
Abstract
We determined whether clinically relevant phosphodiesterase 5 (PDE5) inhibitors interacted with clinically relevant chemotherapies to kill medulloblastoma cells. In medulloblastoma cells PDE5 inhibitors interacted in a greater than additive fashion with vincristine/etoposide/cisplatin to cause cell death. Knockdown of PDE5 expression recapitulated the combination effects of PDE5 inhibitor drugs with chemotherapy drugs. Expression of dominant negative caspase 9 did not significantly inhibit chemotherapy lethality but did significantly reduce enhanced killing in combination with the PDE5 inhibitor sildenafil. Overexpression of BCL-XL and c-FLIP-s suppressed individual and combination drug toxicities. Knockdown of CD95 or FADD suppressed drug combination toxicity. Treatment with PDE5 inhibitors and chemotherapy drugs promoted autophagy which was maximal at ~12 h post-treatment, and in a cell type-dependent manner knockdown of Beclin1 or ATG5 either suppressed or enhanced drug combination lethality. PDE5 inhibitors enhanced the induction of chemotherapy-induced DNA damage in a nitric oxide synthase-dependent fashion. In conclusion, our data demonstrate that the combination of PDE5 inhibitors with standard of care chemotherapy agents for medulloblastoma represents a possible novel modality for future treatment of this disease.
Introduction
Medulloblastoma is a highly malignant central nervous system tumor that predominantly affects children and adolescents. They are the most common primary central nervous system malignancies in children and adolescents, accounting for 18% of primary CNS tumors in this age group.Citation1-Citation3 Medulloblastomas occur predominantly in the cerebellum or posterior fossa of the brain and are commonly associated with the fourth ventricle. Due to the anatomical location, children often present with symptoms of obstructive hydrocephalus and intracranial hypertension. Symptoms and signs include headache, nausea, vomiting, depressed level of consciousness, dysphagia, dysarthria, and ataxia. Current treatment options including aggressive surgical resection followed by high dose chemotherapy and adjuvant neuraxis radiotherapy provide a 5-y progression free survival rate as low as 50%.Citation1-Citation3 Post-surgical treatments demonstrate significant toxicities to patients. Thus there remains an unmet need for more targeted, potent, well tolerated therapies.
The erection dysfunction drugs sildenafil (Viagra), vardenafil (Levitra), and tadalafil (Cialis) inhibit phosphodiesterase 5 (PDE5), the predominant phosphodiesterase enzyme in the corpus cavernosum, which is essential for vascular smooth muscle contraction through elevation of cGMP levels.Citation4 PDE5 inhibitors also protect the heart against ischemia/reperfusion injury and doxorubicin-induced cardiomyopathy. The cardioprotective effect of PDE5 inhibitors is attributed to suppressing apoptosis and necrosis. Downstream of reduced PDE5 function, the effects of elevated cGMP levels include enhanced expression of nitric oxide synthase (NOS) enzymes, particularly endothelial NOS (eNOS) and inducible NOS (iNOS) and activation of protein kinase C isoforms and protein kinase G.Citation5-Citation13
PDE5 expression is also increased in multiple human carcinoma cell types including breast, colon, bladder, and lung cancers.Citation4,Citation14-Citation16 At high non-physiologic levels sildenafil and vardenafil suppress tumor cell growth and induce caspase-dependent apoptosis in B-CLL cells.Citation17 The PDE5 inhibitors sildenafil and vardenafil are also multidrug resistance transporter inhibitors suggesting they may be useful in the treatment of CNS-localized diseases where drug penetration across the blood–brain barrier is an issue.Citation18 More recently in collaboration we have shown in prostate cancer cells and flank tumors that high concentrations of PDE5 inhibitors enhance doxorubicin lethality, and protect the heart from doxorubicin toxicity.Citation19 Mitochondrial reactive oxygen species (ROS) is one key component of anti-tumor activity of doxorubicin in tumor cells. In prostate cancer cells ROS/reactive nitrogen species (ROS/RNS) levels in doxorubicin- and sildenafil-treated cells were greater than those in either PDE5 inhibitor alone or doxorubicin alone.
The present studies were designed to determine whether and how PDE5 inhibitors interacted with standard of care chemotherapeutic agents to kill medulloblastoma cells.
Results
Initial studies examined whether there was a lethal interaction between PDE5 inhibitors and standard of care chemotherapeutic agents for medulloblastoma. In short-term cell killing assays, sildenafil enhanced the lethality of vincristine, cisplatin, and etoposide in two established and two primary medulloblastoma cell lines (). Similar data to that using sildenafil were obtained using the chemically unrelated PDE5 inhibitor tadalafil (). In long-term colony formation assays transient exposure to sildenafil enhanced the lethality of etoposide in a dose-dependent fashion; combination index values for the interaction between sildenafil and etoposide were less than 0.70 indicative of a synergy of interaction ().
Figure 1. PDE5 inhibitors interact with established cytotoxic chemotherapy agents to kill multiple medulloblastoma cell lines. (A–D) D283, DAOY, HOSS1, and VC312 cells were treated with chemotherapy drugs and sildenafil, as indicated. Cells were isolated after 24 h and viability determined by trypan blue exclusion (n = 3, ± SEM) *P < 0.05 greater than corresponding value in vehicle control. (E) DAOY cells were treated with chemotherapy drugs and tadalafil, as indicated. Cells were isolated after 24 h and viability determined by trypan blue exclusion (n = 3, ± SEM) *P < 0.05 greater than corresponding value in vehicle control. (F) DAOY cells, plated as single cells were treated with etoposide and/or sildenafil for 24 h. Colonies were permitted to form (n = 3, ± SEM) #P < 0.05 less than ETO alone value. (G) DAOY cells were transfected with scrambled siRNA or to knock down expression of PDE5. Thirty-six hours after transfection cells were treated with increasing doses of etoposide. Cells were isolated after 24 h and viability determined by trypan blue exclusion (n = 3, ± SEM) *P < 0.05 greater than corresponding value in siSCR control. (H) HOSS1 cells were pre-treated with PBS vehicle (VEH) or the iNOS inhibitor L-NAME (10 μM). Thirty minutes later cells were treated with vehicle, etoposide, sildenafil, or the drugs in combination. Cells were isolated after 24 h and viability determined by trypan blue exclusion (n = 3, ± SEM) #P < 0.05 less than corresponding value in VEH control.
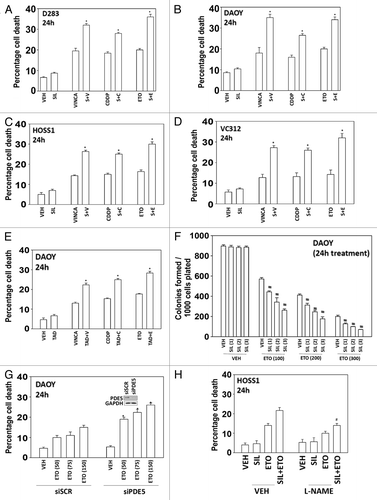
We then determined whether the PDE5 inhibitors were acting in an on-target manner; knockdown of PDE5 enhanced chemotherapy toxicity arguing for an on-target PDE5-dependent effect (). Similar data were obtained in the primary HOSS1 cell isolate (data not shown). One mechanism by which PDE5 inhibitors are thought to act is via cGMP-induced expression of nitric oxide synthase (NOS) and increase intracellular levels of the second messenger NO. Incubation of cells with the NOS inhibitor L-NAME suppressed cell killing, collectively arguing that a cGMP/nitric oxide axis was being stimulated as part of the killing process ().
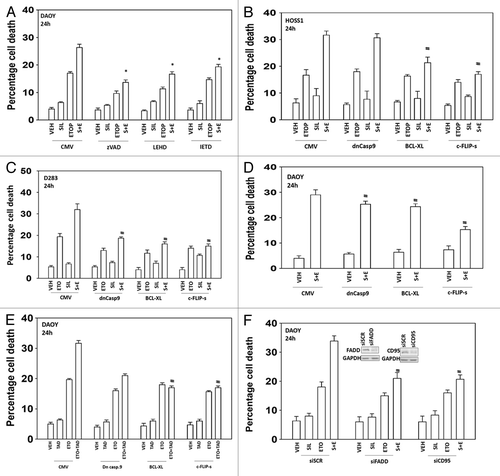
We next attempted to determine the molecular mechanisms by which the drug combination of sildenafil and etoposide killed tumor cells e.g., caspase 9/intrinsic pathway, caspase 8/extrinsic pathway, autophagy, and necrosis. Incubation of cells with the pan-caspase inhibitor zVAD suppressed cell killing by sildenafil and etoposide (). Inhibition of caspase 9 or inhibition of caspase 8 using small molecule inhibitors also suppressed cell killing by sildenafil and etoposide. Expression of a dominant negative caspase 9 protein in a cell type-dependent manner reduced overall killing but did not prevent sildenafil enhancing chemotherapy toxicity (). Overexpression of either the mitochondrial protective protein BCL-XL or the caspase 8 inhibitor c-FLIP-s more consistently prevented sildenafil/PDE 5 inhibitor enhancing chemotherapy toxicity across the cell lines. The data with the caspase 8 inhibitor c-FLIP-s and the mitochondrial protective protein BCL-XL suggests both death receptor and mitochondrial signaling as part of the combinatorial killing process. The combination of sildenafil with chemotherapies increased plasma membrane surface levels of the death receptor CD95 and knockdown of CD95 expression or Fas-associated death domain protein (FADD) expression suppressed the toxic interaction between sildenafil and etoposide (, data not shown).
Figure 2. The toxic interaction between PDE5 inhibitors and chemotherapy is blocked by overexpression of BCL-XL or c-FLIP-s. (A) DAOY cells were treated with zVAD, LEHD, or IETD (all 50 μM) followed by drugs and viability determined by trypan blue exclusion (n = 3, ± SEM). (B–E) HOSS1, D283, and DAOY cells were infected with empty vector adenovirus (CMV) or three other viruses to express dominant negative caspase 9 (dn casp 9), BCL-XL, and c-FLIP-s respectively. Thirty-six hours after infection cells were treated with drugs and viability determined by trypan blue exclusion (n = 3, ± SEM) #P < 0.05 less than corresponding value in CMV control. (F) DAOY cells were transfected with scrambled siRNA (siSCR) or siRNA molecules to knock down expression of CD95 or FADD (siCD95, siFADD). Thirty-six hours after infection cells were treated with drugs and viability determined by trypan blue exclusion (n = 3, ± SEM) #P < 0.05 less than corresponding value in siSCR control.
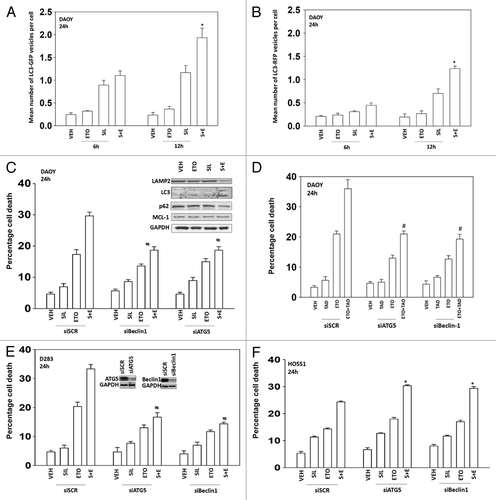
We then determined whether changes in autophagy played any role in sildenafil ± chemotherapy-induced killing. Combined exposure of cells to sildenafil and etoposide resulted initially in enhanced LC3-GFP vesicularization indicative of early autophagosome formation that was temporally followed by LC3-RFP vesicularization indicative of endosome acidification, that correlated with enhanced LC3II protein levels and reduced protein levels of the protein p62 and the protein LAMP2 suggesting that autophagic protein degradation was occurring (). Knock down of the autophagy regulatory proteins Beclin1 or ATG5 protected DAOY and D283 cells from drug combination toxicity (). However, to our surprise, we found that inhibition of autophagy in HOSS1 cells was protective as judged by increased killing with sildenafil and sildenafil + etoposide in cells lacking expression of either ATG5 or Beclin1 (). Thus the appearance of autophagy following sildenafil + etoposide cannot simplistically be associated with being a “toxic” or a “protective” observation.
Figure 3. Sildenafil and chemotherapy-induced lethality is mediated through increased autophagy. (A and B) DAOY cells were transfected to express LC3-GFP-RFP. Twenty-four hours after transfection cells were treated with drugs and cells were examined 6 h and 12 h after treatment using a fluorescent microscope and the mean number of LC3-GFP+ and LC3-RFP+ vesicles determined in >40 cells (n = 3, ± SEM). *P < 0.05 greater than vehicle control. (C–F) DAOY, D283, and HOSS1 cells were transfected with scrambled siRNA (siSCR) or siRNA molecules to knock down expression of ATG5 or Beclin1 (siATG5, siBeclin1). Thirty-six hours after transfection cells were treated with drugs and viability determined by trypan blue exclusion (n = 3, ± SEM) #P < 0.05 less than corresponding value in siSCR control. (C upper) DAOY cells were treated with drugs as indicated. Cells were isolated 12 h after treatment and immunoblotting performed to examine the expression of the indicated proteins (n = 3).
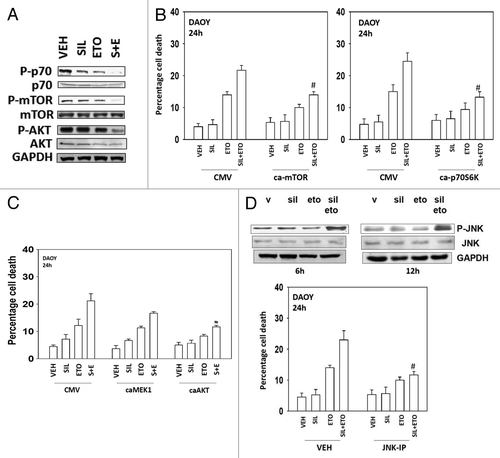
Multiple protein kinases regulate the formation of autophagic vesicles, most notably the kinase mTOR; a kinase downstream of PI3-kinase and AKT. Treatment of cells with sildenafil + etoposide caused inactivation of AKT, mTOR and p70 S6K (). Overexpression of an activated form of the kinase mTOR suppressed drug-induced autophagy and inhibited cell killing (, data not shown). Expression of activated p70S6K also suppressed cell killing, as did expression of an activated form of AKT, but not of MEK1 (). Etoposide treatment activated the JNK1/2 pathway within 6 h, an effect that was prolonged in cells treated in combination with sildenafil (, upper blot). Inhibition of JNK1/2 signaling protected cells from etoposide and sildenafil toxicity (, lower graph).
Figure 4. Modulation of cell signaling pathways controls etoposide and sildenafil lethality. (A) DAOY cells were treated with drugs for 24 h before isolation, SDS PAGE, and determination of the phosphorylation and total expression of the indicated proteins. (B and C) DAOY cells were transfected with empty vector plasmid (CMV) or plasmids to express: an activated form of mTOR (ca-mTOR), an activated form of p70S6K (ca-p70), an activated form of MEK1 (caMEK1), and an activated form of AKT (caAKT). Thirty-six hours after transfection cells were treated with drugs and viability determined by trypan blue exclusion (n = 3, ± SEM) #P < 0.05 less than corresponding value in CMV control. (D) DAOY cells were treated with vehicle or the JNK inhibitory peptide (JNK-IP, 10 μM). Thirty minutes later cells were treated with drugs and viability determined by trypan blue exclusion (n = 3, ± SEM) #P < 0.05 less than corresponding value in vehicle control.
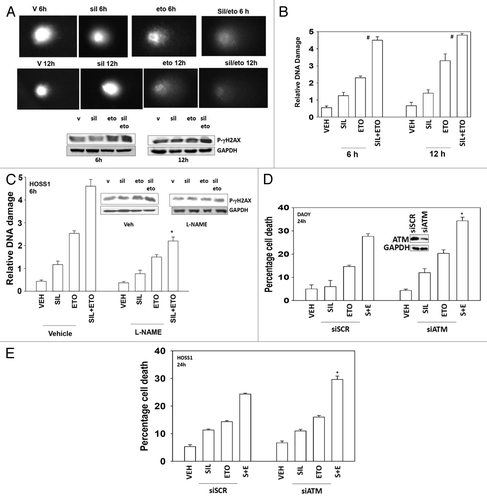
Standard of care chemotherapy agents for the treatment of medulloblastoma are known to cause DNA damage as part of their killing mechanism. Sildenafil rapidly and significantly enhanced the amount of DNA damage caused by etoposide as judged in Comet assays (, images, and B, quantified tail moments). The drug combination increased the phosphorylation of the histone γH2AX 6 and 12 h after exposure (). The phosphorylation of γH2AX is thought to be regulated via the ataxia telangiectasia mutated (ATM) protein. As previously published by ourselves and others, knock down of ATM reduced drug-induced γH2AX phosphorylation (data not shown). Inhibition of NO production, using L-NAME, suppressed the effect of sildenafil on the tail lengthening (DNA damage) caused by etoposide but did not fully obviate the effect (). Knockdown of ATM modestly increased the toxicity of etoposide and further enhanced the toxicity of sildenafil combined with etoposide ().
Figure 5. Sildenafil increases and prolongs chemotherapy-induced DNA damage; knockdown of ATM enhances drug combination toxicity. (A) DAOY cells were grown in soft agar, and treated as indicated with drugs for 6 h and 12 h. Cells were subjected to electrophoresis and stained. Images are a representative (n = 4). Blot: DAOY cells were treated with drugs and were isolated 6 h and 12 h after exposure and the phosphorylation of γH2AX determined, with the fold increase in phosphorylation shown (n = 3, ± SEM). (B) Graph: The tail moments of cells imaged in (A) were measured and plotted (n = 4, ± SEM) #P < 0.05 greater than corresponding value in vehicle-treated cells. (C) HOSS1 cells were grown in soft agar, in the presence or absence of L-NAME (10 mM), and treated as indicated with drugs, as indicated, for 6 h and 12 h. Cells were subjected to electrophoresis and stained. The tail moments of cells were measured and presented below each image (n = 4, ± SEM) *P < 0.05 less than corresponding value in vehicle-treated cells. (D and E) DAOY cells and HOSS1 cells were transfected with scrambled siRNA (siSCR) or to knock down expression of ATM (siATM). Thirty-six hours after transfection cells were treated with drugs and viability determined by trypan blue exclusion (n = 3, ± SEM) *P < 0.05 greater than corresponding value in siSCR control.
Discussion
The present studies were performed to determine whether clinically relevant PDE5 inhibitors interacted with standard of care chemotherapy agents to kill medulloblastoma cells. PDE5 inhibitors interacted with multiple standard of care chemotherapy agents in a greater than additive fashion to kill medulloblastoma cells. One initial concern over our studies was that PDE5 inhibitors were enhancing the toxicity of multiple chemotherapies, implying that we may be observing an off-target phenomenon with respect to PDE5 inhibitor action; however, knockdown of PDE5 argued that loss of PDE5 function was responsible for enhancing chemotherapy toxicity. The interaction between PDE5 inhibitors and standard of care chemotherapy agents was dependent on activation of the extrinsic pathway/death receptors. However, although inhibition of caspase 8, through c-FLIP-s, or mitochondrial protection using BCL-XL abolished the drug interaction, inhibition of caspase 9 only partially reduced overall killing in a cell type-dependent fashion. Molecular inhibition of autophagy was protective in some cell isolates, but in others toxic. Thus, the ability of PDE5 inhibitors to facilitate cytotoxic chemotherapy killing—killing that acts through mitochondrial dysfunction—utilizes the caspase 8 pathway downstream of death receptors.
A frequent mode of cell killing by “traditional” cytotoxic chemotherapies involves the damaging of DNA, and the subsequent inability of cells to fully repair this DNA damage. With etoposide we used two read-outs for DNA damage, the Comet assay and γH2AX phosphorylation. By both measures, drug treatment caused DNA damage. The amount of damage caused by the chemotherapeutic drug was enhanced and prolonged by sildenafil. Sildenafil itself weakly altered the DNA damage read-out as judged by γH2AX phosphorylation, and chemically this drug would not be expected to damage DNA. It is known that protein nitration can alter the formation of DNA repair complexes as well as regulate the phosphatases which regulate protein complex formation and our data argues that sildenafil treatment results in greater levels of DNA damage that are not repaired over time. In general agreement with this hypothesis we note that the amount of cell killing observed in colony formation assays is much greater than that observed in short-term death assays at 24 h, indicating that prolonged DNA damage is resulting in an inability of cells to form colonies. Further studies beyond the scope of the present manuscript will be required to define how changes in protein repair complex formation are altered by sildenafil co-exposure.
The ATM protein controls multiple aspects of cell biology after DNA damage including cell cycle arrest, activation of signaling pathways, activation of apoptosis pathways, and DNA repair protein complex formation (and, as previously described, ROS production). We noted that γH2AX phosphorylation was increased by the drug combination of sildenafil and etoposide compared with DNA damaging agent alone. In general it is believed that, beyond a certain threshold, DNA damage-induced activation of ATM in a cell switches from an arrest/survival outcome to a programmed cell death outcome. For example, genistein can induce apoptosis through ATM signaling.Citation20,Citation21 In T cells etoposide treatment through ATM can also cause apoptosis.Citation22 ATM signaling can also facilitate liver damage in already damaged fibrotic livers.Citation23 Further studies will be required to define how ATM regulates cell viability after sildenafil + DNA damaging agent treatment.
Changes in cell signaling were also important for modulating the lethality of the drug combination. The drug combination reduced mTOR, p70S6K, and AKT activity. Expression of activated forms of these proteins protected cells from the toxic effects of drug combination exposure. At least one portion of the mechanism by which these molecules protected cells was via inhibition of autophagy (see below). The drug combination also enhanced the activity of JNK1/2, a signaling pathway normally associated with tumor cell killing. Inhibition of JNK1/2 protected cells from the toxic effects of the drug combination. Further studies beyond the scope of the present manuscript will be required to understand precisely how PDE5 inhibitors modulate cell signaling processes after exposure to DNA damaging chemotherapy agents.
The role of autophagy in the biology of tumor cells has become an intensely investigated and somewhat controversial area over the last few years. It is now well known that chemotherapy agents can induce autophagy that could be linked to senescence responses that are protective for tumor cells. In our studies we found that sildenafil enhanced etoposide-induced autophagy that was evident within 6 h of drug treatment; based on data examining the expression of p62, LAMP2, and the appearance of GFP+/RFP+ vesicles we assume both autophagosome and acidic endosome formation with protein digestion occurred. Knockdown of either Beclin1 or ATG5 suppressed GFP+/RFP+ vesicle formation and in DAOY and D283 cells reduced the lethality of the sildenafil + etoposide drug combination. To our great surprise we found that in HOSS1 cells autophagy protected cells from the drug combination. Thus in our system the appearance of autophagic vesicles cannot simply be regarded as a “toxic” or “protective” observation; this fact may have general applicability. The molecular reasons why we observe either a protective or a toxic effect of autophagy based on the cell line will require considerable detailed examination that is beyond the scope of the present studies. The precise mechanisms by which sildenafil regulates autophagy in our system(s), possibly by modulating cGMP and NO levels, will also need to be investigated in a future manuscript.
In conclusion, our findings strongly argue that at physiologic concentrations clinically relevant PDE5 inhibitors enhance the lethality of multiple well established chemotherapy agents against established and primary pediatric CNS tumor cells. The drug interaction was dependent in part on expression of death receptors and was observed in multiple medulloblastoma cell types. Further laboratory based and clinical studies will be required to understand more fully the mechanisms of drug interaction and the clinical utility of this therapeutic approach.
Materials and Methods
Materials
Phospho-/total antibodies were purchased from Cell Signaling Technologies and Santa Cruz Biotech. All drugs were purchased from Selleckchem. Commercially available validated short hairpin RNA molecules to knock down RNA/protein levels were from Qiagen. Antibody reagents, other kinase inhibitors, caspase inhibitors cell culture reagents, and non-commercial recombinant adenoviruses have been previously described.Citation24-Citation27 DAOY and D283 cells were obtained from the ATCC where they were previously authenticated. De-indentified VC312 and HOSS1 cells are MCVH-VCU patient derived medulloblastomas obtained after informed consent and surgery, and were not further authenticated beyond the initial post-operative pathology assessment.
Methods
Cell culture and in vitro exposure of cells to drugs
All cancer lines were cultured at 37 °C (5% v/v CO2) in vitro using RPMI supplemented with 5% (v/v) fetal calf serum and 10% (v/v) Non-essential amino acids. For short-term cell killing assays and immunoblotting, cells were plated at a density of 3 × 103 per cm2 and 24 h after plating were treated with various drugs, as indicated. Cells were not cultured in reduced serum media during any study. Unless otherwise stated in the figure legend, cells as indicated in each panel were treated with vincristine (VINCA 100 nM), cisplatin (CDDP, 3 μM), etoposide (ETO, 100 nM), and/or sildenafil (SIL, 2.0 μM) or tadalafil (TAD, 2.0 μM).
Colony formation assay
Tumor cells plated as single cells (250–4000 cells/well) in sextuplicate were treated with etoposide (ETO 100–300 nM) and/or sildenafil (SIL, 1.0–3.0 μM) for 24 h, after which the media was removed and replaced with drug free media. Colonies were permitted to form for the following 10–14 d. Colonies were fixed, stained, and counted (>50 cells per colony).
Cell treatments, SDS-PAGE, and western blot analysis
Cells were treated with various drugs, and isolated at the indicated time points. SDS PAGE and immunoblotting was performed as described in refs. Citation24–Citation27.
Recombinant adenoviral vectors—infection in vitro
We generated and purchased previously noted recombinant adenoviruses as per references Citation24–Citation27. Cells were infected with these adenoviruses at an approximate m.o.i. as indicated in each specific figure/legend (usually an m.o.i. of 50). Cells were incubated for 24 h to ensure adequate expression of transduced gene products prior to drug exposures.
Detection of cell death by trypan blue assay
Cells were harvested by trypsinization with trypsin/EDTA for ~10 min at 37 °C. Harvested cells were combined with the culture media containing unattached cells and the mixture centrifuged (800 rpm, 5 min). Cell pellets were resuspended in PBS and mixed with trypan blue agent. Viability was determined microscopically using a hemocytometer.Citation24-Citation27 Five hundred cells from randomly chosen fields were counted and the number of dead cells was counted and expressed as a percentage of the total number of cells counted. Cell killing was confirmed using the Sceptor instrument (Millipore) with a 60 μm tip, which measured tumor cell size/sub G1 DNA as an indication of tumor cell viability.
Assessment of autophagy
Cells were transfected with a plasmid to express a green and red fluorescent protein (GFP, RFP) tagged form of LC3 (ATG8). Twenty-four hours after transfection cells were treated with drugs for 6h and 12h, as indicated. For analysis of cells transfected with the GFP-LC3-RFP construct, the GFP-LC3-RFP-positive vesicularized cells were examined under the 40× objective of a Zeiss Axiovert fluorescent microscope.
Plasmid transfection
Plasmids
Cells were plated as described above and 24 h after plating, transfected. Plasmids (0.5 μg) expressing a specific mRNA or appropriate vector control plasmid DNA was diluted in 50 μL serum-free and antibiotic-free medium (1 portion for each sample). Concurrently, 2 μL Lipofectamine 2000 (Invitrogen) was diluted into 50 μL of serum-free and antibiotic-free medium. Diluted DNA was added to the diluted Lipofectamine 2000 for each sample and incubated at room temperature for 30 min. This mixture was added to each well/dish of cells containing 200 μL serum-free and antibiotic-free medium for a total volume of 300 μL and the cells were incubated for 4 h at 37 °C. An equal volume of 2× medium was then added to each well. Cells were incubated for 48 h, then treated with drugs. To assess transfection efficiency of plasmids we used a plasmid to express GFP and defined the percentage of cells being infected as the percentage of GFP+ cells. For all cell lines the infection efficiency was >70%.
siRNA
Cells were plated in 60 mm dishes from a fresh culture growing in log phase as described above, and 24 h after plating transfected. Prior to transfection, the medium was aspirated and 1 ml serum-free medium was added to each plate. For transfection, 10 nM of the annealed siRNA, the positive sense control doubled stranded siRNA targeting GAPDH or the negative control (a “scrambled” sequence with no significant homology to any known gene sequences from mouse, rat or human cell lines) were used (predominantly Qiagen; occasional alternate siRNA molecules were purchased from Ambion, Inc.). Ten nanomolar of siRNA (scrambled or experimental) was diluted in serum-free media. Four microliters of Hiperfect (Qiagen) was added to this mixture and the solution was mixed by pipetting up and down several times. This solution was incubated at room temp for 10 min, then added drop-wise to each dish. The medium in each dish was swirled gently to mix, then incubated at 37 °C for 2 h. One milliliter of 10% (v/v) serum-containing medium was added to each plate, and cells were incubated at 37 °C for 24–48 h before re-plating (50 × 103 cells each) onto 12-well plates. Cells were allowed to attach overnight, then treated with drugs (0–24 h). Trypan blue exclusion assays and SDS PAGE/immunoblotting analyses were then performed at the indicated time points.
Data analysis
Comparison of the effects of various treatments was performed using one way analysis of variance and a two tailed Student t test. Differences with a P value of < 0.05 were considered statistically significant. Experiments shown are the means of multiple individual points from multiple experiments (± SEM).
| Abbreviations: | ||
| ERK | = | extracellular regulated kinase |
| MEK | = | mitogen activated extracellular regulated kinase |
| PI3K | = | phosphatidyl inositol 3 kinase |
| MAPK | = | mitogen activated protein kinase |
| ca | = | constitutively active |
| dn | = | dominant negative |
| CMV | = | empty vector plasmid or virus |
| si | = | small interfering |
| SCR | = | scrambled |
| Ad | = | adenovirus |
| TUNEL | = | terminal deoxynucleotidyl transferase UTP nick end labeling |
| VEH | = | vehicle |
| SIL | = | sildenafil |
| VAR | = | vardenafil |
| TAD | = | tadenafil |
| DOX | = | doxorubicin |
| MITO | = | mitomycin C |
| Gemzar | = | gemcitabine |
| CDDP | = | cisplatinum |
| PDE | = | phosphodiesterase |
| NECRO | = | necrostatin 1 |
| FADD | = | Fas-associated death domain protein |
| CD95 | = | cluster of determinants 95 |
| AIF | = | apoptosis inducing factor |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Support for the present study was funded from PHS grants from the National Institutes of Health (R01-CA141704, R01-CA150214, R01-DK52825). Thanks to Mrs Grizzard for her support. P.D. is the holder of the Universal Inc. Professorship in Signal Transduction Research.
References
- Alexiou GA, Vartholomatos G, Stefanaki K, Patereli A, Dova L, Karamoutsios A, Lallas G, Sfakianos G, Moschovi M, Prodromou N. Expression of heat shock proteins in medulloblastoma. J Neurosurg Pediatr 2013; 12:452 - 7; http://dx.doi.org/10.3171/2013.7.PEDS1376; PMID: 23992239
- De Braganca KC, Packer RJ. Treatment Options for Medulloblastoma and CNS Primitive Neuroectodermal Tumor (PNET). Curr Treat Options Neurol 2013; 15:593 - 606; http://dx.doi.org/10.1007/s11940-013-0255-4; PMID: 23979905
- American Cancer Society. Cancer Facts and Figures 2013. Atlanta, Ga: American Cancer Society, 2013. Available online.
- Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 2006; 58:488 - 520; http://dx.doi.org/10.1124/pr.58.3.5; PMID: 16968949
- Das A, Ockaili R, Salloum F, Kukreja RC. Protein kinase C plays an essential role in sildenafil-induced cardioprotection in rabbits. Am J Physiol Heart Circ Physiol 2004; 286:H1455 - 60; http://dx.doi.org/10.1152/ajpheart.01040.2003; PMID: 15020304
- Das A, Xi L, Kukreja RC. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem 2005; 280:12944 - 55; http://dx.doi.org/10.1074/jbc.M404706200; PMID: 15668244
- Das A, Salloum FN, Xi L, Rao YJ, Kukreja RC. ERK phosphorylation mediates sildenafil-induced myocardial protection against ischemia-reperfusion injury in mice. Am J Physiol Heart Circ Physiol 2009; 296:H1236 - 43; http://dx.doi.org/10.1152/ajpheart.00100.2009; PMID: 19286961
- Das A, Xi L, Kukreja RC. Protein kinase G-dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J Biol Chem 2008; 283:29572 - 85; http://dx.doi.org/10.1074/jbc.M801547200; PMID: 18723505
- Ockaili R, Salloum F, Hawkins J, Kukreja RC. Sildenafil (Viagra) induces powerful cardioprotective effect via opening of mitochondrial K(ATP) channels in rabbits. Am J Physiol Heart Circ Physiol 2002; 283:H1263 - 9; PMID: 12181158
- Salloum FN, Abbate A, Das A, Houser JE, Mudrick CA, Qureshi IZ, Hoke NN, Roy SK, Brown WR, Prabhakar S, et al. Sildenafil (Viagra) attenuates ischemic cardiomyopathy and improves left ventricular function in mice. Am J Physiol Heart Circ Physiol 2008; 294:H1398 - 406; http://dx.doi.org/10.1152/ajpheart.91438.2007; PMID: 18223185
- Salloum F, Yin C, Xi L, Kukreja RC. Sildenafil induces delayed preconditioning through inducible nitric oxide synthase-dependent pathway in mouse heart. Circ Res 2003; 92:595 - 7; http://dx.doi.org/10.1161/01.RES.0000066853.09821.98; PMID: 12637371
- Salloum FN, Ockaili RA, Wittkamp M, Marwaha VR, Kukreja RC. Vardenafil: a novel type 5 phosphodiesterase inhibitor reduces myocardial infarct size following ischemia/reperfusion injury via opening of mitochondrial K(ATP) channels in rabbits. J Mol Cell Cardiol 2006; 40:405 - 11; http://dx.doi.org/10.1016/j.yjmcc.2005.10.002; PMID: 16480739
- Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation 2005; 111:1601 - 10; http://dx.doi.org/10.1161/01.CIR.0000160359.49478.C2; PMID: 15811867
- Karami-Tehrani F, Moeinifard M, Aghaei M, Atri M. Evaluation of PDE5 and PDE9 expression in benign and malignant breast tumors. Arch Med Res 2012; 43:470 - 5; http://dx.doi.org/10.1016/j.arcmed.2012.08.006; PMID: 22960860
- Eggen T, Sager G, Berg T, Nergaard B, Moe BT, Ørbo A. Increased gene expression of the ABCC5 transporter without distinct changes in the expression of PDE5 in human cervical cancer cells during growth. Anticancer Res 2012; 32:3055 - 61; PMID: 22843873
- Zhang X, Yan G, Ji J, Wu J, Sun X, Shen J, Jiang H, Wang H. PDE5 inhibitor promotes melanin synthesis through the PKG pathway in B16 melanoma cells. J Cell Biochem 2012; 113:2738 - 43; http://dx.doi.org/10.1002/jcb.24147; PMID: 22441938
- Sarfati M, Mateo V, Baudet S, Rubio M, Fernandez C, Davi F, Binet JL, Delic J, Merle-Beral H. Sildenafil and vardenafil, types 5 and 6 phosphodiesterase inhibitors, induce caspase-dependent apoptosis of B-chronic lymphocytic leukemia cells. Blood 2003; 101:265 - 9; http://dx.doi.org/10.1182/blood-2002-01-0075; PMID: 12393651
- Chen JJ, Sun YL, Tiwari AK, Xiao ZJ, Sodani K, Yang DH, Vispute SG, Jiang WQ, Chen SD, Chen ZS. PDE5 inhibitors, sildenafil and vardenafil, reverse multidrug resistance by inhibiting the efflux function of multidrug resistance protein 7 (ATP-binding Cassette C10) transporter. Cancer Sci 2012; 103:1531 - 7; http://dx.doi.org/10.1111/j.1349-7006.2012.02328.x; PMID: 22578167
- Das A, Durrant D, Mitchell C, Mayton E, Hoke NN, Salloum FN, Park MA, Qureshi I, Lee R, Dent P, et al. Sildenafil increases chemotherapeutic efficacy of doxorubicin in prostate cancer and ameliorates cardiac dysfunction. Proc Natl Acad Sci U S A 2010; 107:18202 - 7; http://dx.doi.org/10.1073/pnas.1006965107; PMID: 20884855
- Zhang Z, Wang CZ, Du GJ, Qi LW, Calway T, He TC, Du W, Yuan CS. Genistein induces G2/M cell cycle arrest and apoptosis via ATM/p53-dependent pathway in human colon cancer cells. Int J Oncol 2013; 43:289 - 96; PMID: 23686257
- Wang Y, Wang H, Zhang W, Shao C, Xu P, Shi CH, Shi JG, Li YM, Fu Q, Xue W, et al. Genistein sensitizes bladder cancer cells to HCPT treatment in vitro and in vivo via ATM/NF-κB/IKK pathway-induced apoptosis. PLoS One 2013; 8:e50175; http://dx.doi.org/10.1371/journal.pone.0050175; PMID: 23365634
- Korwek Z, Sewastianik T, Bielak-Zmijewska A, Mosieniak G, Alster O, Moreno-Villanueva M, Burkle A, Sikora E. Inhibition of ATM blocks the etoposide-induced DNA damage response and apoptosis of resting human T cells. DNA Repair (Amst) 2012; 11:864 - 73; http://dx.doi.org/10.1016/j.dnarep.2012.08.006; PMID: 23058634
- Daugherity EK, Balmus G, Al Saei A, Moore ES, Abi Abdallah D, Rogers AB, Weiss RS, Maurer KJ. The DNA damage checkpoint protein ATM promotes hepatocellular apoptosis and fibrosis in a mouse model of non-alcoholic fatty liver disease. Cell Cycle 2012; 11:1918 - 28; http://dx.doi.org/10.4161/cc.20259; PMID: 22544329
- Bareford MD, Park MA, Yacoub A, Hamed HA, Tang Y, Cruickshanks N, Eulitt P, Hubbard N, Tye G, Burow ME, et al. Sorafenib enhances pemetrexed cytotoxicity through an autophagy-dependent mechanism in cancer cells. Cancer Res 2011; 71:4955 - 67; http://dx.doi.org/10.1158/0008-5472.CAN-11-0898; PMID: 21622715
- Cruickshanks N, Tang Y, Booth L, Hamed H, Grant S, Dent P. Lapatinib and obatoclax kill breast cancer cells through reactive oxygen species-dependent endoplasmic reticulum stress. Mol Pharmacol 2012; 82:1217 - 29; http://dx.doi.org/10.1124/mol.112.081539; PMID: 22989520
- Park MA, Zhang G, Martin AP, Hamed H, Mitchell C, Hylemon PB, Graf M, Rahmani M, Ryan K, Liu X, et al. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol Ther 2008; 7:1648 - 62; http://dx.doi.org/10.4161/cbt.7.10.6623; PMID: 18787411
- Zhang G, Park MA, Mitchell C, Hamed H, Rahmani M, Martin AP, Curiel DT, Yacoub A, Graf M, Lee R, et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin Cancer Res 2008; 14:5385 - 99; http://dx.doi.org/10.1158/1078-0432.CCR-08-0469; PMID: 18765530