, and P-Akt (S473 and T308) were detected by western blotting; the blots were stripped and re-incubated with an anti-Akt1 antibody. GAPDH was used as a loading control. (C and D) Cells were plated in 6-well plates for a clonogenic assay; after 24 h, the cells were treated the indicated concentrations of MEK inhibitor PD98059 (PD), PI3K inhibitor PI-103 (PI), or combination of PI and PD. Colonies that formed after 10 d were counted, and PE was calculated and graphed. The data points shown represent the mean PE ± SD of 12 data from two independent experiments. The statistical analysis indicated that the combination of PI and PD significantly increased the anti-clonogenic activity compared with PI alone (*P < 0.05; **P < 0.01; ***P < 0.001). (E) A model illustrating the signaling pathways involved in proliferation and survival of tumor cells with K-RAS mutation or cells overexpressing K-RASwt. The densitometric values represent the ratios of P-Akt (S473 and T308)/Akt1, P-PARA40/PRAS40, and P-ERK2/GAPDH normalized to 1 in the corresponding controls. n.d., non-detectable.
Discussion
Using a panel of 5 non-small cell lung cancer (NSCLC) and 5 head and neck squamous cell carcinoma (HNSCC) cell lines, we here demonstrate that constitutive high K-RAS activity due either to K-RAS mutation or the overexpression of the wild-type K-RAS protein leads to resistance against the EGFR-TK inhibitor erlotinib. Similar to previous reports on the autocrine production of EGFR ligands by K-RASmut tumor cells,Citation19,Citation20 stimulated AREG production was also observed in overexpressing HNSCC tumor cells, which exhibit a high constitutive activity of K-RASwt. K-RAS activity induces Akt activation, which has EGFR/PI3K-dependent and EGFR/PI3K-independent components. In cells with enhanced K-RAS activity, the short-term (2 h) inhibition of EGFR or PI3K results in the downregulation of EGFR/PI3K-dependent Akt activation. In contrast, the long-term (24 h) inhibition of EGFR or PI3K leads to the EGFR/PI3K-independent but MAPK/ERK pathway-dependent reactivation of Akt.
Among the various factors associated with the sensitivity of tumor cells to EGFR-TK inhibitors, exon 19 deletion and the L858R point mutation of EGFR in NSCLC are the most important thus far. As the alterations lead to ligand-independent EGFR-TK activity,Citation22,Citation23 these mutations are predictive markers for selecting NSCLC patients who would most likely benefit from treatment with EGFR-TK inhibitors.Citation24,Citation25 In addition, mutations in pathways downstream of EGFR, such as RAS and PI3K, have been proposed as markers for predicting the response to EGFR-targeting strategies. Within this context, the mutational activation of K-RAS in NSCLC and colon cancers is of prime importance for the lack of a response to both EGFR-TK inhibitorsCitation26,Citation27 and EGFR antibodies.Citation28 High constitutive K-RAS activity due to K-RAS mutation was confirmed for the NSCLC cell lines used in the present study, and elevated constitutive K-RAS activity was correlated with the erlotinib resistance demonstrated by the A549 and H460 cells, resistance that could be overcome by siRNA-mediated repression of the K-RAS-protein. Therefore, enhanced K-RAS activity is causative for a lack of response to erlotinib. Sunaga et al.Citation29 demonstrated that the knockdown of oncogenic K-RAS sensitizes NSCLC cells to gefitinib and cetuximab,Citation29 together with our results, demonstrating that the role of K-RAS mutation in the resistance to EGFR-TK inhibitors is independent of the targeting approach used to antagonize EGFR.
In contrast to NSCLC, exon 19 deletion and the L858R point mutation, which result in sensitivity to EGFR-TK inhibitors, are very rare in HNSCC. Conversely, deletion of the extracellular domain of EGFR, known as EGFRvIII, is rather frequent in HNSCC cells and contributes to resistance to cetuximab either administered alone or in combination with chemotherapy.Citation14,Citation16 Because the HNSCC cells used in the present study express wild-type EGFR, the differential response to erlotinib must be due to other alterations. Thinhofer et al.Citation16 observed that, in addition to EGFRvIII mutation, the level of AREG expression identifies HNSCC patients who are not responsive to combined cetuximab and docetaxel treatment. In agreement with this observation,Citation16 we have recently reported cetuximab resistance in the HNSCC cell lines SAS and UT5R, a subline of the UT5 cells that are resistant to cetuximab.Citation30 We also previously reported that NSCLC cells with an endogenous K-RAS mutationCitation19 or wild-type K-RAS HNSCC cells with induced overexpression of mutated K-RAS demonstrate elevated AREG production.Citation20 In the present study, we also found that K-RASwt-overexpressing HNSCC cells have high K-RAS activity and show enhanced expression of AREG. As K-RASmut cells with AREG overexpression show enhanced activation of PI3K-Akt signaling,Citation20 this pathway might be the major pathway for the clonogenic activity of K-RAS-mutated NSCLC cells and K-RASwt-overexpressing HNSCC cells. The strong inhibition of clonogenic activity by the PI3K inhibitor PI-103 in comparison to the effect of erlotinib supports this conclusion in both K-RASmut-NSCLC cells and K-RASwt-overexpressing HNSCC cells.
It is known that the K-RAS protein does not directly interact with PI3K to activate Akt; rather, when mutated, K-RAS enhances the autocrine production of EGFR ligands, e.g., AREG, which can stimulate Akt activation through EGFR/PI3K signaling.Citation19 In the present study, we showed that elevated AREG production is also observed in SAS and UT5R cells presenting overexpressed wild-type K-RAS protein and high K-RAS enzyme activity. Thus, as summarized in , the high constitutive activity of K-RAS can lead to EGFR ligand production and autocrine stimulation of EGFR/PI3K signaling to enhance Akt activity (, pathway I).
In tumor cells with oncogenic K-RAS, the production of EGFR ligands depends on the enhanced activation of wild-type H-RAS.Citation31 H-RAS, in parallel to its activation of the MAPK-ERK1/2 pathway through Raf kinase, directly interacts with the P110 subunit of PI3K and stimulates the PI3K-Akt survival pathway.Citation32 Thus, H-RAS-dependent PI3K activity is a potential second pathway by which oncogenic K-RAS leads to the activation of Akt and other downstream PI3K targets involved in clonogenic cell survival, a pathway that can shift the dependency of the PI3K/Akt pathway on EGFR signaling to EGFR-independent H-RAS signaling. The inhibition of Akt after 2 h of erlotinib treatment and its reactivation after 24 h of treatment supports this hypothesis. Thus, it can be concluded that targeting PI3K in tumor cells with constitutively high K-RAS activity is a more efficient approach than targeting EGFR to inhibit clonogenic activity.
The PI3K/Akt and MAPK/ERK pathways are the major effectors of oncogenic RAS. Due to the crosstalk between these two pathways, the inhibition of one pathway can lead to the activation of the other. Constitutive MEK signaling restores the expression of the phosphatase and tensin homolog (PTEN), both in vitro and in vivo;Citation33 as a consequence of MEK inhibition, recruitment of PTEN to the cell membrane is reduced, resulting in increased PI3K accumulation and Akt activation.Citation33,Citation34 In contrast, the inhibition of PI3K results in a compensatory activation of the ERK signaling pathway.Citation35 This phenomenon was observed at least in A549 cells. In the present study the pharmacological inhibition of MEK or siRNA knockdown of ERK2 led to elevated Akt phosphorylation, and enhanced ERK2 phosphorylation was observed when the cells were treated with the PI3K inhibitor PI-103 for 24 h.
Based on the above-described crosstalk, activation of PI3K/Akt is the major escape mechanism leading to MEK inhibitor resistance. In the present study, we showed that a short-term (2 h) treatment with a PI3K inhibitor led to the complete inhibition of Akt activation, whereas a long-term treatment (24 h) did not affect Akt activity. Thus, restimulation of Akt activity most likely occurred through a compensatory switch of pathways, which was able to reactivate Akt to the level of the untreated controls. Because the specific MEK kinase inhibitor PD98059 completely blocked the reactivation of Akt, it can be assumed that Akt reactivation under the conditions applied was MEK dependent. However, as long-term treatment (24 h) with PI-103 did not markedly affect ERK phosphorylation, it can be postulated that the basal activity of MEK is necessary for the phosphorylation of Akt; indeed, MEK1 has been described as a regulatory protein for the PI3K-dependent reactivation of Akt after treatment with MEK inhibitors.Citation34 To our knowledge, the PI3K-independent reactivation of Akt after treatment with a PI3K inhibitor is a novel pathway and has not been reported previously. The activation of this pathway (, pathway III) in K-RASmut cells and in cells overexpressing K-RASwt indicates that this is a pathway that is specifically regulated in cells with constitutively high K-RAS activity. The activation of this pathway appears to be essential to diminish the anticlonogenic activity of PI3K inhibitors. Thus, detailed analyses of this pathway can provide specific insight into how combined treatments with MEK and PI3K inhibitors can be used to more effectively target tumor cells with constitutively high K-RAS activity.
Materials and Methods
Materials
Anti-phospho-PRAS40 (2997), -PRAS40 (2691), -phospho-GSK3α-S21 (9316), -GSK3α (9338), -phospho-ERK1/2 (4377), -ERK1/2 (4695), and -phospho-Akt-S473 (9271) antibodies were purchased from Cell Signaling. Non-targeting siRNA (D-001810-10), ERK2-siRNA (NM-002745), K-RAS-siRNA (M-005069) were purchased from Theroscientific. Akt1 antibody (610877) and EGFR (610016) were purchased from BD Transduction laboratories. PI-103 (Calbiochem, 528100) and PD98059 (Calbiochem, 513000) were purchased from Calbiochem. The EGFR-TK inhibitor erlotinib was provided by Hoffmann-La Roche Ltd. GST-conjugated Raf1-RBD (Millipore, 14-278) and K-RAS (Sigma-Aldrich, WH0003845M1) were used. The EGFP-C1 control and EGFP/K-RAS(V12) plasmids were described previously.Citation36
Cell lines
Established NSCLC cell lines (A549, H460, SK-MES-1, H661, and HTB-182) and HNSCC cells (FaDu, UT-SCC-5 [UT5], UT5R, UT-SCC-15 [UT15], and SAS) were used. UT5R is a subline of UT5 that presents acquired resistance to cetuximab, as described previously.Citation30 Briefly, UT5 cells were continuously treated with increasing concentrations of cetuximab, from 5 nM and gradually doubled to 100 nM after every cell culture passage; acquired resistance to cetuximab was tested by proliferation and clonogenic assays.Citation30
Cells were cultured in DMEM (A549, SK-MES-1, HTB-182, UT5, UT5R, UT15, SAS, and FaDu) or RPMI-1640 (H460 and H661) routinely supplemented with 10% FCS and 1% penicillin–streptomycin and incubated in a humidified atmosphere with 93% air/7% CO2 at 37 °C. Mycoplasma testing was performed regularly on the cells used for this study.
Sequencing of EGFR, PIK3A, K-RAS, and TP53
Total RNA was isolated from frozen cell pellets of the SAS, UT15, FaDu, UT5, UT5R, and A549 cell lines using the RNeasy mini kit (Qiagen) and reverse transcribed with the Reverse-iT 1st strand synthesis kit (Abgene) using anchored oligo-dT primers. The PCR amplification of specific sequences was performed from cDNA using ReddyMix PCR Master Mix (Abgene). The complete coding sequence of EGFR was amplified in four overlapping fragments using the following primer pairs (5′/3′):
GAGCTCTTCG GGGAGCAG/TCCTCCATCT CATAGCTGTC G,
TCCGCAAGTG TAAGAAGTGC/TTGGACAGCC TTCAAGACCT,
GCCATCCAAA CTGCACCTAC/TGGTACATAT GGGTGGCTGA,
and TCCATCCTGG AGAAAGGAGA/TCGGTGTAAA CGTTGCAAAA.
The PIK3CA gene was amplified using the following primer pairs (5′/3′):
GACAAAGAAC AGCTCAAAGC AA/GCCGTAAATC ATCCCCATTT
and AGAGTTACTG TTTCAGAACA ATGAGA/TCAGTTATCT TTTCAGTTCA ATGC.
Exons 1 to 3 of K-RAS were amplified with primers (5′/3′) GAGAGGCCTGCT GAAAATGA/TGGTGAATAT CTTCAAATGA TTTAGT.
The amplicons were isolated using QIAquick columns (Qiagen), and both strands were sequenced by a commercial subcontractor (SeqLab).
Mutations of TP53 in the UT15, FaDu, and UT5 cell lines were previously published.Citation37 The mutation status of the SAS, A549, H460, H661, SK-MES-1, and HTB-182 cell lines was obtained from the Sanger Institute Catalogue of Somatic Mutations in Cancer website, http://www.sanger.ac.uk/cosmic.Citation38
Proliferation kinetics and clonogenic assay
Anti-proliferative effects were examined over a growth period of 5 d. Cells (5 × 104) were seeded in 60-mm culture dishes and treated or not with inhibitors after 24 h. The cells from 4 parallel cultures were counted within 5 d after treatment.
To analyze clonogenic survival, cells were plated in 6-well plates at a density of 250 to 500 cells per well (depending on the cell line) in a medium containing 20% serum. After 24 h, the cells were treated with the indicated concentration of the inhibitors or vehicle; 10 to 13 d later, the culture dishes were stained with Coomassie blue. Colonies with more than 50 cells were counted, and the plating efficiency (number of colonies/number of seeded cells) was calculated and graphed.
RAS activity assay, protein extraction, western blotting, and enzyme-linked immunosorbent assay
The assays were performed according to the supplier’s instruction and as reported previously.Citation39 To analyze protein expression and activity after the indicated treatments in each experiment, cells were washed twice with phosphate-buffered saline and lysed with lysis buffer.Citation39 Western blotting was performed as described previously.Citation36 Densitometry was performed where appropriate using ImageJ software (http://rsbweb.nih.gov/ij/). The enzyme-linked immunosorbent assay (ELISA) was performed as described previously.Citation19
siRNA transfection and K-RAS(V12) overexpression
Cells were transfected with 50 nM non-targeting siRNA or specific siRNA using the Lipofectamine 2000 transfection reagent according to the protocol of the manufacturer, as described.Citation36 Briefly, cells were apportioned into 6-well plates and transfected 24 h later with 50 nM control siRNA or specific siRNA. At 48 h after transfection, the cells were distributed into 6-well plates, and a clonogenic assay was performed. In parallel, protein samples were isolated, and the efficiency of transfection was analyzed.
To overexpress K-RAS(V12), sub-confluent K-RASwt-FaDu cells expressing a low level of endogenous K-RAS were transiently transfected with the control vector or vector expressing K-RAS(V12), as described.Citation36 After 24 h, the efficiency of transfection was tested by fluorescent microscopy of green fluorescent protein (GFP). Thereafter, the media were changed, and the cells were used for the experiments after another 24 h.
Statistics and densitometry
The Student t test was used to compare the data between two groups. The values are expressed as the mean ± SD. P < 0.05 was considered statistically significant (*P < 0.05; **P < 0.01; ***P < 0.001). Densitometric quantification analyses of the immunoblots were performed with ImageJ computer software (http://rsbweb.nih.gov/ij/).
Additional material
Download Zip (241.7 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (Ro527/5-1 and SFB-773-TP B02) and the Federal Ministry of Research and Education (BMBF grants 0258416 and 03NUK006D) awarded to H.P.R. as well as GRK 1302/2 (T11) awarded to M.T. and H.P.R.
References
- Sharafinski ME, Ferris RL, Ferrone S, Grandis JR. Epidermal growth factor receptor targeted therapy of squamous cell carcinoma of the head and neck. Head Neck 2010; 32:1412 - 21; http://dx.doi.org/10.1002/hed.21365; PMID: 20848399
- Rodemann HP, Dittmann K, Toulany M. Radiation-induced EGFR-signaling and control of DNA-damage repair. Int J Radiat Biol 2007; 83:781 - 91; http://dx.doi.org/10.1080/09553000701769970; PMID: 18058366
- Chen G, Noor A, Kronenberger P, Teugels E, Umelo IA, De Grève J. Synergistic effect of afatinib with su11274 in non-small cell lung cancer cells resistant to gefitinib or erlotinib. PLoS One 2013; 8:e59708; http://dx.doi.org/10.1371/journal.pone.0059708; PMID: 23527257
- Oxnard GR, Arcila ME, Sima CS, Riely GJ, Chmielecki J, Kris MG, Pao W, Ladanyi M, Miller VA. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011; 17:1616 - 22; http://dx.doi.org/10.1158/1078-0432.CCR-10-2692; PMID: 21135146
- Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, Ince WL, Jänne PA, Januario T, Johnson DH, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 2005; 23:5900 - 9; http://dx.doi.org/10.1200/JCO.2005.02.857; PMID: 16043828
- Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757 - 65; http://dx.doi.org/10.1056/NEJMoa0804385; PMID: 18946061
- Wu YL, Zhong WZ, Li LY, Zhang XT, Zhang L, Zhou CC, Liu W, Jiang B, Mu XL, Lin JY, et al. Epidermal growth factor receptor mutations and their correlation with gefitinib therapy in patients with non-small cell lung cancer: a meta-analysis based on updated individual patient data from six medical centers in mainland China. J Thorac Oncol 2007; 2:430 - 9; http://dx.doi.org/10.1097/01.JTO.0000268677.87496.4c; PMID: 17473659
- Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304:1497 - 500; http://dx.doi.org/10.1126/science.1099314; PMID: 15118125
- Nguyen KS, Kobayashi S, Costa DB. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer 2009; 10:281 - 9; http://dx.doi.org/10.3816/CLC.2009.n.039; PMID: 19632948
- Ludovini V, Bianconi F, Pistola L, Chiari R, Minotti V, Colella R, Giuffrida D, Tofanetti FR, Siggillino A, Flacco A, et al. Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. J Thorac Oncol 2011; 6:707 - 15; http://dx.doi.org/10.1097/JTO.0b013e31820a3a6b; PMID: 21258250
- Yamamoto H, Shigematsu H, Nomura M, Lockwood WW, Sato M, Okumura N, Soh J, Suzuki M, Wistuba II, Fong KM, et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res 2008; 68:6913 - 21; http://dx.doi.org/10.1158/0008-5472.CAN-07-5084; PMID: 18757405
- Bissada E, Abboud O, Abou Chacra Z, Guertin L, Weng X, Nguyen-Tan PF, Tabet JC, Thibaudeau E, Lambert L, Audet ML, et al. Prevalence of K-RAS Codons 12 and 13 Mutations in Locally Advanced Head and Neck Squamous Cell Carcinoma and Impact on Clinical Outcomes. Int J Otolaryngol 2013; 2013:848021; http://dx.doi.org/10.1155/2013/848021; PMID: 23737793
- Hama T, Yuza Y, Suda T, Saito Y, Norizoe C, Kato T, Moriyama H, Urashima M. Functional mutation analysis of EGFR family genes and corresponding lymph node metastases in head and neck squamous cell carcinoma. Clin Exp Metastasis 2012; 29:19 - 25; http://dx.doi.org/10.1007/s10585-011-9425-5; PMID: 21953075
- Sok JC, Coppelli FM, Thomas SM, Lango MN, Xi S, Hunt JL, Freilino ML, Graner MW, Wikstrand CJ, Bigner DD, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res 2006; 12:5064 - 73; http://dx.doi.org/10.1158/1078-0432.CCR-06-0913; PMID: 16951222
- Mukherjee B, McEllin B, Camacho CV, Tomimatsu N, Sirasanagandala S, Nannepaga S, Hatanpaa KJ, Mickey B, Madden C, Maher E, et al. EGFRvIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res 2009; 69:4252 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-08-4853; PMID: 19435898
- Tinhofer I, Klinghammer K, Weichert W, Knödler M, Stenzinger A, Gauler T, Budach V, Keilholz U. Expression of amphiregulin and EGFRvIII affect outcome of patients with squamous cell carcinoma of the head and neck receiving cetuximab-docetaxel treatment. Clin Cancer Res 2011; 17:5197 - 204; http://dx.doi.org/10.1158/1078-0432.CCR-10-3338; PMID: 21653686
- Hoa M, Davis SL, Ames SJ, Spanjaard RA. Amplification of wild-type K-ras promotes growth of head and neck squamous cell carcinoma. Cancer Res 2002; 62:7154 - 6; PMID: 12499248
- Murugan AK, Hong NT, Fukui Y, Munirajan AK, Tsuchida N. Oncogenic mutations of the PIK3CA gene in head and neck squamous cell carcinomas. Int J Oncol 2008; 32:101 - 11; PMID: 18097548
- Toulany M, Baumann M, Rodemann HP. Stimulated PI3K-AKT signaling mediated through ligand or radiation-induced EGFR depends indirectly, but not directly, on constitutive K-Ras activity. Mol Cancer Res 2007; 5:863 - 72; http://dx.doi.org/10.1158/1541-7786.MCR-06-0297; PMID: 17699110
- Minjgee M, Toulany M, Kehlbach R, Giehl K, Rodemann HPK-RAS. K-RAS(V12) induces autocrine production of EGFR ligands and mediates radioresistance through EGFR-dependent Akt signaling and activation of DNA-PKcs. Int J Radiat Oncol Biol Phys 2011; 81:1506 - 14; http://dx.doi.org/10.1016/j.ijrobp.2011.05.057; PMID: 21985943
- Sunayama J, Matsuda K, Sato A, Tachibana K, Suzuki K, Narita Y, Shibui S, Sakurada K, Kayama T, Tomiyama A, et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells 2010; 28:1930 - 9; http://dx.doi.org/10.1002/stem.521; PMID: 20857497
- Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007; 7:169 - 81; http://dx.doi.org/10.1038/nrc2088; PMID: 17318210
- Soria JC, Mok TS, Cappuzzo F, Jänne PA. EGFR-mutated oncogene-addicted non-small cell lung cancer: current trends and future prospects. Cancer Treat Rev 2012; 38:416 - 30; http://dx.doi.org/10.1016/j.ctrv.2011.10.003; PMID: 22119437
- He C, Liu M, Zhou C, Zhang J, Ouyang M, Zhong N, Xu J. Detection of epidermal growth factor receptor mutations in plasma by mutant-enriched PCR assay for prediction of the response to gefitinib in patients with non-small-cell lung cancer. Int J Cancer 2009; 125:2393 - 9; http://dx.doi.org/10.1002/ijc.24653; PMID: 19530244
- Mack PC, Holland WS, Burich RA, Sangha R, Solis LJ, Li Y, Beckett LA, Lara PN Jr., Davies AM, Gandara DR. EGFR mutations detected in plasma are associated with patient outcomes in erlotinib plus docetaxel-treated non-small cell lung cancer. J Thorac Oncol 2009; 4:1466 - 72; http://dx.doi.org/10.1097/JTO.0b013e3181bbf239; PMID: 19884861
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005; 2:e17; http://dx.doi.org/10.1371/journal.pmed.0020017; PMID: 15696205
- Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, Liu DD, Bekele BN, Herbst RS, Wistuba II. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res 2007; 13:2890 - 6; http://dx.doi.org/10.1158/1078-0432.CCR-06-3043; PMID: 17504988
- Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 2009; 27:2091 - 6; http://dx.doi.org/10.1200/JCO.2009.21.9170; PMID: 19188670
- Sunaga N, Shames DS, Girard L, Peyton M, Larsen JE, Imai H, Soh J, Sato M, Yanagitani N, Kaira K, et al. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol Cancer Ther 2011; 10:336 - 46; http://dx.doi.org/10.1158/1535-7163.MCT-10-0750; PMID: 21306997
- Saki M, Toulany M, Rodemann HP. Acquired resistance to cetuximab is associated with the overexpression of Ras family members and the loss of radiosensitization in head and neck cancer cells. Radiother Oncol 2013; 108:473 - 8; http://dx.doi.org/10.1016/j.radonc.2013.06.023; PMID: 23891090
- Cengel KA, Voong KR, Chandrasekaran S, Maggiorella L, Brunner TB, Stanbridge E, Kao GD, McKenna WG, Bernhard EJ. Oncogenic K-Ras signals through epidermal growth factor receptor and wild-type H-Ras to promote radiation survival in pancreatic and colorectal carcinoma cells. Neoplasia 2007; 9:341 - 8; http://dx.doi.org/10.1593/neo.06823; PMID: 17460778
- Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 2007; 129:957 - 68; http://dx.doi.org/10.1016/j.cell.2007.03.051; PMID: 17540175
- Ciuffreda L, Di Sanza C, Cesta Incani U, Eramo A, Desideri M, Biagioni F, Passeri D, Falcone I, Sette G, Bergamo P, et al. The mitogen-activated protein kinase (MAPK) cascade controls phosphatase and tensin homolog (PTEN) expression through multiple mechanisms. J Mol Med (Berl) 2012; 90:667 - 79; http://dx.doi.org/10.1007/s00109-011-0844-1; PMID: 22215152
- Zmajkovicova K, Jesenberger V, Catalanotti F, Baumgartner C, Reyes G, Baccarini M. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell 2013; 50:43 - 55; http://dx.doi.org/10.1016/j.molcel.2013.01.037; PMID: 23453810
- Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M, Rodriguez S, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011; 30:2547 - 57; http://dx.doi.org/10.1038/onc.2010.626; PMID: 21278786
- Toulany M, Schickfluss TA, Eicheler W, Kehlbach R, Schittek B, Rodemann HP. Impact of oncogenic K-RAS on YB-1 phosphorylation induced by ionizing radiation. Breast Cancer Res 2011; 13:R28; http://dx.doi.org/10.1186/bcr2845; PMID: 21392397
- Eicheler W, Zips D, Dörfler A, Grénman R, Baumann M. Splicing mutations in TP53 in human squamous cell carcinoma lines influence immunohistochemical detection. J Histochem Cytochem 2002; 50:197 - 204; http://dx.doi.org/10.1177/002215540205000207; PMID: 11799138
- Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer 2004; 91:355 - 8; PMID: 15188009
- Toulany M, Dittmann K, Baumann M, Rodemann HP. Radiosensitization of Ras-mutated human tumor cells in vitro by the specific EGF receptor antagonist BIBX1382BS. Radiother Oncol 2005; 74:117 - 29; http://dx.doi.org/10.1016/j.radonc.2004.11.008; PMID: 15734199