
Abstract
A novel pan ERBB inhibitor PF-00299804 (dacomitinib) is currently in phase II clinical trials in glioblastoma multiforme (GBM) patients; however its pre-clinical efficacy in GBMs has not been tested. In this study, we evaluated the efficacy of dacomitinib alone or in combination with PI3K/mTOR dual inhibitor PF-05212384 in GBM and assessed the mechanisms of resistance and the molecular determinants of response. A panel of established and patient derived primary GBM lines that present different molecular profiles and also the GBM lines engineered to express EGFRvIII mutant or PTEN were treated with either dacomitinib, PF-05212384, or combination and assessed for their viability and changes in EGFR/PI3K/mTOR signaling. We show that dacomitinib significantly reduced phosphorylated EGFR in all the GBM lines but did not show a dose-dependent response on cell viability in a majority of the lines tested. Multiple lesions in the receptor tyrosine kinases (RTKs) pathway including PTEN mutation, co-activation of RTKs, and EGFRvIII mutation resulted in unaltered active status of PI3K/mTOR in the GBM lines even in the presence of EGFR inhibition. Blocking PI3K/mTOR dramatically inhibited cell proliferation in most GBM lines and enhanced dacomitinib induction of apoptosis in a GBM line that has both EGFR amplification and EGFR-independent PI3K activation. These data suggest molecular profiling of EGFR/PI3K/PTEN status to select GBM patients for EGFR or/and PI3K/mTOR targeted therapies.
Introduction
Glioblastoma multiforme (GBM) is the most common primary tumor in adults. Standard treatment for glioblastoma patients includes surgery followed by radiotherapy with concomitant or adjuvant chemotherapy, such as temozolomide.Citation1 Nevertheless, GBM shows intrinsic resistance to those therapies and has a poor prognosis with median survival of approximately 1 y after diagnosis. Epidermal growth factor receptor (EGFR), also called ErbB1/HER1, is known to play a significant role in GBM tumorigenesis, and high EGFR expression has been shown to be associated with resistance to concomitant chemoradiotherapy in GBM.Citation2 EGFR, therefore, represents an attractive target for GBM therapy. Although small molecule EGFR tyrosine kinase inhibitors gefinitib (Iressa, Astra-Zeneca) and erlotinib (Tarceva, OSI Pharmaceuticals) have an established role in the treatment of non-small cell lung cancer (NSCLC), monotherapy with erlotinib or gefitinib has shown modest therapeutic benefit in GBM patients.Citation3
PF-00299804 (dacomitinib), an irreversible second generation pan-ERBB inhibitor has been shown to inhibit both wild-type and different mutant variants of EGFR. Furthermore, preclinical studies have shown that dacomitinib is highly effective in a variety of human xenograft models that overexpress ERBB family members or contain EGFR mutations associated with resistance to gefitinib.Citation4,Citation5 Currently, there are a number of clinical trials evaluating dacomitinib in patients with different types of ERBB-driven advanced solid tumors and patients with recurrent GBM (Table S1). Loss of functional phosphatase and tensin homolog (PTEN) drives PI3K, AKT, and mTOR activation in an EGFR-independent manner and confers resistance to inhibition of EGFR.Citation6 GBM patients that bear PTEN mutations do not respond to EGFR inhibitor erlotinib.Citation7 A potent pan-class I PI3K/mTOR inhibitor, PF-05212384, has been shown efficacy in different tumor models and currently also in phase II clinical trials (Table S2) in patients with different solid tumors.
Given the use of both dacomitinib and PF-05212384 in ongoing clinical trials in patients with GBM and other cancer types, it is critical to evaluate their therapeutic efficacy alone or in combination in GBM lines that present different molecular profiles. In this study, we examined the efficacy of dacomitinib in a panel of established and patient derived GBM cell lines that present different molecular profiles. To further understand the role of PI3K and mTOR in GBM cell growth, we utilized PF-05212384 alone or in combination with dacomitinib and evaluated the molecular determinants of response.
Results
Response of established and primary GBM cells to dacomitinib
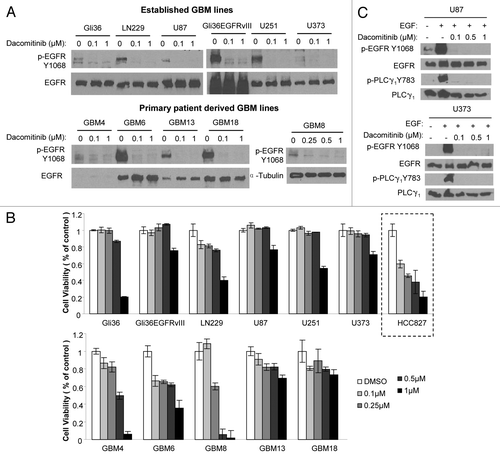
To investigate dacomitinib efficacy in GBM cells, we first evaluated the ability of dacomitinib to inhibit EGFR phosphorylation and its subsequent effect on the viability of different established (Gli36, Gli36EGFRvIII, LN229, U87, U251, and U373) and patient derived primary (GBM4, GBM6, GBM8, GBM13, and GBM18) GBM cell lines. Dacomitinib effectively inhibited EGFR phosphorylation but did not have a dose dependent effect on the GBM cell viability in a majority of the lines tested. At high dacomitinib dose (1 µM), a significant decrease in cell viability was seen in some the GBM lines (). Caspase3/7 assays and GBM cell morphology changes post-dacomitinib treatment indicated apoptosis mediated cell death (Figs. S1 and S2). In comparison, a dose dependent decrease in cell viability to dacomitinib was seen in HCC827 (EGFR Del E746-A750), a lung cancer line which has been previously shown to respond to dacomitinibCitation4 (). To confirm whether EGFR kinase activity in GBM lines is inhibited by dacomitinib, we also examined one of EGFR direct effectors, PLCγ1. Dacomitinib completely blocked EGFR-dependent activation of PLCγ1 (). These results reveal that dacomitinib inhibits phosphorylation of EGFR in both established and primary GBM lines but does not significantly influence the viability of GBM cells.
Figure 1. Established and primary GBM lines in response to PF299804 (dacomitinib). (A) Dacomitinib suppresses EGFR phosphorylation. All GBM cells were treated with dacomitinib for overnight (except that GBM8 cells were treated for 4 h), and cell lysates were analyzed by western blot. (B) Cell viability of GBM lines in response to 72 h treatment of different doses of dacomitinib. Mean ± SD. (C) Western blot analysis shows PLCγ1 activation is dependent on EGFR activation, and dacomitinib inhibits EGFR and its downstream effector PLCγ1 activation.
Differential PI3K/mTOR signalings in response to dacomitinib in GBMs
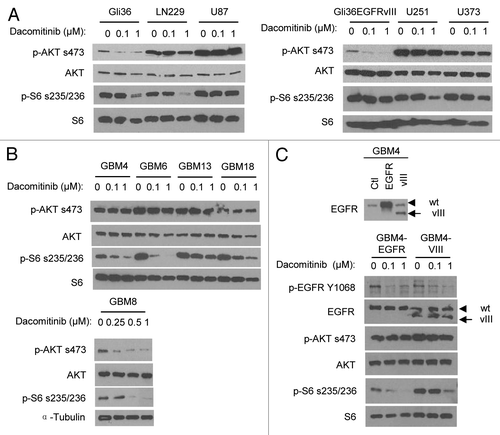
Previous studies have shown that downregulation of PI3K/AKT signaling pathway is necessary for EGFR inhibitors to inhibit the growth of EGFR/ERBB-driven tumor cells.Citation8 The downstream effector of mammalian target of rapamycin 1 (mTOR1) pathway ribosomal protein S6 (rpS6) has been recently shown as another biomarker of EGFR inhibition.Citation9 To understand the varied responses of GBM cells to dacomitinib, we assessed the effect of dacomitinib on the PI3K/mTOR signaling. lists the known oncogenic characteristic of RTKs (PI3K/PTEN; RAS/RAF) in all the lines tested in this study. PTEN is the negative regulator of PI3K/AKT pathway and PTEN mutation or deletion occurs in more than 30% GBM patients.Citation10 Among the 6 established lines, 3 lines (U87, U251, and U373) have been characterized for loss of functional PTEN.Citation11 Dacomitinib treatment reduced PI3K/AKT signaling in PTEN intact (Gli36, LN229, and Gli36EGFRvIII) GBM cells, but had no significant effect on p-AKT in PTEN mutated (U87, U251, and U373) GBM cells. A significant suppression of mTOR1/rpS6 by dacomitinib at 1 µM was observed in Gli36, LN229, and U251 cells (). Since dacomitinib IC50 for cellular EGFR was shown at nanomolar range,Citation4 this could be an off-target effect. Although there is no genomic mutation information for primary GBM lines, it is still possible to examine whether the EGFR/PI3K/mTOR pathway is functional and analyze its response to dacomitinib treatment. Among the 5 primary patient derived lines, treatment with dacomitinib reduced p-AKT and p-rpS6 in GBM8 cells and had no effect on p-AKT in the rest of the primary lines. Surprisingly, despite unaltered status of activated AKT, treatment with dacomitinib resulted in the suppression of mTOR/rpS6 in those primary GBM cells (). These results suggest that dacomitinib can suppress EGFR/ERBB-family dependent mTOR pathway which is independent of AKT. Overall, these results reveal that dacomitinib can inhibit EGFR activity in all GBM lines, while the effect on PI3K/AKT and mTOR1/rpS6 signalings is not always correlated with suppression of EGFR. Although, the majority GBM cell lines do not show a dose-dependent response to dacomitinib mono-therapy, these data reflect the extent of how dacomitinib affects both AKT-mediated cell survival and mTOR-regulated cell proliferation.
Table 1. Comparison of oncogenic RTKs pathways in GBM cell lines
Figure 2. Differential PI3K/mTOR signalings in response to dacomitinib in GBMs. Western blot analysis shows differential EGFR/PI3K/AKT/mTOR signalings in established GBM lines (A), patient-derived primary GBM lines (B), and engineered GBM4-EGFR or GBM4-EGFRvIII cells (C) in response to dacomitinib. All GBM cells were treated with indicated doses of dacomitinib and cell lysates were analyzed by western blot.
EGFRvIII mutant in GBMs is less responsive than wild-type EGFR to dacomitinib
EGFR is amplified in 50% of all GBM cases and 40% of these cases express a constitutively active mutant, EGFRvIII.Citation12 To examine the response of GBMs expressing EGFRvIII to dacomitinib, we engineered wild-type EGFR and EGFRvIII mutant lentiviral vectors and introduced wild-type EGFR or EGFRvIII mutant into a patient-derived GBM line, GBM4 which has the minimal endogenous EGFR levels compared with other patient-derived GBM lines. Western blot analysis on the lysates of engineered GBM4-EGFR and GBM4-EGFRvIII confirmed the overexpression of wild-type EGFR or mutant EGFRvIII in these lines (). Similar to the parental GBM4 ( and ), treatment with dacomitinib significantly attenuated EGFR phosphorylation and EGFR-dependent ribosomal S6 phosphorylation in GBM4-EGFR, while AKT did not respond to dacomitinib treatment. In contrast, overexpression of EGFRvIII increased baseline level of rpS6 phosphorylation, and dacomitinib had less effect on EGFRvIII induced p-rpS6 compared with wild-type EGFR (). This data suggests that the presence of EGFRvIII mutant in GBM cells could lead to increased resistance to dacomitinib through hyperactivating the mTOR/rpS6 pathway.
Targeting PI3K/mTOR in GBM by dual inhibitor PF-05212384 is efficacious than targeting EGFR by dacomitinib
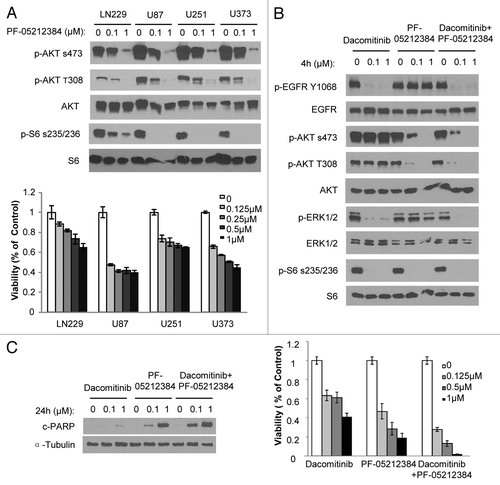
In addition to EGFR amplification and mutation, co-activation of RTKs is commonly found in GBMs.Citation10,Citation13 More than half of the established lines (LN229, U87, U251, and U373) tested in this study have been shown to have co-activating multiple RTKs (). Therefore, it is possible that co-activation of RTKs results in dacomitinib resistance in GBMs through the common downstream pathways PI3K and mTOR, particularly in PTEN mutant lines. Next, we tested whether blocking PI3K and mTOR pathways has better efficacy in GBM lines characterized with co-activating RTKs. We utilized a dual PI3K/mTOR kinase inhibitor PF-05212384, currently in phase II clinical trials in patients with solid tumors, which is a potent pan-class I PI3K/mTOR inhibitor and has been shown efficacy in multiple preclinical tumor models.Citation14 PF-05212384 was able to inhibit PI3K/mTOR pathway as measured by decreased p-AKT and p-S6 and resulted in reduced cell viability in LN229, U87, U251, and U373 GBM cells (). These results show that GBM lines with co-activation of RTKs are sensitive to PI3K/mTOR dual inhibition in comparison to EGFR inhibition alone.
Figure 3. PI3K/mTOR dual inhibitor PF-05212384 inhibits EGFR-independent PI3K and mTOR activation in GBM cells. (A) Upper panel: GBM cells were exposed to indicate concentrations of PF-05212384 and analyzed by western blotting of PI3K and mTOR pathway markers. Bottom panel: Viability of cells in response to different doses of PF-05212384 for 48 h. (B) Western blot analysis of EGFR signaling in the presence of dacomitinib or/and PF-05212384 in GBM6 cells. (C) Western blot analysis of cleaved-PARP and cell viability of GBM6 cells in response to treatment of dacomitinib or/and PF-05212384 (96 h). Mean ± SD.
PF-05212384 enhances dacomitinib effect in GBM with amplified EGFR and EGFR-independent PI3K activation
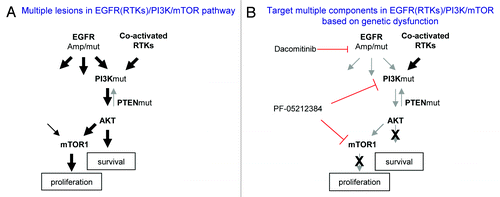
Based on the observation that PI3K/AKT and mTOR1/rpS6 pathways are critical for dacomitinib effect on GBM cell survival and proliferation, we tested if the suppression of PI3K/mTOR could sensitize GBM cells to EGFR inhibition. In most GBM lines, blocking EGFR and PI3K/mTOR by dacomitinib and PF-05212384 together did not show increased efficacy than blocking PI3K/mTOR alone. In a primary GBM line, GBM6, which has EGFR amplification and a constitutively activated PI3K,Citation15 dacomitinib (0.1–1 µM) completely inhibited EGFR and its downstream RAS/MAPK and mTOR1/rpS6, indicating that both pathways depend on EGFR in GBM6. However, PI3K/AKT is constitutively active even in the presence of dacomitinib (1 µM). Treatment of GBM6 cells with PF-05212384 inhibits both PI3K and mTORs, but has no effect on EGFR and MAPK. The combination of dacomitinib and PF-05212384 completely blocks EGFR, PI3K, MAPK, and mTOR () in GBM6 resulting in the increased suppression of GBM cell growth and induction of apoptosis as compared with the treatment with dacomitinib or PF-05212384 alone (). This result suggests that blocking EGFR in EGFR driving GBMs has substantial effect on EGFR-dependent cell growth. However, constitutively active PI3K/AKT is independent of EGFR in GBM6, and targeting EGFR by dacomitinib is not sufficient to block the PI3K/AKT pathway. The partial suppression of GBM6 cell growth by dacomitinib indicates that even if there is EGFR amplification, targeting EGFR alone is not sufficient due to its downstream pathway dysfunctions. Combined targeting PI3K enhances EGFR targeted therapeutic efficacy in this cellular context. Together these results suggest that multiple lesions in the EGFR (RTKs)/PI3K/mTOR pathway determine GBM cell response to EGFR and PI3K/mTOR inhibitors ().
Figure 4. A simplified model of dysregulated EGFR (RTKs)/PI3K/mTOR pathway in GBM and corresponding target therapies. (A) Multiple lesions cause constitutive activation of EGFR (RTKs)/PI3K/mTOR pathway. (B) Targeting different components in EGFR(RTKs)/PI3K/mTOR pathway based on patient tumor genetic profile.
Discussion
In this study, we evaluated the efficacy of dacomitinib, an irreversible pan-ERBB inhibitor, in a panel of established and patient derived primary GBM cell lines. We show that dacomitinib significantly reduced phosphorylated EGFR in all the GBM lines, but did not show a dose-dependent response on cell viability in a majority of the lines tested. Furthermore, we show that both PI3K and mTOR status contributed to the dacomitinib response in GBMs and blocking PI3K/mTOR with dual inhibitor PF-05212384 had dominant effect in most lines tested. PF-05212384 enhances the efficacy of EGFR inhibition in GBMs that have both EGFR amplification and independent PI3K activation, resulting in significantly decreased GBM cell survival and induction of apoptosis.
Dacomitinib, an irreversible EGFR inhibitor has been shown very effective in lung cancer models that are resistant to gefitinib and erlotinib. In line with this observation in NSCLC model, our results showed that lung cancer cell line HCC827 has dose dependent response to dacomitinib, while the majority GBM cell lines lack response to dacomitinib monotherapy. Based on available information of GBM genetic lesion in RTK pathway, we concluded that multiple lesions in RTK pathway contribute to PF299 resistance in GBMs.
Genetic mutations in PI3K and PTEN pathways occur in about 50% of GBM patients.Citation10 Most dacomitinib-resistant GBM cells we tested have PTEN loss of function or constitutively active p-AKT suggesting that dysregulation of PTEN/PI3K pathway may cause lack of response to EGFR inhibition. To investigate whether PTEN loss contributes to dacomitinib resistance in GBM cells, we introduced wild-type PTEN into U373 GBM cells. Introducing functional PTEN resulted in a reduction of p-AKT (Fig. S3A) and sensitized U373 GBM lines response to dacomitinib (Fig. S3B). This result suggests that functional PTEN is important for negatively regulating PI3K signaling and introducing functional PTEN in PTEN mutant GBM cells can sensitize EGFR inhibition. However, introducing PTEN resulted in U373 cell apoptosis even without dacomitinib (Fig. S3C). It is not clear why overexpression of wild-type PTEN causes dramatic apoptotic effect, although it has been previously shown that subtle variations of PTEN dose determine tumor susceptibility. PTEN has been shown to play multiple roles in regulating not only PI3K, mTOR activities, but also EGFR stabilityCitation16 and phosphorylation.Citation6 It is very possible that overexpression of PTEN sensitizes GBM cells to dacomitinib via multiple pathways.
PI3K and mTOR pathways are known to regulate cell survival and proliferation and serve as downstream effectors of EGFR and other RTKs. Our results showed that GBMs with co-activated RTKs are more sensitive to PI3K/mTOR dual inhibitor than EGFR inhibitor. This could be attributed to the fact that targeting EGFR alone without shutting down alternative activation of PI3K/mTOR through other RTKs has a role to play in the resistance of GBM cells to EGFR monotherapy.
RAS/RAF/MEK/MAPK pathway is another major EGFR downstream pathway involved in cell growth and proliferation and has also been shown mutated in about 2% of GBM patients.Citation10 Among GBM lines we tested in this study, U87 and U373 lines have been verified to have RAS/RAF activating mutations. We expect that activating mutations of RAS/RAF in conjunction with PTEN mutation abrogate upstream EGFR inhibition in those lines. Previous studies have shown that combined targeting of PI3K/mTOR and MEK in a preclinical colon cancer model has better therapeutic effect than targeting PI3K/mTOR and MEK individually.Citation14 Based on RAS and RAF mutations in GBMs, targeting RAS/RAF/MEK/MAPK pathway simultaneously could offer a potential therapeutic benefit.
EGFRvIII is the most common EGFR mutation found in GBM. EGFRvIII is constitutively active and previous studies using NR6 (murine fibroblasts) model has shown that introducing EGFRvIII results in gefitinib resistance.Citation17 Quantitative analysis of EGFRvIII signaling using U87 GBM model indicated that overexpression of EGFRvIII preferentially utilizes PI3K pathway over MAPK and STAT3 pathways, and cross-activates other RTKs like c-Met, Axl, and EphA2.Citation18 In our patient-derived GBM model, we observed different signaling patterns driven by EGFRvIII in GBM4 () vs. GBM6 (Fig. S4). It is plausible to suspect that EGFRvIII activates multiple RTKs in a cell context dependent manner and further studies need to be done to understand the EGFRvIII-mediated differential downstream signaling.
A recent study has shown that active EGFR suppresses autophagy via Beclin1 and EGFR inhibition by erlotinib induces autophagic cell death in NSCLC cells.Citation19 In addition to EGFR, PI3K, AKT, and mTOR all have been shown to play a role as negative regulators of Beclin1 and autophagy.Citation20,Citation21 Our data indicates that dacomitinib induces cell death via apoptosis in GBM cells. Furthermore, we have shown that multiple lesions in RTK pathway activate PI3K, AKT, and mTOR independently of EGFR in GBM. It would not be surprising that Beclin1 and autophagy are independent of EGFR in GBMs and are regulated by other pathways. It is plausible that suppression of autophagy contributes to tumor progression and chemoresistance in GBMs, so it would be interesting to further investigate autophagy regulation and its role in GBM in future studies.
Unlike most solid tumors, GBM harbors multiple genetic lesions.Citation10 Our results suggest that multiple genetic lesions in RTK pathway candidates contribute to EGFR targeted therapeutic resistance and highlight the potential importance of applying diagnostic genomic screening for individual GBM patients. As genomic sequencing becomes available for tumor patients in the future, targeted tumor therapy should be personalized based on individuals' tumor genomic profile.
Materials and Methods
Antibodies and reagents
Antibodies against phospho-AKT(Ser-473), phospho-AKT(Thr-308), AKT, EGFR, cleaved PARP, phospho-S6 ribosomal protein(Ser-235/236), S6 ribosomal protein, phospho-p-44/42MAPK(ERK1/2) (Thr202/Tyr204), and p-44/42MAPK(ERK1/2) were obtained from Cell Signaling. Anti-phospho-EGFR(Tyr-1068) was purchased from AbCam. Anti-α-tubulin was purchased from Sigma. Dacomitinib and PF-05212384 were kind gifts from Pfizer. Stock solutions of dacomitinib (10 mmol/L) and PF-05212384 (1 mmol/L) were prepared in DMSO and stored at −20 °C.
Cell lines
Established human GBM cell lines (Gli36, Gli36EGFRvIII, LN229, U87, U251, and U373) were cultured as described.Citation22 Patient-derived primary GBM cells (GBM4, GBM6, GBM8, GBM13, GBM18) were collected from surgical specimens at Massachusetts General Hospital with approval of the Institutional Review Board and grown in supplemented neural basal medium (Invitrogen) as described previously.Citation15,Citation23 Lung cancer cell line HCC827 (ATCC) was grown in RPMI-1640 supplemented with 10% (vol/vol) FBS and 1% (vol/vol) penicillin/streptomycin.
Lentiviral transductions and engineering stable cell lines
Lentivral packaging was performed by transfection of 293T cells as previously described.Citation24 GBM4 cells were transduced with lentiviral vectors LV-EGFR, or LV-EGFRvIII at multiplicity of infection (M.O.I.) = 2.
Cell viability and caspase assays
GBM cells were plated on 96-well plates and treated with different doses of dacomitinib or/and PF-05212384 for indicated times. Cell viability was measured using an ATP-dependent luminescent reagent (CellTiterGlo, Promega) and caspase activity was determined using a DEVD-aminoluciferin (CaspaseGlo 3/7, Promega) according to manufacturer’s instructions. All experiments were performed in triplicates.
Immunoblotting
Following treatment, cells were washed twice with cold PBS, then lysed with 20 mM TRIS-HCl pH 8.0, 137 mM NaCl, 10% glycerol, 1% NP-40, 2 mM EDTA plus protease inhibitors (Roche), and phosphatase inhibitors (Phosphatase Inhibitor Cocktail I and Phosphatase Inhibitor Cocktail II from Sigma-Aldrich) at 4 °C. Cell lysates were clarified by centrifugation at 16 000 × g for 10 min, and the supernatant protein concentrations were determined by using a Bio-Rad protein assay kit. 6× SDS-sample buffer (Boston BioProducts) was added to protein samples and heated at 100 °C for 3 min. Ten to 30 micrograms of protein were resolved on SDS-PAGE gel and transferred to nitrocellulose membrane, and probed with primary antibodies.
| Abbreviations: | ||
| EGFR | = | epidermal growth factor receptor |
| GBM | = | glioblastoma multiforme |
| PI3K | = | phosphatidylinositide 3-kinase |
| mTOR | = | mammalian target of rapamycin |
| PTEN | = | functional phosphatase and tensin homolog |
| RTKs | = | receptor tyrosine kinases |
| NSCLC | = | non-small cell lung cancer |
| PLCγ1 | = | Phospholipase C isotype γ1 |
| rpS6 | = | ribosomal protein S6 |
Additional material
Download Zip (861.6 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Ethics Statement
Patient-derived primary GBM cells (GBM4, GBM6, GBM8, GBM13, and GBM18) were collected from surgical specimens at Massachusetts General Hospital (MGH) with approval of the MGH Institutional Review Board (IRB). The IRB-approved protocol is to collect excess tissue materials and informed consent from patients is exempt. The ethics committees/IRBs approve this consent procedure.
Acknowledgments
This work was supported by Pfizer Inc. (K.S.) and NIH-R01CA138922 (K.S.).
References
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al, European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups, National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352:987 - 96; http://dx.doi.org/10.1056/NEJMoa043330; PMID: 15758009
- Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MC, et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol 2008; 26:3015 - 24; http://dx.doi.org/10.1200/JCO.2007.15.7164; PMID: 18565887
- Rich JN, Rasheed BK, Yan H. EGFR mutations and sensitivity to gefitinib. The New England journal of medicine. 2004; 351:1260 1
- Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov GN, Bradner JE, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res 2007; 67:11924 - 32; http://dx.doi.org/10.1158/0008-5472.CAN-07-1885; PMID: 18089823
- Gonzales AJ, Hook KE, Althaus IW, Ellis PA, Trachet E, Delaney AM, Harvey PJ, Ellis TA, Amato DM, Nelson JM, et al. Antitumor activity and pharmacokinetic properties of PF-00299804, a second-generation irreversible pan-erbB receptor tyrosine kinase inhibitor. Mol Cancer Ther 2008; 7:1880 - 9; http://dx.doi.org/10.1158/1535-7163.MCT-07-2232; PMID: 18606718
- Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res 2009; 69:3256 - 61; http://dx.doi.org/10.1158/0008-5472.CAN-08-4055; PMID: 19351834
- Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 2005; 353:2012 - 24; http://dx.doi.org/10.1056/NEJMoa051918; PMID: 16282176
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316:1039 - 43; http://dx.doi.org/10.1126/science.1141478; PMID: 17463250
- Fan QW, Cheng C, Knight ZA, Haas-Kogan D, Stokoe D, James CD, McCormick F, Shokat KM, Weiss WA. EGFR signals to mTOR through PKC and independently of Akt in glioma. Sci Signal 2009; 2:ra4; http://dx.doi.org/10.1126/scisignal.2000014; PMID: 19176518
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455:1061 - 8; http://dx.doi.org/10.1038/nature07385; PMID: 18772890
- Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, Van Meir EG. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol 1999; 9:469 - 79; http://dx.doi.org/10.1111/j.1750-3639.1999.tb00536.x; PMID: 10416987
- Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A 1990; 87:8602 - 6; http://dx.doi.org/10.1073/pnas.87.21.8602; PMID: 2236070
- Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007; 318:287 - 90; http://dx.doi.org/10.1126/science.1142946; PMID: 17872411
- Mallon R, Feldberg LR, Lucas J, Chaudhary I, Dehnhardt C, Santos ED, Chen Z, dos Santos O, Ayral-Kaloustian S, Venkatesan A, et al. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res 2011; 17:3193 - 203; http://dx.doi.org/10.1158/1078-0432.CCR-10-1694; PMID: 21325073
- Wakimoto H, Mohapatra G, Kanai R, Curry WT Jr., Yip S, Nitta M, Patel AP, Barnard ZR, Stemmer-Rachamimov AO, Louis DN, et al. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol 2012; 14:132 - 44; http://dx.doi.org/10.1093/neuonc/nor195; PMID: 22067563
- Vivanco I, Rohle D, Versele M, Iwanami A, Kuga D, Oldrini B, Tanaka K, Dang J, Kubek S, Palaskas N, et al. The phosphatase and tensin homolog regulates epidermal growth factor receptor (EGFR) inhibitor response by targeting EGFR for degradation. Proc Natl Acad Sci U S A 2010; 107:6459 - 64; http://dx.doi.org/10.1073/pnas.0911188107; PMID: 20308550
- Learn CA, Hartzell TL, Wikstrand CJ, Archer GE, Rich JN, Friedman AH, Friedman HS, Bigner DD, Sampson JH. Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clin Cancer Res 2004; 10:3216 - 24; http://dx.doi.org/10.1158/1078-0432.CCR-03-0521; PMID: 15131063
- Huang PH, Mukasa A, Bonavia R, Flynn RA, Brewer ZE, Cavenee WK, Furnari FB, White FM. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc Natl Acad Sci U S A 2007; 104:12867 - 72; http://dx.doi.org/10.1073/pnas.0705158104; PMID: 17646646
- Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013; 154:1269 - 84; http://dx.doi.org/10.1016/j.cell.2013.08.015; PMID: 24034250
- Botti J, Djavaheri-Mergny M, Pilatte Y, Codogno P. Autophagy signaling and the cogwheels of cancer. Autophagy 2006; 2:67 - 73; PMID: 16874041
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012; 338:956 - 9; http://dx.doi.org/10.1126/science.1225967; PMID: 23112296
- Bagci-Onder T, Wakimoto H, Anderegg M, Cameron C, Shah K. A dual PI3K/mTOR inhibitor, PI-103, cooperates with stem cell-delivered TRAIL in experimental glioma models. Cancer Res 2011; 71:154 - 63; http://dx.doi.org/10.1158/0008-5472.CAN-10-1601; PMID: 21084267
- Wakimoto H, Kesari S, Farrell CJ, Curry WT Jr., Zaupa C, Aghi M, Kuroda T, Stemmer-Rachamimov A, Shah K, Liu TC, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res 2009; 69:3472 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-08-3886; PMID: 19351838
- Shah K, Hingtgen S, Kasmieh R, Figueiredo JL, Garcia-Garcia E, Martinez-Serrano A, Breakefield X, Weissleder R. Bimodal viral vectors and in vivo imaging reveal the fate of human neural stem cells in experimental glioma model. J Neurosci 2008; 28:4406 - 13; http://dx.doi.org/10.1523/JNEUROSCI.0296-08.2008; PMID: 18434519
- Meléndez B, García-Claver A, Ruano Y, Campos-Martín Y, Rodríguez de Lope A, Pérez-Magán E, Mur P, Torres S, Lorente M, Velasco G, Mollejo M. Copy Number Alterations in Glioma Cell Lines. In: Ghosh A, ed. Glioma - Exploring Its Biology and Practical Relevance. Rijeka Croatia: InTech; 2011.