Abstract
Ewing tumor is driven by the oncogenic EWS-FLI1 fusion protein that functions as an aberrant transcription factor. The identification of EWS-FLI1 protein partners is essential to enhance its vulnerability as a therapeutic target. We utilized phage display library screening against recombinant EWS-FLI1 protein. We identified 27 unique Ewing sarcoma binding peptides. The cytotoxicity evaluation of these peptides with in EWS-FLI1 containing cell lines yielded one potent peptide called ESAP1 (TMRGKKKRTRAN). ESAP1 binds EWS-FLI1 with 0.202 micromolar affinity as measured in surface plasmon resonance. The minimal interaction region of ESAP1 is characterized and found that the lysine residues are critical for cellular cytotoxicity. ESAP1 reduces the transcriptional activity of EWS-FLI1 as well as disrupts cell cycle kinetics in Ewing Tumor cells. These findings provide both a novel experimental probe and a potential therapeutic scaffold for Ewing Tumor.
Introduction
Ewing tumor (ET) is a highly malignant tumor of the bone and soft tissue that is typically diagnosed in the second decade of life, but is found in patients from newborns to octogenarians. Despite chemotherapy, 30% of patients with localized disease and 75–80% of patients with metastatic disease will die within five years of diagnosis.Citation1 The EWS-FLI1 oncogenic fusion results from a chromosomal translocation t(11;22)(q24;q12) in 95% of cases.Citation2 The fusion protein is the combination of 6–10 N-terminal exons of the Ewing's sarcoma gene (EWS) on chromosome 22 with 4–5 of the carboxy terminal of the ets family gene, either friend leukemia insertion 1 (FLI1) or ets-related gene (ERG).Citation3 While there is some variation in exon combinations across tumors, these translocation variants are not clinically prognostic.Citation1,Citation4 There is a growing list of EWS-FLI1 target genes, whose combined activities contribute to oncogenesis and tumor maintenance.Citation5,Citation6
EWS-FLI1 binds to DNA through the conserved ets binding domain to modulate transcription through direct binding to promoters and RNA splicing.Citation7 Protein-protein interactions are critical for transcriptional and splicing complexes, yet only limited protein interactions with EWS-FLI1 have been validated including RNA Helicase A (RHA).Citation8–Citation12 Disruption of the protein complex between EWS-FLI1 and RHA leads to rapid ET cell death.Citation13 Protein-protein disruption is possible because of the favorable thermodynamics of peptides binding to intrinsically disordered proteins.Citation14 EWS-FLI1 has been predicted to have a significant intrinsic disorderCitation15 and proven to require disorder for function.Citation16 Peptides can be novel reagents to block proteinprotein interactions based upon significant specificity for their binding to targets.Citation17 In order to resolve the functional protein partners of EWS-FLI1, we used phage display screening to identify peptides that might lead to specific protein interactions. In a previous report, we described the peptide E9 as having homology to a region of RHA and a modified E9 peptide inhibiting Ewing tumor cell monolayer growth and anchorage independent colony formation in soft agar.Citation13 We report a novel peptide that both binds directly to EWS-FLI1 and alters its function.
Results
Phage display reveals 27 novel peptides using EWS-FLI1 as bait.
We previously described the purification of recombinant EWS-FLI1 from a bacterial expression system.Citation15 Recombinant EWS-FLI1 was determined to have a physiologic conformation based upon DNA binding and transcript activation assays.Citation15 This recombinant EWS-FLI1 was utilized in a phage display assay to identify novel binding peptides. Three cycles of phage enrichment led to approximately 300 individual phages. These phages were evaluated for EWS-FLI1 binding using ELISA and those having a binding ratio of greater than 2.0, compared with albumin, were selected for sequencing. The corresponding peptide sequences demonstrated sequence 1 (TMR GKK KRT RAN) in 30% of the 96 phage clones, which is heretofore called Ewing Sarcoma Antagonist Peptide 1 (ESAP1) ().
These 27 unique peptides were synthesized along with the N-terminal-16 amino acid Antennapedia (Penetratin) sequence for cell penetration.Citation18 We identified that Antennapedia was superior to the TAT sequence for intracellular delivery of peptides into ET cell lines (data not shown). These peptides were evaluated for their effects upon growth of the ET cell line TC32 (EWS-FLI1 containing cell line) and the neuroblastoma cell line SKNAS (lacking EWS-FLI1) over 4 d. Six out of 27 peptides inhibited growth >50% in TC32 while none of the peptides inhibited growth >50% in SKNAS cell lines (Fig. S1). ESAP1, peptide number 1, (30 µM) consistently inhibited ET cell growth by >90%. The assay was repeated at least twice per cell line. We therefore sought to identify the candidate proteins that might be represented by sequence homology with ESAP1.
Proteome analysis identifies four potential ESAP1 containing proteins.
A protein BLAST against all known proteins resulted in no matches with greater than seven out of 12 consecutive ESAP1 amino acids. By reducing the stringency to seven or fewer amino acids, four candidate proteins were identified: Bromodomain containing 9 (BRD9), DEAD (Asp-Glu-Ala-Asp) box polypeptide 27 (DDX27), myeloid/lymphoid or mixedlineage leukemia 3 (MLL3), and thioredoxin domain containing 9 (TXNDC9) (Fig. S2). Unfortunately, we did not identify any of these proteins in complex with EWS-FLI1 using a series of immunoprecipitations from ET cell lysate (Fig. S3). We therefore decided to explore the toxicity and mechanism of ESAP1, as a novel agent, against ET cells independently of specific partner proteins.
ESAP1 directly binds EWS-FLI1.
While the initial phage display was performed with recombinant EWS-FLI1 as bait, we validated that ESAP1 directly binds to EWS-FLI1 using both a direct peptide binding ELISA-style assay and surface plasmon resonance (SPR). The ELISA-style assay used alkaline phosphatase (colorimetric readout) conjugated peptide (ESAP1-AP) to confirm ESAP1 binding to EWS-FLI1 in a dose-dependent manner (). In a separate experiment, synthetic ESAP1 was shown to compete away the interaction between ESAP1-AP and EWS-FLI1 (). SPR technology further confirmed direct binding of the ESAP1 to EWS-FLI1. ESAP1 bound to recombinant EWS-FLI1 with a high affinity (KD = 0.202 ± 0.04 µM) in the SPR experiments. This KD value is the average of 10 different binding experiments in SPR ().
Lysine residues in the ESAP1 appear most critical for EWS-FLI1 binding.
The bacterially expressed ESAP1-AP allowed us to quickly create a series of peptides with alanine substitution point mutations. These alanine substitution mutant peptides identified the central lysine and C-terminal residues as most critical for binding to EWS-FLI1 (Fig. S4). When any of the three lysines are replaced with alanine, the percentage of ESAP1-AP that is bound to EWS-FLI1 decreases dramatically as a percentage of wild-type peptide. The C-terminal of the peptide appears more critical for binding than the N-terminal and shows a greater decrease in binding to EWS-FLI1 when those amino acids are mutated. The N-terminal does not show a decrease in binding when mutated to alanine.
We mutated ESAP1 at the 5th position to alanine (ESAP1-K5A), which seemed the most critical amino acid for EWS-FLI1 binding in the ESAP1-AP experiment (). However, the binding of the single amino acid substituted ESAP1-K5A to EWS-FLI1 had a similar binding affinity by SPR as the wildtype peptide (KD = 0.245 ± 0.09 µM, ). Thereafter, additional mutations were made, including removing all amino acids except the lysine core (ESAP1+K) and removing 2 central lysines (ESAP1-K). The binding affinity of ESAP1-K was 8.5-fold less (KD = 1.7 ± 0.8 µM) than the wild-type ESAP1 and was similar to the antennapedia control sequence affinity. The ESAP1+K affinity (KD = 0.388 ± 0.45 µM) was just under 2-fold less than the wild-type ESAP1 (). KD values of mutant peptides are the averages reported from between four and seven independent experiments using different batches of recombinant EWS-FLI1. Deletion of 3 residues at either end of the peptide (ESAP1-N, GKK KRT RAN, ESAP1-C, TMR GKK KRT) did not alter binding (data not shown).
The mutant peptides were then synthesized fused with FITC to monitor uptake and tested for their effects upon ET monolayer cell growth. We confirmed that there was no effect of FITC tag, based upon similar cellular toxicity, upon IC50 levels of ESAP1-treated cells with or without FITC (p value = 0.74) (). A shortened peptide with the central core of GKKKRT (ESAP1+K) demonstrated similar IC50 (15 µM) to that of the full length ESAP1 IC50 (16 µM); however there was a 2-fold reduction in IC90 from 42 to 25 µM. The deletion mutant (ESAP1-K) was much less potent based upon a doubling of the IC50 (34 µM, ). The ESAP1-K5A mutant, that showed similar binding to EWS-FLI1, as the wild type peptide, had a similar IC50 = 14 µM to ESAP1 (). To assure that all peptides achieved equal access to the nucleus, confocal microscopy was performed on TC-71 cells treated with wild-type FITC-tagged ESAP1, FITC-tagged +K (GKKKRT-OH), and FITC-tagged −K (TMRGKTRAN-OH). All three peptides showed similar localization in TC-71 cells with some cytoplasmic, but mostly nuclear localization. The peptides were observed in the heterochromatic regions of the nucleus (). The loss of two of the lysines and an arginine did not affect the localization of the peptide, but reduced cytotoxicity.
ESAP1 specifically reduces ET monolayer and anchorage-independent growth.
We expanded our evaluation of ESAP1 to a larger panel of ET cell lines with different fusion protein translocations and a larger panel of non-EWS-FLI1 containing cell lines including: TC-32, TC71 (EWS-FLI1, fusion type 1), A4573 (EWS-FLI1, fusion type 3), SKNAS (neuroblastoma cell line lacking EWS-FLI1), and PANC1 (pancreatic epithelioid carcinoma). The ET cell lines showed the highest levels of cytotoxicity from ESAP1, while the SKNAS and PANC1 cells did not show evidence of cytotoxicity until 50 µM (). The difference between the ET cells and the controls was statistically significant (p < 0.001). Peptide uptake was confirmed using confocal microscopy (Fig. S5). We noted that cells containing EWS-FLI1 showed more peptide in the nucleus than cells lacking EWS-FLI1, which could indicate peptide retention in the nucleus is due to EWS-FLI1 binding.
TC71 cells transfected with EGFP-fused ESAP1 were grown for 5 d in G418 antibiotic selection media and then seeded in soft agar. The cells expressing ESAP1 were identified by EGFP expression, which was generally consistent across a colony (). Cells expressing ESAP1 demonstrated significant reduction (4.45-fold, p = 0.0158) in anchorage independent growth compared with EGFP alone colonies ().
EWS-FLI1-dependent transcription is reduced by ESAP1.
A current standard for EWS-FLI1 transcription activity is based upon cotransfection of EWS-FLI1 along with the NR0B1-luciferase reporter plasmid. EWS-FLI1 increased transcription for the plasmid 27.5-fold above non-EWS-FLI1 expressing control, while the addition of ESAP1 reduced this activity by 55% (). The EWS-FLI1 inhibitory small molecule YK-4-279, used as a control, completely abrogated transcriptional activity. Exogenously expressed EWS-FLI1 levels were shown in the protein gel blot ().
In order to evaluate the effect of ESAP1 on endogenously expressed EWS-FLI1 direct targets, we used synchronized cells and performed quantitative RT-PCR. GLI1, cyclin E were previously shown as a direct targets of EWS-FLI1.Citation19,Citation20 GLI1 and cyclin E transcript levels were decreased significantly at the 8 h of ESAP1 treatment in synchronized cells when compared with non-treated synchronized cells ( and D).
ESAP1-treated ET cells become blocked at S-G2 phase of cell cycle.
We initially explored apoptosis as a mechanism of cell death by ESAP1, but discovered that caspase-3 activation level did not change upon ESAP1 treatment (Fig. S6). The results indicated that ESAP1 exerts its cytotoxic effect through a mechanism other than caspase-3-dependent apoptosis. We therefore evaluated effects of ESAP1 on the cell cycle. Different synchronization strategies were tested, including double-thymidine and nocodazole induced arrest. We achieved the best synchronization with TC71 using nocodazole, which gives a potent G2/M phase blockade; with the release of this arrest cells proceed through the cell cycle and undergo mitosis.
Without any synchronizations, TC71 cells in log phase growth demonstrated 25% G2/M, 40% S and 35% G1 phases of cell cycle (). Treatment of ESAP1 without synchronization did not cause major disturbances of the TC71 cell cycle (). When nocodazole was removed after a 16 h incubation, 56.5% of the treated cells were in the G2/M phase. Sixty percent of cells then progressed into G1 phase at the two hour time point (). Eight hours following nocodazole removal, 90% of cells were duplicating their DNA in S phase (). In contrast, 36% cells of treated with ESAP1 at the time of nocodazole remained in G2/M phase, while only 27% progressed to G1 (). ESAP1 appeared to freeze cells, without progression through mitosis and re-entry into G1. There was a gradual increase in S phase from 20% to 30% at 24 h. Forty-eight hours after removal of nocodazole, in the presence of ESAP1, S phase increased to 35%, similar to cells without ESAP1 treatment. Coincident with this, the fraction of cells in G1 phase decreased and those in G2/M phase increased (). This indicates that a small fraction of G2/M arrested cells were completing their cycle and entering G1 phase. A small number of cells in G1 phase were progressing to S phase, where cell cycle dynamics appeared to slow with accumulation in S phase (). Cells appeared healthy in the no treatment group () and 24 h of ESAP1 treatment without synchronization (). After 24 h following release of synchronization there were no signs of cytotoxicity (). However, blocked cells treated with ESAP1 were dysmorphic and fragmented (). ESAP1 treatment of synchronized cells revealed disruption in cell cycle progression that lead to a caspase-3 independent cell death.
ESAP1-treated cells were further analyzed for viability, based upon propidium iodide exclusion. The fractions having incomplete genetic content usually associated with dead cells was calculated, and we observed the amount of genetic content skewed toward the left indicating reduced DNA content per cell. This death fraction was different than the usual apoptotic cell DNA content called sub-G0. Thus, some cell death occurred in ESAP1-treated cells that was independent of cell cycle. Cell death percentages were 11 and 8% in synchronized and unsynchronized cells, respectively at 48 h of treatment (Fig. S7). ESAP1-treated synchronized cells showed this increased death fraction, based upon DNA content, at 28 h (). However, those cells with reduced DNA content disappeared by hour 48, likely due to cell death (). Coincident with the cell population having less DNA content was a population indicated by the arrow (–K) that had hyperdiploid DNA content, consistent with cells having dysregulated mitosis. In cells treated only with ESAP1, but not synchronized, this population of cells was observed at 48 h, which was similar to the 28 h peak observed in the synchronized cells ().
Expression pattern of ESAP1-treated cells consistent with growth arrest.
In order to identify the mechanism underlying the growth arrest upon ESAP1 treatment, we analyzed the mRNA expression pattern of ESAP1-treated cells in cell cycle specific real time PCR array. An almost 4-fold increase in p21 mRNA at 8 hours following ESAP1 treatment was measured in nocodazole synchronized cells treated with ESAP1 compared with untreated controls (based on the average of fold difference calculations of five individual housekeeping genes) (). Other transcript changes that support the cell cycle profile observed include increase cyclin D1 and cyclin H expression more than 2-fold while the expression of cyclin G2 was reduced. Rad51 increased the second highest among those analyzed, increasing 2.89-fold, which supports a DNA damage element that is also consistent with the abnormal DNA content we observed. Nibrin, a part of multiprotein complex in DNA damage response function, was also reduced to 0.62-fold. ESAP1-treated cells yielded very little total protein, thus immunoblotting studies to corroborate with protein levels were not successful.
Discussion
Cancer results from dysregulated transcription and this transcription requires a finely tuned balance of protein interactions. Some tumors such as ET, contain a tumor-specific fusion protein, EWS-FLI1, that is a central transcriptional regulator of tumorigenesis as evidenced by gene expression investigations.Citation21 Thus, EWS-FLI1 protein interactions are critical to define in order to understand its function. Phage display identified ESAP1 as a peptide that directly bound EWS-FLI1 (). ESAP1 also reduced ET cell growth ( and ) and inhibited EWS-FLI1 function (). While ESAP1 did not lead us to identify a new partner of EWS-FLI1, ESAP1 has demonstrated a novel ability to disrupt EWS-FLI1 function. ESAP1 treatment alters cell cycle kinetics through dysregulation of cell division in ET cells causing cell death.
Investigators recognized the importance of the EWS-FLI1 transcriptional complexes and the potential to reveal the mysteries of this disease if these complexes could be resolved.Citation9,Citation22 Limited data exists for those proteins that are directly complexed with and regulated by EWS-FLI1.Citation7 Resolving transcriptional protein complexes is challenging given their transient occurrence in cells.Citation23 Phage display is a powerful, unbiased strategy to identify protein interaction motifs.Citation24 This approach has the advantage of screening diverse libraries and enrichment of bound phage. We minimized bias by screening against full-length EWS-FLI1 rather than an isolated domain or peptide, given its intrinsic disorder.Citation8,Citation15,Citation16 Disadvantages of phage display include the dependency of target protein structure and stringency of screening. Given the nature of phage display, phage binding is limited to protein surfaces, which is consistent with protein-protein interactions. A further caveat of phage display is that non-physiologic epitopes may form when proteins are adhered to an ELISA plate, either within or between individual proteins. Finally, a protein interaction may be missed because of inadequate antibody reagents for confirmation of binding.
We fused antennapedia peptides to each of the 27 phage-identified peptides to measure cell growth. While ESAP1 was selected for further study since it clearly reduced cell growth, the remaining 26 peptides that had little or no growth effects, may still reveal informative protein binding properties. Our deeper investigations have not included the 26 peptides, which may have lacked growth effects based upon their cellular access. Since peptides identified from the screening must be validated independently from phage display to consider as interacting peptides, the peptide data set could be explored in future work. To fully validate peptide interaction with EWS-FLI1, surface plasmon resonance provided kinetic data demonstrating direct binding with ESAP1.
The nuclear uptake of peptides is also critical for their function because EWS-FLI1 localizes in the nucleus. If the peptide has cytoplasmic retention, less peptide would be available in the nucleus to inhibit EWS-FLI1 protein function. This might explain the discrepancy between the IC50 level of ESAP1 for the cytotoxicity (14 µM) and its KD value (0.2 µM) for EWS-FLI1 binding. As seen in the fluorescent peptide uptake experiments, a considerable amount of peptide is found in the cytoplasm (). While antennapedia is a cell permeable peptide that facilitates macromolecule entry into cells, the final subcellular localization depends on the nature of the macromolecule. Since ESAP1 is a peptide lacking functional domains or motifs, homogenous distribution of peptide in the cell could be expected. It is important to note that we observed a skewed distribution of ESAP1 based upon the localization of EWS-FLI1, those cells with nuclear EWS-FLI1 appeared to retain ESAP1 in the nucleus. This may also explain the localization of ESAP1 mostly in the cytoplasm in neuroblastoma and pancreatic carcinoma cell line (Fig. S5).
ESAP1 clearly alters the function of EWS-FLI1 yet, no novel protein partner was confirmed in these investigations. The top four candidates were explored to the best of our abilities with existing antibody reagents, and could not be convincingly found in EWS-FLI1 complexes in ET cells. As explained above, this may not fully exclude BRD9, TXNDC9, MLL3 and DDX27 as directly interacting proteins. Despite not identifying any cognate protein, ESAP1 has informed the oncogenic mechanism of EWS-FLI1. The ESAP1 antiproliferative effect on ET cells was identified through the delayed cell cycle progression. A careful exploration of cell cycle, with synchronized cells showed prolonged S and G2 arrest due to ESAP1 treatment, such that cells may reenter the cell cycle with two different cellular populations. This is similar to a recently reported novel radiation induced cell cycle suspension.Citation25
ESAP1 treatment of the TC71 cell line caused 4-fold increase of p21 expression compared with nocodazole synchronized cells. The increased expression of p21 might be the molecular mechanism for inhibiting the cell cycle. Increased expression of p21 could also be an indicator of disrupted EWS-FLI1 transcription because antisense reduction of EWS-FLI1 increased p21 protein levels.Citation26 Also, reduction of EWS-FLI1 protein levels can lead to G1 arrest by increasing the cyclin-dependent kinase inhibitors (p27Kip1 and p57Kip2) expression and increasing pRB protein.Citation27 EWS-FLI1 affects the transcription of a large number of proteins that function in the regulation of the cell cycle,Citation28 supporting our findings that ESAP1 interrupts cell cycle kinetics at important downstream pathways from EWS-FLI1.
Deregulation of the cell cycle could be due to reduced expression of cell cycle regulators however we found only Nibrin and cyclin G2 were reduced, out of the limited number of cell cycle related genes represented in the PCR array. Other platforms might reveal more ESAP1 reduced genes. The presence of DNA damage or stress response genes, e.g., Rad51, BRCA1, RAD1 and NBN, in the both upregulated or downregulated transcripts groups could be indicative of an ESAP1 molecular mechanism or the nature of EWS-FLI1 functional inhibition.
Peptides have been used to probe, treat, target, deliver and diagnose immunologic conditions.Citation29,Citation30 In cancer, the systemic use of therapeutic peptides of greater than 4 or 5 amino acids has not been achieved due to delivery and stability issues. While some work suggests that peptide modifications can overcome these challenges, these are not proven in the clinic.Citation31 In addition, the biochemical peptide interactions may be affected by stapling.Citation32 Some peptide-based compounds like Mifamutide, liposomal muramyl tripeptide phoshatidyl ethanolamide (MTPPE), have been used clinically as an immune system activator in the lung lesions of osteosarcoma.Citation33 Another successful example of using peptides to inhibit protein function requires intracellular delivery of inhibitory peptide (rPP-c8) that prevents STAT3 binding to PIAS3.Citation34 The inhibitory peptide reduced STAT3 target gene expression, proliferation and migration of glioblastoma cells. While the importance of using peptides to inhibit interactions of two proteins has been gaining significance, future stabilizing and targeting modifications are required.
In summary, these experiments provide a panel of novel peptides that potentially interact with EWS-FLI1. One of these, ESAP1, is a peptide that directly binds to EWS-FLI1 and alters its function leading to cell death that is caused by a cell cycle/cytokinetic disruption. Future explorations of the EWS-FLI1 transcriptome may be enhanced by ESAP1. While ESAP1 may not directly become a therapeutic reagent for patients with ET, as peptide technology improves, therapeutic opportunities may arise.
Experimental Procedures
Cell lines.
All of the ET cell lines (TC32, TC71, A4573) were grown in RPMI (GIBCO 11875) with 10% fetal bovine serum (FBS-HyClone Thermo Scientific SH30396.03) and 1% HEPES (GIBCO 15630). SKNAS and PANC1 cell lines were grown in DMEM (GIBCO 11965) with 10% FBS.
Phage display and peptides.
Phage Display was performed as previously reported in reference Citation8. Twenty-seven peptides, each 12 amino acids long, derived from phage display were commercially synthesized (Biosyn) to the Antennapedia sequence (NH2-RQI KIW FQN RRM KWK K) to ensure cellular uptake. Additional ESAP1 (TMR GKK KRT RAN-OH), ESAP1 mutants (GKK KRT RAN-OH; GKK KRT-OH; TMR GKT RAN-OH; TMR GKK KRT-OH), and FITC tagged ESAP1 and mutants were commercially synthesized to Antennapedia by Biomatik. All peptides were dissolved in sterile water.
BLAST.
A BLAST search was performed against all known proteins in the NCBI database (www.ncbi.nlm.nih.gov) with the following parameters: Query TMRGKKKRTRAN sequence, organism as Homo sapiens, protein-protein blast with the adjusted for short protein sequence algorithm. BLOSUM62 matrix was used as a scoring parameter.
Immunoprecipitation.
Immunoprecipitation assays were performed by TC71 and TC32 ET cell lysates with MLL3 (Santa Cruz, sc-13073), DDX27 (Santa Cruz, sc-81074), BRD9 (Santa Cruz, sc-102170), TXNDC9 (Santa Cruz, sc-100599) and FLI-1 (Santa Cruz C-19) antibodies according to the manufacturer's suggested protocol.
ESAP1-AP.
Oligonucleotides coding for the ESAP1 were cloned into pMY101 vectors to create ESAP1-alkaline phosphatase (ESAP1-AP) fusion proteins in bacteria.Citation35 DH5α-F' bacteria was transformed with the ESAP1-AP construct and protein expression was induced by 1 mM IPTG. The supernatant from the bacterial culture containing the ESAP1-AP fusion protein was used in direct binding assays. Recombinant EWS-FLI1 protein was immobilized on a 96-well plate and incubated with the ESAP1-AP fusion protein. Unbound ESAP1-AP fusion protein was washed and bound ESAP1-AP fusion protein was detected by adding the alkaline phosphatase substrate p-nitrophenyl phosphate. A colorimetric assay was performed to confirm binding to EWS-FLI1. For a separate experiment, ESAP1-AP conjugates were prepared with alanine substitutions throughout the peptide. One amino acid was mutated at a time.
Surface plasmon resonance.
Amine-coupling methodology was used to immobilize EWS-FLI1 to CM5 chip to a density of 350–400 maximum response units (RUmax). ESAP1 and mutant ESAP1 at varying concentrations ranging from 0–2 µM in a HBS-P buffer (GE-BR 1006-71) at pH 8.0 were injected. Affinity was calculated assuming first order binding between EWS-FLI1 and ESAP1 using Biacore T100 evaluation software (Uppsala, Sweden).
Cellular toxicity.
Initial cellular toxicity for the 27 ESAPs was assessed by triplicate plating at a density of 5,000 cells/well for SKNAS and 10,000 cells/well for TC32 cell lines in a 96 well plate. The cell lines were treated with 30 µM concentrations of each peptide. Cell viability was evaluated using a MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (Sigma M5655) or WST-1 (Roche 11644807001) assay according to manufacturer's protocol after 4 d. Later, ten thousand cells of TC32, TC71, A4573 cells and five thousand cells of SKNAS and PANC1 were plated for the assessment of ESAP1 cytotoxicity. All experiments were performed at least twice. Half maximal inhibitory concentrations (IC50) were calculated using GraphPad Prism software.
Confocal microscopy.
20 µM FITC-tagged wild type ESAP1 (TMR GKK KRT RAN-OH), −K (TMR GKT RAN-OH), and +K (GKK KRT-OH) was added two hours before microscopy in the TC71 cell line. Cells were also treated with Hoechst 3342 (Invitrogen-H3570) for nuclear detection. At the end of two hours treatment cells were washed with PBS to remove excess FITC-tagged peptides and Hoechst. Phenolred free RPMI (GIBCO 11835) with 10% FBS and 1% HEPES media were used during image acquisition. Olympus FV300 confocal microscope and 60x/1.4 N.A oil lens was used for imaging. Images acquired and merged with Fluoview 300 software.
Soft agar assays.
Soft agar assays were performed in TC71 and A4573 cell lines as previously reported in reference Citation36. Each of the cell lines were electroporated with 30 µg of EGFP fused ESAP1 encoding plasmid (pGESAP1) or empty vector (pG1).Citation37 After selecting cells for five days with G418 (Calbiochem 345812), soft agar colony formation assay was set up.Citation8 The colonies were stained with MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] for 2 h at 37°C. Microscopic images of EGFP-fusion protein expressed colonies were taken with a Nikon epifluorescence microscope.
Luficerase reporter assay.
Cos 7 cells were transfected with pciNeo-EWS-FLI1 or empty vectors along with NR0B1 reporter plasmid. Antennapedia fused ESAP1 was added to culture media after two hours of transfection. Cells were lysed 18 h after transfection with passive lysis buffer and luciferase activity was assessed according to manufacturer's suggestion (Promega Luciferase Assay Systems E1501). Exogenously expressed EWS-FLI1 protein levels were measured with protein gel blot.
Cell cycle arrest and cell cycle analysis.
TC71 cells were arrested at G2/M phase by 20 nM Nocadozole (Sigma M1404) treatment for 16 h. Media were replenished and arrest was released. After certain time points, cells were trypsinized and fixed with ethanol. Fixed single cell suspensions were analyzed for their DNA content by FACS to determine cell cycle status. ModFit software was used to evaluate cell cycle status of analyzed cells.
Quantitative RT-PCR and PCR array.
RNA was isolated from ESAP1-treated cells at indicated time points by using Qiagen RNA isolation kit. First strand synthesis (SABiosciences, C-03) and real time PCR analysis of cell cycle related genes (PAHS-020A-2) were performed as suggested in Qiagen's protocols in Eppendorf Real Time PCR equipment. Five different housekeeping genes were used as normalizers. The fold-difference in each gene was calculated by 2−ΔΔCt method respectively for each housekeeping gene. The average of five different folddifferences was reported as the fold-difference for genes. Taqman based primers and probe sets for GLI1 and GAPDH were purchased from ABI (Invitrogen-hs00171790_M1) and used as previously reported in reference Citation38. SYBR Green based primers from cyclin E and GAPDH were synthesized from Invitrogen.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| ET | = | ewing tumor |
| RHA | = | RNA helicase A |
| ESAP1 | = | ewing sarcoma antagonist peptide 1 |
| BRD9 | = | bromodomain containing 9 |
| DDX27 | = | DEAD (Asp-Glu-Ala-Asp) box polypeptide 27 |
| MLL3 | = | myeloid/lymphoid or mixed-lineage leukemia 3 |
| TXNDC9 | = | thioredoxin domain containing 9 |
| SPR | = | surface plasmon resonance |
Figures and Tables
Figure 1 ESAP1 directly binds to EWS-FLI1. (A) ELISA type assay using bacterial expressed and secreted alkaline phosphatase conjugated peptides with direct binding to full length recombinant EWS-FLI1. Vector only expressed and secreted alkaline phophatase conjugated peptide (■ V-AP) and ESAP1 encoded and secreted alkaline phosphatase conjugated peptide (▲ ESAP1-AP) were placed recombinant EWS-FLI1 coated wells, respectively. (● V-AP and □ ESAP1-AP peptides were placed on BSA coated wells). (B) Dose-dependent competition of synthetic ESAP1 against EWS-FLI1 and ESAP1-AP complexes measured using ELISA type assay with recombinant EWS-FLI1. (■ and ▲ represent EWS-FLI1 coated wells that were treated with ESAP1-AP and 1:10 diluted ESAP1-AP peptides, respectively. ▼ represents EWS-FLI1 coated wells that were treated with vector only-AP peptide. ◆ represents BSA coated wells that were treated with ESAP1-AP peptide). (C) Binding kinetics and the affinities of Antennapedia-fused synthetic ESAP1 is examined using SPR in Biacore T100. For this representative experiment, Ka = 1.436 × 107, Kd = 2.849 and KD = 1.984 × 10−7 M were calculated. X-axis is time (s), y-axis is arbitrary real-time binding unit (response unit-RU). Chi square was 152 and U was 4 for this specific example.
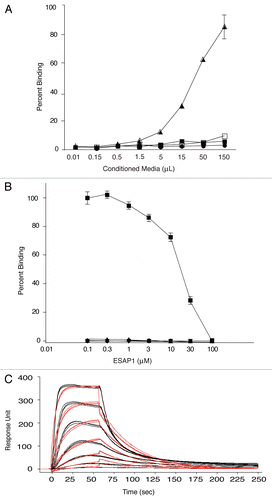
Figure 2 Lysine-rich core of ESAP1 required for cytotoxicity. (A) A dose-response experiment of FITC-tagged antennapedia fused ESAP1 (△), Antennapedia fused ESAP1 without FITC (■) and fragments (□-FITC-Antp-ESAP1+K, ○-FITC-Antp-ESAP1−K) were exposed to the TC32 cell line in monolayer growth. (B) FITC-tagged antenniapedia fused ESAP1, ESAP1+K and ESAP1−KI peptides entered TC71 cells in 20 min. Hoeschst 3342 is counter-stain for nucleus in the left column. Cellular peptide uptake is shown in middle column. Right column presents merged image of nuclear staining and FITC peptides.
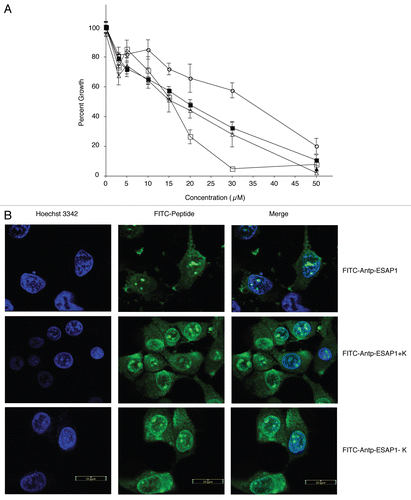
Figure 3 ESAP1 inhibits ET cell lines growth. (A) ET (▲, TC71 and ■, TC32) Type I fusion protein bearing ET cell lines, (▼, A4573) Type III fusion protein bearing ET cell line and cell lines without EWS-FLI1 (○, SKNAS-neuroblastoma and □, PANC1-Pancreas) were treated with ESAP1. Cells were incubated for 4 d followed by formazan dye-reduction assay to determine the number of viable cells. The assay was repeated between 2–4 times depending on the cell line. (B) An EGFP-ESAP1 fusion protein was expressed in ET (TC71) cells and impaired the anchorage independent growth by 4.45-fold. Fluorescent colonies were counted under an epifluorescence microscope. (C) Soft agar colony numbers significantly reduced in EGFP-ESAP1 expressing cell line (p = 0.0158).
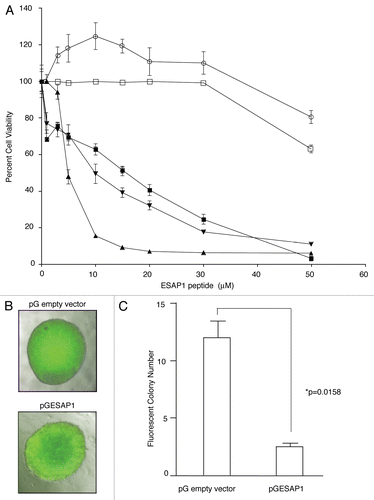
Figure 4 ESAP1 reduces EWS-FLI1 regulated transcript activation. (A) The NR0B1 promoter luciferase activity was evaluated for EWS-FLI1-dependent transcriptional activity in Cos-7 cells. (B) Exogenously expressed EWS-FLI1 levels were confirmed by immunoblotting. (C and D) GLI1 and cyclin E transcript levels were measured with qRT-PCR. mRNAs of synchronized cells (open bars) and the 8 h ESAP1-treated cells (gray bars) were isolated and subjected to the qRT-PCR with respective primers. GAPDH was used as a normalizer for fold difference calculation by 2−ΔΔCt method. Student's t-test was used to evaluate statistical significance of fold difference of the ESAP1 treatment. p values were given in the graph.
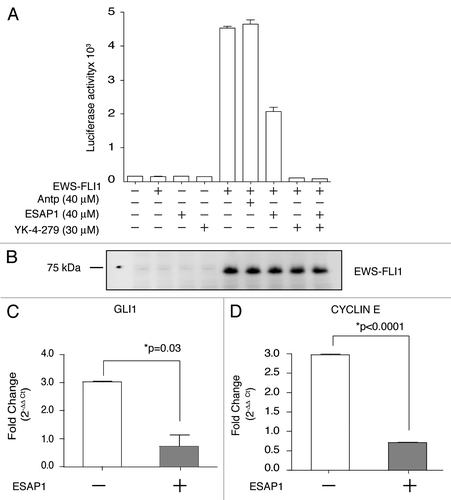
Figure 5 ESAP1 blocks cell cycle progression in a Ewing tumor cell line. Unsynchronized (A and B) or nocodazole synchronized (C and D) TC71 cells were treated with ESAP1 (B and D) or untreated in order to evaluate cell cycle kinetics. Cells were harvested at times indicated and treated with propidium iodide followed by flow-cytometric analysis. Cell phase percentages calculated in ModFit software. This experiment was representative of four repetitions. (E–H) The light microscope with 20x final magnification captured the representative morphologic appearance of the cells at the 24 h of the treatment respective to cell cycle evaluation. Sizing bar is equal to 100 µm. (I–L) Cell cycle histograms showed the examples of the DNA content aberrations. (M) Cell cycle real time PCR array results. Cellular mRNA was analyzed from the 8-h time point of ESAP1 treatment. The fold differences of mRNA expression were calculated with the five different housekeeping gene's averages for each gene using a 2−ΔΔCt method.
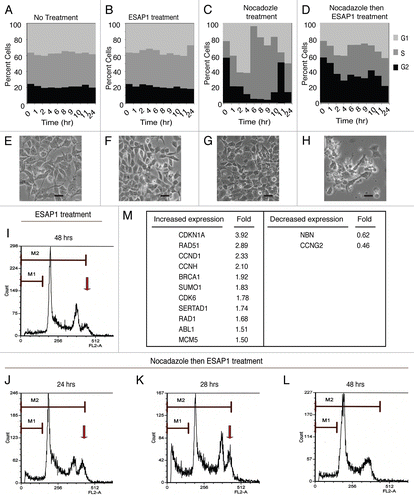
Table 1 Ewing sarcoma antagonist peptide sequences revealed in phage display
Table 2 KD values of peptides measured in SPR (µM)
Additional material
Download Zip (679.7 KB)Acknowledgments
This work was generously supported by the Children's Cancer Foundation of Baltimore, MD (J.T. and A.Ü.), Go4theGoal Foundation (J.T.), Dani's Foundation of Denver, CO (J.T.), Alex's Lemonade Stand Foundation (J.T.) the Liddy Shriver Sarcoma Initiative (J.T.), the Amschwand Sarcoma Cancer Foundation (J.T.), Burroughs-Wellcome Clinical Scientist Award in Translational Research (J.T.), NIH R01CA138212 (J.T.), R01CA133662 (J.T.), and RC4 (J.T.). NIH support through the following grants Cancer Center Support Grant P30 CA051008 for use of Biacore Molecular Interaction Shared Resource, Flow Cytometry and Cell Sorting Shared Resource and Microscopy and Imaging Shared Resource. We also thank Drs. Stephen Lessnick and J.V. Frangioni for providing reagents, for Ms. Veronica Rodriguez for technical assistance, Mrs. Kerry Adam for editorial assistance.
References
- Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003; 348:694 - 701; PMID: 12594313; http://dx.doi.org/10.1056/NEJMoa020890
- Üren A, Toretsky JA. Ewing's sarcoma oncoprotein EWS-FLI1: the perfect target without a therapeutic agent. Future Oncol 2005; 1:521 - 528; PMID: 16556028; http://dx.doi.org/10.2217/14796694.1.4.521
- Delattre O, Zucman J, Melot T, Garau XS, Zucker JM, Lenoir GM, et al. The Ewing family of tumors—a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994; 331:294 - 299; PMID: 8022439; http://dx.doi.org/10.1056/NEJM199408043310503
- Le Deley MC, Delattre O, Schaefer KL, Burchill SA, Koehler G, Hogendoorn PC, et al. Impact of EWS-ETS fusion type on disease progression in Ewing's sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol 2010; 28:1982 - 1988; PMID: 20308673; http://dx.doi.org/10.1200/JCO.2009.23.3585
- Kovar H. Downstream EWS/FLI1—upstream Ewing's sarcoma. Genome Med 2010; 2:8; PMID: 20156317; http://dx.doi.org/10.1186/gm129
- Burdach S, Plehm S, Unland R, Dirksen U, Borkhardt A, Staege MS, et al. Epigenetic maintenance of stemness and malignancy in peripheral neuroectodermal tumors by EZH2. Cell Cycle 2009; 8:1991 - 1996; PMID: 19502792; http://dx.doi.org/10.4161/cc.8.13.8929
- Erkizan HV, Uversky VN, Toretsky JA. Oncogenic partnerships: EWS-FLI1 protein interactions initiate key pathways of Ewing's sarcoma. Clin Cancer Res 2010; 16:4077 - 4083; PMID: 20547696; http://dx.doi.org/10.1158/1078-0432.CCR-09-2261
- Toretsky JA, Erkizan V, Levenson A, Abaan OD, Parvin JD, Cripe TP, et al. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Res 2006; 66:5574 - 5581; PMID: 16740692; http://dx.doi.org/10.1158/0008-5472.CAN-05-3293
- Petermann R, Mossier BM, Aryee DN, Khazak V, Golemis EA, Kovar H. Oncogenic EWS-Fli1 interacts with hsRPB7, a subunit of human RNA polymerase II. Oncogene 1998; 17:603 - 610; PMID: 9704926; http://dx.doi.org/10.1038/sj.onc.1201964
- Spahn L, Petermann R, Siligan C, Schmid JA, Aryee DN, Kovar H. Interaction of the EWS NH2 terminus with BARD1 links the Ewing's sarcoma gene to a common tumor suppressor pathway. Cancer Res 2002; 62:4583 - 4587; PMID: 12183411
- Nakatani F, Tanaka K, Sakimura R, Matsumoto Y, Matsunobu T, Li X, et al. Identification of p21WAF1/CIP1 as a direct target of EWS-Fli1 oncogenic fusion protein. J Biol Chem 2003; 278:15105 - 15115; PMID: 12560328; http://dx.doi.org/10.1074/jbc.M211470200
- Kim S, Denny CT, Wisdom R. Cooperative DNA binding with AP-1 proteins is required for transformation by EWS-Ets fusion proteins. Mol Cell Biol 2006; 26:2467 - 2478; PMID: 16537893; http://dx.doi.org/10.1128/MCB.26.7.2467-2478.2006
- Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing's sarcoma. Nat Med 2009; 15:750 - 756; PMID: 19584866; http://dx.doi.org/10.1038/nm.1983
- Metallo SJ. Intrinsically disordered proteins are potential drug targets. Curr Opin Chem Biol 2010; 14:481 - 488; PMID: 20598937; http://dx.doi.org/10.1016/j.cbpa.2010.06.169
- Üren A, Tcherkasskaya O, Toretsky JA. Recombinant EWS-FLI1 oncoprotein activates transcription. Biochemistry 2004; 43:13579 - 13589; PMID: 15491164; http://dx.doi.org/10.1021/bi048776q
- Ng KP, Potikyan G, Savene RO, Denny CT, Uversky VN, Lee KA. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proc Natl Acad Sci USA 2007; 104:479 - 484; PMID: 17202261; http://dx.doi.org/10.1073/pnas.0607007104
- Plescia J, Salz W, Xia F, Pennati M, Zaffaroni N, Daidone MG, et al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell 2005; 7:457 - 468; PMID: 15894266; http://dx.doi.org/10.1016/j.ccr.2005.03.035
- Mäe M, Langel U. Cell-penetrating peptides as vectors for peptide, protein and oligonucleotide delivery. Curr Opin Pharmacol 2006; 6:509 - 514; PMID: 16860608; http://dx.doi.org/10.1016/j.coph.2006.04.004
- Zwerner JP, Joo J, Warner KL, Christensen L, Hu-Lieskovan S, Triche TJ, et al. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene 2008; 27:3282 - 3291; PMID: 18084326; http://dx.doi.org/10.1038/sj.onc.1210991
- Beauchamp E, Bulut G, Abaan O, Chen K, Merchant A, Matsui W, et al. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J Biol Chem 2009; 284:9074 - 9082; PMID: 19189974; http://dx.doi.org/10.1074/jbc.M806233200
- Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med 2001; 7:673 - 679; PMID: 11385503; http://dx.doi.org/10.1038/89044
- Knoop LL, Baker SJ. The splicing factor U1C represses EWS/FLI-mediated transactivation. J Biol Chem 2000; 275:24865 - 24871; PMID: 10827180; http://dx.doi.org/10.1074/jbc.M001661200
- Fuxreiter M, Simon I, Bondos S. Dynamic protein-DNA recognition: beyond what can be seen. Trends Biochem Sci 2011; In press
- Pande J, Szewczyk MM, Grover AK. Phage display: concept, innovations, applications and future. Biotechnol Adv 2010; 28:849 - 858; PMID: 20659548; http://dx.doi.org/10.1016/j.biotechadv.2010.07.004
- He J, Li J, Ye C, Zhou L, Zhu J, Wang J, et al. Cell cycle suspension: A novel process lurking in G2 arrest. Cell Cycle 2011; 10:1468 - 1476; PMID: 21455017; http://dx.doi.org/10.4161/cc.10.9.15510
- Matsumoto Y, Tanaka K, Nakatani F, Matsunobu T, Matsuda S, Iwamoto Y. Downregulation and forced expression of EWS-Fli1 fusion gene results in changes in the expression of G(1)regulatory genes. Br J Cancer 2001; 84:768 - 775; PMID: 11259090; http://dx.doi.org/10.1054/bjoc.2000.1652
- Hu HM, Zielinska-Kwiatkowska A, Munro K, Wilcox J, Wu DY, Yang L, et al. EWS/FLI1 suppresses retinoblastoma protein function and senescence in Ewing's sarcoma cells. J Orthop Res 2008; 26:886 - 893; PMID: 18271016; http://dx.doi.org/10.1002/jor.20597
- Kauer M, Ban J, Kofler R, Walker B, Davis S, Meltzer P, et al. A molecular function map of Ewing's sarcoma. PLoS ONE 2009; 4:5415; PMID: 19404404; http://dx.doi.org/10.1371/journal.pone.0005415
- Khati M. The future of aptamers in medicine. J Clin Pathol 2010; 63:480 - 487; PMID: 20360137; http://dx.doi.org/10.1136/jcp.2008.062786
- Taraboletti G, Rusnati M, Ragona L, Colombo G. Targeting tumor angiogenesis with TSP-1-based compounds: rational design of antiangiogenic mimetics of endogenous inhibitors. Oncotarget 2010; 1:662 - 673; PMID: 21317461
- Verdine GL, Walensky LD. The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin Cancer Res 2007; 13:7264 - 7270; PMID: 18094406; http://dx.doi.org/10.1158/1078-0432.CCR-07-2184
- Joseph TL, Lane D, Verma CS. Stapled peptides in the p53 pathway: computer simulations reveal novel interactions of the staples with the target protein. Cell Cycle 2010; 9:4560 - 4568; PMID: 21088491; http://dx.doi.org/10.4161/cc.9.22.13816
- Anderson PM, Tomaras M, McConnell K. Mifamurtide in osteosarcoma—a practical review. Drugs Today (Barc) 2010; 46:327 - 337; PMID: 20517534
- Borghouts C, Tittmann H, Delis N, Kirchenbauer M, Brill B, Groner B. The intracellular delivery of a recombinant peptide derived from the acidic domain of PIAS3 inhibits STAT3 transactivation and induces tumor cell death. Mol Cancer Res 2010; 8:539 - 553; PMID: 20371673; http://dx.doi.org/10.1158/1541-7786.MCR-09-0417
- Yamabhai M, Kay BK. Examining the specificity of Src homology 3 domain—ligand interactions with alkaline phosphatase fusion proteins. Anal Biochem 1997; 247:143 - 151; PMID: 9126384; http://dx.doi.org/10.1006/abio.1997.2040
- Toretsky JA, Kalebic T, Blakesley V, LeRoith D, Helman LJ. The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J Biol Chem 1997; 272:30822 - 30827; PMID: 9388225; http://dx.doi.org/10.1074/jbc.272.49.30822
- Voss SD, DeGrand AM, Romeo GR, Cantley LC, Frangioni JV. An integrated vector system for cellular studies of phage display-derived peptides. Anal Biochem 2002; 308:364 - 372; PMID: 12419351; http://dx.doi.org/10.1016/S0003-2697(02)00268-3
- Beauchamp EM, Ringer L, Bulut G, Sajwan KP, Hall MD, Lee YC, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J Clin Invest 2011; 121:148 - 160; PMID: 21183792; http://dx.doi.org/10.1172/JCI42874