Abstract
Heterochromatin in S. pombe is associated with gene silencing at telomeres, the mating locus and centromeres. The compact heterochromatin structure raises the question how it unpacks and reforms during DNA replication. We show that the essential DNA replication factor Cdc18 (CDC6) associates with heterochromatin protein 1 (Swi6) in vivo and in vitro. Biochemical mapping and mutational analysis of the association domains show that the N-terminus of Cdc18 interacts with the chromoshadow domain of Swi6. Mutations in Swi6 that disrupt this interaction disrupt silencing and delay replication in the centromere. A mutation cdc18-I43A that reduces Cdc18 association with Swi6 has no silencing defect at the centromere, but changes Swi6 distribution and accelerates the timing of centromere replication. We suggest that fine tuning of Swi6 association at replication origins is important for negative as well as positive control of replication initiation.
Introduction
The fission yeast Cdc18 protein (ScCdc6) is an essential, conserved replication initiation factor found from archaea to eukaryotes (reviewed in ref. Citation1 and Citation2) Cdc18 binds to the origin recognition complex ORC at the replication origins, and is required for loading the MCM helicase to assemble the pre-replication complex (preRC). Following origin activation, Cdc18 is turned over so that the origins cannot be re-used until the next cell cycle. Cdc18 is regulated by multiple mechanisms including cell cycle dependent transcription, phosphorylation and ubiquitylation, and is thus ensures that individual replication origins are competent to fire only once per cell cycle.
In fission yeast, three large centromeres contain numerous replication origins that are activated early in the cell cycle.Citation3–Citation6 Each centromere contains an essential central core region, which is defined by the presence of the histone H3 variant, Cnp1 (H3Cen, Cenp-A) and by association with the kinetochore (reviewed in ref. Citation7). This central core is flanked by large, repetitive sequences in the pericentromere which are organized into inner and outer repeats. These repeats contain methylated histone H3K9 (reviewed in refs. Citation7 and Citation8). H3K9me recruits the heterochromatin protein Swi6 (HP1). Swi6 is highly conserved from fission yeast to mammals and consists of a chromodomain that binds methylated histones and a chromoshadow domain that is required for dimerization and binding to additional proteins.Citation9–Citation13 Binding of Swi6 to H3K9me causes repression of gene expression in the pericentromeric heterochromatin (reviewed in refs. Citation7–Citation9). In turn, Swi6 recruits additional Clr4 methylase, which leads to expansion of histone methylation and further Swi6 binding, until it reaches boundary regions that prevent further spreading. Swi6 is also required for silencing in the mating type locus and at the telomeres.
In addition to silencing the pericentromeric heterochromatin, Swi6 recruits cohesin proteins to this region.Citation14,Citation15 It also contributes to proper assembly of the central core histone Cnp1, and the kinetochore (reviewed in ref. Citation7). In the absence of Swi6, fission yeast cells are viable, but are defective in silencing and in centromere cohesion.Citation14–Citation16 This results in lagging chromosomes, causing an anaphase delay and increased rates of chromosome loss (reviewed in ref. Citation17).
Numerous observations suggest that there is a specific S-phase component to Swi6 function. Centromere heterochromatin is uniquely dynamic during S phase. In mammalian cellsCitation18,Citation19 and in fission yeast,Citation20,Citation21 Swi6 binding in the pericentromeric heterochromatin is disrupted in M phase of each cell cycle as a result of phosphorylation of the H3S10 residue, and rebinds by the end of the subsequent S phase. In S. pombe, this cyclical disruption allows a window of transcription from the pericentromere during S phase, which is used to generate siRNAs to target the Clr4 methyltransferase back to the centromere in order to modify new histones and restore silencing.Citation20,Citation21
However, Swi6 recruitment during S phase may not be solely dependent on methylated histone. First, association of the Swi6 homologue HP1 with heterochromatin in Drosophila requires association with the origin recognition complex ORC.Citation22–Citation25 In mouse, Orc1, Orc2 and Cdc6 also associate with HP1.Citation26 Second, the replication-specific chromatin assembly factor CAF1 interacts with HP1.Citation27–Citation29 CAF1 is associated with the elongating replication fork, and its association with HP1 may provide a mechanism to propagate already established heterochromatin onto newly synthesized DNA (reviewed in refs. Citation30 and Citation31). The Pob3 protein, a component of the FACT elongation complex that is associated with replication forks, is also required for heterochromatin assembly.Citation32,Citation33 Third, Swi6 assembly in fission yeast requires DNA polymerase alpha (Swi7/Pol1,Citation34). Both Swi7 and the pola associated protein Mcl1 are required for centromere silencing at the outer repeat and at the central core.Citation35,Citation36 Fourth, the S. pombe DDK kinase Hsk1, which is active only during S phase, associates with Swi6.Citation37 Recent studies suggest that Swi6 also recruits Hsk1 to ensure early replication timing of the centromere.Citation38 Swi6 association with both replication initiation factors (ORC, Cdc6, Hsk1) and with elongation factors (CAF, FACT, polα) suggests that Swi6 may contribute to regulation of replication, or that its silencing activity may depend on replication-specific functions. These possibilities are not mutually exclusive.
In this study, we identified S. pombe Cdc18 as a binding partner of Swi6 in vivo and in vitro, and biochemically mapped the binding domains in the two proteins. We used site-directed mutations in each protein to disrupt the interaction. These mutations do not affect overall chromosome replication or segregation, and do not affect bulk Swi6 localization. However, they cause modest silencing defects, changes in distribution of Swi6 in specific centromere regions, and changes in the timing of centromere replication. These results suggest that recruitment of Swi6 to the centromere is linked to preRC assembly, and implicate Swi6 in the regulation of replication initiation independent of its role in recruiting Hsk1.
Results
Isolation of Swi6 as a Cdc18-interacting protein.
In order to identify Cdc18-interacting proteins, we performed a yeast two-hybrid screen in duplicate. Each screen of 1 × 107 fission yeast cDNA clones yielded on average 30 positives. The most common isolate was Orc2, a component of the ORC complex, previously shown to interact with Cdc18 and Cdc2.Citation39 However, we also identified Swi6, the orthologue of metazoan Heterochromatin Protein 1 HP1.Citation16,Citation40 We recovered two isoforms of Swi6: a large fragment (Swi6 94–328) which is nearly full length, and a shorter truncated form (Swi6 267–328) which contains only the C-terminal chromo-shadow domain. An amino-terminally truncated Cdc18 (Cdc18-169-577) was still competent for association with Orc2, but unable to interact with Swi6, suggesting the amino terminus of Cdc18 is required for Swi6 association. We verified the results of the two hybrid screen by performing a co-immunoprecipitation experiment in vivo in a strain expressing tagged versions of Cdc18 and Swi6 from plasmids (). Thus, the two-hybrid result reflects a protein-protein interaction that occurs in vivo.
Mapping and mutational analysis of Swi6 binding domains.
To map the domains of Swi6 required for binding to Cdc18, we constructed a series of truncation and point mutations in Swi6 and tested them for their association with wild type Cdc18 by immunoprecipitation assays (). Consistent with the two hybrid data, we mapped the interaction of Cdc18 to the chromoshadow domain (CSD) of Swi6. Using deletion analysis, we identified the region of Swi6 that interacts with Cdc18 to the extreme C-terminus of the CSD. A substitution F324A prevented binding of Swi6 to Cdc18 in vitro. Structural studies have shown that this residue is part of the pocket in the CSD that binds to other proteins. The binding pocket is formed at the dimer interface of two CSD domains.Citation11–Citation13 We observed that mutation of a residue involved in homo-dimerization of the CSD (L315K) also abolished the interaction between Swi6 and Cdc18 ().
To assess the effects of mutations that disrupt the interaction between Swi6 and Cdc18 (and perhaps other proteins), we constructed GFP fusions of Swi6-L315K and Swi6-F324A. As controls we made similar constructs containing wild type Swi6 or Swi6 with substitutions in residues adjacent to those that disrupt Swi6-Cdc18 interactions (Swi6-T323A and Swi6-R325A). The constructs were each integrated into the chromosome under the control of the thiamine-repressible nmt1-81 promoter in Δswi6 cells (see materials and methods). All strains were viable and in the absence of thiamine, all expressed the Swi6 derivatives at a level similar to that of the endogenous gene in wild-type cells (Sup. Fig. 1).
Swi6-L315K and Swi6-F324A levels are reduced at the centromere.
We examined the distribution of Swi6 mutant proteins in the cells. Wild type Swi6 is normally localized to heterochromatin which clusters in 1–3 spots, corresponding to centromeres and telomeres, at the periphery of the nucleus.Citation16 This localization can be disrupted by loss of the methylation on histone H3K9, which is the binding site for Swi6 or by disruption of Swi6 dimerization.Citation11,Citation41 We determined the distribution and number of GFP-Swi6 foci in wild type and mutant strains (). Most mutants showed wild-type distribution of foci. However, Swi6-L315K severely disrupted focus formation, showing general nuclear staining with sporadic single focus in a small number of cells. This result suggests that most of the Swi6 mutations are competent for general heterochromatin, and suggests that Swi6-L315K may indeed be defective for dimerization.
We examined dimerization of Swi6 derivatives in vivo. The Flag-Swi6 plasmid was transformed into the GFP-Swi6 integrated strain with the same mutation (e.g., the Flag-Swi6-L315K plasmid was transformed into the GFP-Swi6-L315K integrant), and the dimerization was tested by coimmuniprecipitation. We saw that wild-type Flag-Swi6 coimmunoprecipitated with GFP-Swi6, as expected. No co-immunoprecipitation was detected in Swi6-L315K or Swi6-F324A under antibody concentrations sufficient to detect wild type (). Using increased antibody levels and long exposure times, there was a signal apparent in Swi6-F324A, but due to higher background levels, this result was not conclusive (data not shown); therefore we concluded that both L315K and L324A derivatives of Swi6 are somewhat impaired in dimerization, although probably not to the same degree.
swi6-F324A mutant disrupts silencing only at cen1.
The level of Swi6 at the centromere affects both heterochromatin structure and silencing.Citation16 We examined silencing using strains that contain a ura4+ or ade6+ reporter gene integrated at three regions of centromere 1: the outer repeats cen1L(dh) and cen1R(dg) and the inner repeat cen1L(imr). We also examined silencing in the outer repeats of cen2(dg) and cen3(dg) ( and D). Cells defective in silencing ura4+ can grow in the absence of uracil, but are killed by the suicide substrate 5-fluoro-orotic acid (5-FOA). Conversely, cells that silence ura4+ expression are unable to grow on media lacking uracil, but are resistant to 5-FOA. Cells deficient in adenine expression form pink or red colonies and are unable to grow on media lacking adenine. Consistent with the disrupted localization of GFP-Swi6-L315K foci, we found that swi6-L315K is defective for silencing of all the markers tested in all three centromeres ( and D). In contrast, swi6-F324A has a silencing defect primarily at the three insertions of centromere 1 and little or no defect in cen2 or cen3 ( and D).
We used chromatin immunoprecipitation (ChIP) with primers to the integrated ura4+ transgene to examine Swi6 occupancy at specific regions of the centromeres: cen1 otr1L(dh)::ura4+, cen2 otr2K″(dg)::ura4+ or cen3 otr2K3′(dg)::ura4+. These were normalized against a ura4-DS/E mini-gene at the endogenous ura4+ euchromatic locus, which is a non-functional allele with an internal deletion. As expected, Swi6-L315K localization was low at all sites ( and D), consistent with its impaired focus formation and defects in silencing. We detected Swi6-F324A at the reporters in all three centromeres, although levels were modestly reduced relative to wild type ( and D). To verify that these effects were not dependent on the integrated transgenes, we also performed ChIP using primers that recognize the dg repeats in all three centromeres (). We saw little reduction of Swi6-F324A, but strikingly reduced levels of Swi6-F315K. Therefore, the silencing phenotype reflects the amount of Swi6 at the centromere, and cen1 appears to be particularly sensitive.
Mutations that destabilize the centromere typically cause increased sensitivity to microtubule destabilizing agent thiabendazole (TBZ). We observed that Swi6-L315K and Swi6-F324A were both sensitive to TBZ, similar to Δswi6 (Sup. Fig. 2A). We also examined the frequency of lagging chromosomes during mitosis. These result from merotelic attachment of a single centromere to both spindle poles, which causes the attached sister chromatid to lag on the mitotic spindle. We synchronized the cells in hydroxyurea in early S phase and released into mitosis. We collected time points and stained the nuclei with DAPI. Unlike Δswi6, few of the mutations caused lagging chromosomes; surprisingly, even Swi6-L315K, which showed the most severe disruption of Swi6 localization, caused only a slight increase in the frequency of lagging chromosomes (Sup. Fig. 2B). This indicates that the central core of the centromere and the kinetochore are functionally intact in these mutant strains.
Identification of a Cdc18 allele defective for binding Swi6.
Using in vitro binding assays, we identified the region in Cdc18 required for Swi6 binding (). Again, consistent with the two-hybrid data, deletion of the first 50 amino acids of Cdc18 prevented its binding to Swi6 ( and B; Sup. Fig. 3) whereas deletion of first 40 amino acids barely affected the interaction. We used alanine scanning mutagenesis to narrow the region of Cdc18 that binds to Swi6 to residues 42–47. Finally we observed that the single substitution I43A was sufficient to reduce Cdc18 binding to Swi6 in vitro. This residue falls outside of the conserved part of the protein and lies in a relatively unstructured domain of the protein.Citation42 This region of Cdc18 is not required for its function in DNA replication. We integrated cdc18-I43A with an HA tag into the chromosome and observed no significant difference in Cdc18-I43A expression or cell growth compared to a control strain Cdc18-HA. The kinetics of bulk DNA replication in synchronized cell populations were also similar for mutant and wild type strains (Sup. Fig. 4). Next, we examined centromere function by monitoring TBZ sensitivity and fraction of lagging chromosomes in cdc18-I43A compared to the swi6 mutants. By itself, cdc18-I43A showed no defects and, when combined with swi6 mutations, did not exhibit any synthetic effects in these assays. These observations suggest that chromosome cohesion and mitotic function of the centromere are generally intact in cdc18-I43A, and further suggest that cdc18-I43A is epistatic with the swi6 mutants (Sup. Fig. 5).
cdc18-I43A affects Swi6 distribution.
We examined the effects of cdc18-I43A on expression of a normally silenced transgene integrated at three heterochromatin domains: the centromere subdomains, a telomere located on a minichromosome, and the mating type locus on chromosome II ( and Sup. Fig. 6). We observed no defect in silencing in any heterochromatin regions in cdc18-I43A. Chromosome segregation and Swi6 localization measured by GFP focus formation were unaffected by the cdc18-I43A mutation, suggesting that cdc18-I43A does not generally disrupt Swi6 throughout the centromere (Sup. Fig. 5 and data not shown). However, when we used chromatin immunoprecipitation to examine Swi6 at the ura4+ transgene in cen1(dg) repeat, we saw a modest, but statistically significant increase in its levels (). We next used primers that recognized the total dg and dh repeats. Again, we observed a trend towards increased Swi6 at dg repeats (across all three centromeres) and reduced Swi6 in total dh repeats (). This suggests that Cdc18 contributes to Swi6 distribution in subdomains of the centromere, and may help recruit Swi6 specifically to the dh regions.
Cdc18-I43A accelerates centromeric DNA replication.
Cdc18 is a rate-limiting factor required for replication origin assembly and firing (reviewed in ref. Citation1 and Citation2). In S. pombe, replication origins at the centromere fire early in the cell cycle and this is dependent on Swi6.Citation4,Citation6 However, Swi6 is dislodged from the chromatin in mitosis and only regains access during S phase.Citation20,Citation21 One possibility is that interaction between Cdc18 and Swi6 contributes to the recruitment of one of these factors. Therefore, we investigated how Swi6 and Cdc18 mutations affect replication within the centromere.
Replication can be monitored by the incorporation of the thymidine analogue BrdU into the DNA using a strain engineered to express thymidine kinase and a nucleoside transporter.Citation43,Citation44 We released cdc18-I43A and cdc18+ cells from G1 phase (nitrogen starvation) into hydroxyurea (HU), which allows early origins to fire but blocks completion of DNA synthesis and prevents firing of late origins.Citation5,Citation6,Citation45 We compared the timing of initiation at a known euchromatic early origin, ars2004, in wild-type and mutant cells. In wild-type cells, BrdU incorporation at ars2004 was detected beginning at 4 hours after release into HU, but was not observed at a control, non-origin sequence. The profile was similar for wild type, Δswi6, cdc18-I43A and swi6-F324A, indicating that under this protocol, replication timing in the euchromatin is unaffected by these mutations ().
Next, we used the same ChIP strategy to examine replication timing in the centromeres. We used one pair of primers specific for the dh repeats in all three centromeres, and another pair specific for the dg sequences in all three centromeres. A third pair of primers was specific for the inner repeat at cen1 (imr1). The final pair of primers identifies a central core sequence (cnt) in cen1 and cen3 (). Thus, most of our primers recognized repetitive sequences in all three centromeres. In wild-type cells, we saw BrdU incorporation in dg and dh repeats at low levels at 4 hr, reaching a maximum at 5 hr after release from nitrogen starvation. The inner repeat and central core were somewhat slower. In Δswi6 cells there was little incorporation of BrdU following HU arrest, suggesting that these otherwise early origins are unable to fire with early timing in Δswi6. This is consistent with a previous report using a slightly different method, and confirms that Swi6 is required for early replication of the centromeres.Citation38 Interestingly, the swi6-F324A strain showed slower replication timing at the dg repeat, although not as severe as Δswi6; other domains were not examined.
Finally, we examined incorporation of BrdU in cdc18-I43A cells. We observed a modest, but reproducible acceleration of centromere replication across all three centromeres, indicating that disruption of Swi6-Cdc18 binding actually speeds up DNA replication in the centromere domains.
Discussion
In this study, we used two-hybrid screening to identify fission yeast Swi6 and Orc2 proteins as binding partners of the essential replication factor Cdc18 (ScCdc6). Wild-type Cdc18 is essential for DNA replication and assembly of a pre-replication complex.Citation2 We show that the interaction of Cdc18 is disrupted by mutations in the Swi6 chromoshadow domain: F324A and L315K. Swi6-L315 corresponds to I165 in human HP1 which is predicted to lie at the dimerization interface, while Swi6-F324 corresponds to W174, which is important for binding other proteins.Citation11–Citation13 Swi6-L315K severely disrupts Swi6 localization at all heterochromatic loci, which is consistent with previous studies showing that dimerization through the chromoshadow domain is required for Swi6 assembly on the chromatin.Citation11–Citation13 Swi6-L315K also causes defects in silencing and chromosome segregation. Because Swi6-L315K mutation disrupts Swi6 localization and dimerization (), we conclude that these phenotypes are unlikely to be specific for the Cdc18 association, but probably represent defects in overall Swi6 structure and function.
Dimerization of the CSD is thought to form a pocket that binds a number of proteins, mediated by the F324 residue;Citation12,Citation13,Citation46 therefore, we consider it likely that Swi6-F324A is defective in a number of different protein associations. Although the Swi6-F324A is also defective in dimerization as detected by our in vivo co-immunoprecipitation, its phenotype is substantially different from swi6-L315K. This suggests that it is a hypomorphic allele, with an attenuation rather than complete disruption of Swi6 activity. First, focus formation is not disrupted in swi6-F324A cells, although its association at the centromere is modestly reduced. Second, swi6-F324A cells do not have silencing defects in cen2 or cen3, but curiously, only in cen1.
cen1 is the smallest centromere, and contains single dh and dg repeats on either side of the central core. In contrast, cen2 and cen3 have multiple copies of dh and dg on each side of their central cores. Additionally, the relative orientation of dh and dg in cen1 is different from that in cen2 and cen3: The cen1 dh repeat is the distal element in cen1, lying the furthest away from the central core. It is arranged in a head-to-head orientation with its adjacent dg repeat. In cen2 and cen3, a dg element is the most distal repeat, and dh-dg pairs are arranged head-to-tail. Thus, the boundary between dg and dh repeats is different in cen1, compared to cen2 and cen3, as is the boundary between the pericentromere heterochromatin domains and the adjacent euchromatin ().
We propose that cen1 is more sensitive to the level of Swi6 than cen2 and cen3 because of its unusual structure. We further suggest that the mechanism of centromeric heterochromatin formation is slightly different among centromeres, depending on the numbers and orientation of the repeats. Region-specific effects on centromere silencing and Swi6 localization were previously observed in mutants lacking the histone deacetylase Sir2, which specifically disrupts silencing at the inner repeat imr.Citation47,Citation48 Similarly, mutation of the tas3 gene specifically affects silencing at the imr, not the otr of cen1.Citation49 These reports suggest that recruitment, spreading or maintenance of Swi6 is differentially affected in different heterochromatin repeats within the centromere. Most of these studies have analyzed domains of cen1, so it is possible that these other phenotypes are also cen1-specific.
We identified the cdc18-I43A mutation by in vitro screening and found that does not cause any replication-associated defects or damage sensitivity in vivo. This region does not obviously match the PXVXL motif that is found in many Swi6-interacting proteins.Citation11,Citation46 However, the PXVXL sequence is not essential. Mammalian PML body protein SP100 also binds the HP1 chromodomain and does not have a clear match to this motif.Citation50 cdc18-I43A has little effect on Swi6 function in heterochromatin. There was no increase of TBZ sensitivity nor lagging chromosomes in cdc18-I43A, indicating that overall centromere function is unperturbed by cdc18-I43A. However, there is a modest change in Swi6 distribution in cdc18-I43A compared to wild type: it is slightly increased at cen1 dg repeats, but reduced at dh repeats.
Current models for Swi6 assembly suggest that the protein can spread from its initial nucleation into adjacent domains along methylated chromatin. Since we see a change in the distribution of Swi6 at dg and dh repeats in cdc18-I43A, we suggest that nucleation of Swi6 after mitosis also depends on the pre-RC at the centromere, particularly in early S phase, when H3K9me is limited. Swi6 accumulation thus partly depends on origin binding via Cdc18 in the dh repeat. We propose that the assembled replication origin may provide an initial nucleation event to localize Swi6 in the vicinity of origins during early S phase, independent of histone methylation. It has been shown that there are replication origins with the dh and dg repeats.Citation3 The abnormal Swi6 distribution at dh and dg repeats in cdc18-I43A mutant implies that replication factors other than Cdc18 may contribute to Swi6 nucleation during early S phase. Consistent with a role for origin proteins in recruiting Swi6, we have also observed association between ORC and Swi6 in fission yeast (Li PC and Forsburg SL, unpublished). This may be a generally important mechanism, because HP1 also interacts with ORC and Cdc6 in metazoans.Citation23,Citation26,Citation51 Both S. pombe and S. cerevisiae replication origins are associated with initiation of silent heterochromatin domains (reviewed in ref. Citation52); additionally, mutations in S. cerevisiae ORC genes are specifically associated with silencing defects.Citation53–Citation55
Does this interaction affect DNA replication? Centromere heterochromatin in fission yeast replicates early.Citation4–Citation6 In budding yeast, early centromere replication has been suggested to be important for establishment of proper centromere orientation and function.Citation56 In fission yeast, we observed that centromere replication in Δswi6 cells was severely delayed relative to wild type, indicating that Swi6 has a positive role in replication. Our results are consistent with recent work,Citation38 which also showed that fusing the Swi6 chromodomain to the DDK kinase subunit Dfp1 is sufficient to rescue early centromere replication timing in Δswi6 cells. DDK is known to be required for the initiation of DNA replication (reviewed in ref. Citation1 and Citation2). This suggests that Swi6 plays a positive role in promoting DNA replication initiation, by recruiting DDK.
Our data suggest that Swi6 also has an inhibitory effect on replication initiation, because specifically disrupting the interaction between Swi6 and Cdc18 accelerates centromere replication. This suggests that the interaction between the two has a negative role in replication timing. Such an inhibition could operate either at the level of origin timing, or at the level of number of origins fired. Thus, we propose that Swi6 has both positive and negative roles in centromere replication (). The positive effect reflects the Swi6-dependent enrichment of DDK kinase in the heterochromatin region,Citation38 which may act directly to promote origin firing. The most parsimonious model suggests that Swi6 binding to Cdc18 may antagonize DDK and prevent it from acting prematurely at the origin ( and ii). In the absence of Swi6, the H3K9me chromatin is refractory to early replication, possibly because of other methyl-histone binding complexes that delay origin activation or unwinding (). If the interaction with Cdc18 is selectively disrupted, the Swi6-DDK recruited to the adjacent histones is sufficient to activate all the origins with accelerated timing (). This implies that Swi6-DDK leads to a “hyper-activation” phenotype, perhaps due to increased concentration of the replication-inducing kinase in the vicinity of origins. Relief of the Cdc18-Swi6 inhibition may depend on accumulating a certain level of DDK. Since swi6-F324A is expected to disrupt additional interactions, not just Cdc18, it is likely that it also disrupts DDK binding; this would be consistent with its delayed replication timing. Future experiments will examine how DDK recruitment contributes to these phenotypes.
Combined with the data from reference Citation20 and Citation21, investigating the timing of Swi6 association during S phase, our findings support a model in which Swi6 recruitment in S phase occurs in two waves: an initial recruitment of Swi6 to the preRC and to residual H3K9me residues at the beginning of S phase, which contributes to replication timing, followed by a significant increase of Swi6 at the end of S phase, as the RNAi-driven histone methylation provides additional binding sites for a final burst of Swi6 recruitment. Thus, the phenotypes we observe reflect changes in distribution of Swi6 required for formation or maintenance of contiguous heterochromatin and regulated origin activity. Further investigation will be required to dissect the components of regulated heterochromatin replication and the effects of histone methylation on replication timing.
Materials and Methods
Yeast strains and media
Strains used in this study are listed in Supplemental Table 1. Fission yeast strains were grown and maintained on yeast extract plus supplements (YES) or Edinburgh minimal media (EMM) with appropriate supplements, using standard techniques.Citation57 Mating was performed on synthetic sporulation agar (SPAS) or Malt Extract (ME) plates for 2–3 days at 25°C. Temperature sensitive cells were grown at 25°C and non-ts cells at 32°C. Transformations were carried out by electroporation. G418 plates were YES supplemented with 100 µg/ml G418 (Sigma). Double mutants were constructed by standard tetrad analysis or random spore analysis. TBZ plates were EMM supplemented with 17 µg/ml TBZ (Sigma). For silencing assay, glutamate was used to replace NH4Cl as nitrogen source and 0.1% FOA was used.
Yeast two-hybrid screen.
A two-hybrid screen was performed with S. cerevisae reporter strain PJ694A, two S. pombe cDNA libraries (provided by Dr. G. Hannon and Dr. S. Elledge) and the pDBTrpcdc18 bait plasmid. The screen was performed according to Clontech Matchmaker protocol. The pDBTrpcdc18 plasmid was constructed as follow; the ORF encoding Cdc18 protein was amplified by PCR and cloned as a Sal1/Not1 fragment into the pBDTrp vector (Invitrogen Life Technologies).
Expression and purification of fusion proteins.
For the fusion plasmid MAL-Myc-tagged Cdc18, the Myc epitope tag was first cloned as a BamHI/XbaI fragment into the pMAL-c2X vector (New England Biolabs) followed by the insertion of the cdc18+ gene (using Stu1/Xba1 digestion sites). Deletion mutants of Cdc18 were generated by PCR and also inserted as Stu1/Xba1 fragments. Site-directed mutagenesis was performed using QuickChange PCR (Stratagene). Constructs were transfected in the E. coli derived BL21-Gold(DE3) competent cells (Stratagene) and the fusion proteins were purified using amylose resin as directed by the manufacturer (New England Biolabs (NEB) pMAL™ ProteinPurification System protocols). Briefly, the expression strains were grown in LB and protein expression was induced with 0.35 mM IPTG for 5 hrs at 20°C. Cells were harvested and the pellet was washed and resuspended in Amylose Column Buffer (20 mM Tris-HCl pH 7.5, 0.5 M NaCl, 1 mM EDTA pH 8.0, 10 mM β-mercaptoethanol, 10% glycerol, 1% Triton-X100, 2.5 µg/ml soybean trypsin inhibitor, 4 µg/ml aprotinin, 2 mM pefabloc, 20 µg/ml leupeptin, 20 µg/ml bestatin, 50 µg/ml TLCK, 2 µg/ml pepstatin) and then frozen at −20°C. For the purification, cells were broken and the crude extracts were added to amylose resin. Beads were washed with Amylose Column Buffer and the protein was eluted with 10 mM maltose. Purified protein was stored at −80°C.
The fusion plasmid 6 his-FLAG-Swi6 was constructed as follow: the FLAG epitope tag was first cloned as a Xho1/BamH1 fragment into the pET15b vector (Novagene) followed by the insertion of the Swi6+ gene (using Stu1/BamH1 digestion sites). Deletion mutants of Swi6 were generated by PCR and also inserted as Stu1/BamH1 fragments. Site-directed mutagenesis was performed using QuickChange PCR (Stratagene). Recombinant proteins were expressed in E. coli derived BL21-Gold(DE3) competent cells (Stratagene) and purified on a nickel resin column according to instructions provided by the manufacturer (Qiagen, Inc.). Briefly, the expression strains were grown in LB and protein expression was induced with 0.35 mM IPTG for 5 hrs at 20°C. Cells were harvested and the pellet was washed and resuspended in Nickel Column Buffer (50 mM Hepes pH 7.5, 0.5 M NaCl, 2.5 mM MgCl2, 10% glycerol, 2.5 µg/ml soybean tryspsin inhitor, 4 µg/ml aprotinin, 2 mM pefablock, 20 µg/ml leupeptin, 20 µg/ml bestatin, 40 µg/ml TLCK, 2 µg/ml pepstatin) and then frozen at −20°C. For the purification, cells were broken and crude extracts were added to Ni-NTA resin. Beads were washed with Nickel Column Buffer containing 5 mM imidazole and the protein was eluted with buffer containing 250 mM imidazole. Purified protein was stored at −80°C.
Construction of tagged strains.
To construct strains with tagged episomal versions of Cdc18 and Swi6, the cdc18+ open reading frame was amplified from S. pombe cDNA using PCR and cloned as a Xho1/Sma1 fragment into the pSLF172 vectorCitation58 downstream of the nmt1 promoter, generating a Cdc18 tagged with the triple HA epitope sequence at the C-terminus. The FLAG-tagged Swi6 open reading frame was amplified from the S. pombe plasmid pET15b FLAG-tagged Swi6 (see below) as a Xho1/BamH1 fragment and cloned into pREP81XCitation59 downstream of the nmt promoter. The two resulting plasmid were transformed into the haploid S. pombe TK3 (leu1-32 ura4-D18 ade6-M210) strain. Ura+ Leu+ transformants were selected and screened for expression of HA and FLAG tags; Cdc18 was detected by immunoblotting with anti-HA monoclonal antibody (F7) and FLAG-Swi6 was detected with anti-FLAG monoclonal antibody (M2). Transformants expressing both epitope tags were selected.
To construct strains with endogenously tagged Cdc18 mutants, the cdc18+ promoter and 3′UTR were isolated by PCR of genomic DNA and cloned into the S. pombe plasmid pIRT2U.Citation60 The tagged cdc18 gene (cdc18-3HA) was amplified by PCR from S. pombe plasmid pSLF172 cdc18-3HA and cloned into the pIRT2U vector in between the cdc18+ promoter and 3′UTR. Site-directed mutagenesis of pIRT2U-cdc18-3HA to generate single base substitution mutations (I43A) was performed using QuickChange PCR (Stratagene). The 3HA-tagged cdc18+ gene was excised from the construct by Sma1/Sal1 digestion and transformed into haploid S. pombe AR 5504 strain carrying a chromosomal disruption of the cdc18+ gene (cdc18::ura4+) and a wild-type copy of cdc18+ on the plasmid prep81X. Transformants were grown in presence of FOA (50 µg/ml Ura, 0.1% FOA). Ura4-minus transformants were selected and screened by PCR for the cdc18 3HA+ gene. The plasmid was cured by outgrowth on nonselective media and all subsequent experiments were carried out in presence of 5 ug/ml of thiamine.
To construct strains with GFP-Swi6 mutations, two step cloning was performed. First, pET15a-6 his-Flag-Swi6 mutants (see below) were digested with NotI/BglII and cloned into pBAR1253-GFP vector. The GFP-swi6 mutant constructs were further digested with PstI/SacI and cloned into integration plasmid pJK148. The plasmid was linearized by NruI and integrated into the leu1-32 locus in strain FY2228, which has no endogenous swi6+. Leu+ transformants were selected and confirmed by western blot using Swi6 antibody.
Immunoprecipitation.
Cells (1L) co-expressing Cdc18 tagged-3HA and FLAG-tagged Swi6 proteins were grown to log phase and then blocked in G1 with 25 mM hydroxyurea (HU) for 4 h. Cells were harvested and washed first with ice-cold water followed by a wash with 1x Lysis Buffer (LB) (50 mM Hepes pH 7.5, 1 mM EDTA pH 8.0, 200 mM NaCl, 1 mM β-mercaptoethanol, 10% glycerol, 0.5% Triton X-100). Cells were pelleted, weighed and resuspended in an equal volume 2x LB (containing the following protease inhibitors 2 mM pefabloc, 10 mM benzamidine, 10 µg/ml leupeptin, 20 µg/ml bestatin, 2 µg/ml pepstatin A, 10 µg/ml aprotinin, 2 mM soybean trypsin inhibitor, 40 µg/ml TLCK). Cells in suspension were dropped into liquid nitrogen. The frozen cell suspension was stored at −80°C. To prepare extracts, cells were broken with a coffee mill. Briefly, 60 g dry ice was reduced to powder in a pre-chilled coffee mill. The frozen cell suspension was added to the chamber and grounded for 5 minutes with constant agitation. The extract was collected, placed on ice and the dry ice was left to sublimate. The extract was centrifuged in a tabletop Eppendorf centrifuge at 14,000 rpm for 20 minutes at 4°C. The resulting supernatant was spun at 100,000 g for 45 minutes at 4°C. The supernatant was recovered, added to 500 µl anti-FLAG (M2) agarose beads (Sigma) and incubated for 1 h at 4°C with gentle agitation. The beads were pelleted and washed with 20 beads volume of 1 X LB. The beads were finally resuspended in 1x LB and the bound proteins were analysed by SDS-PAGE. Cdc18 was detected by immunoblotting with anti-HA monoclonal antibody (F7). FLAG-Swi6 was detected with anti-FLAG monoclonal antibody (M2) and the endogenous Swi6 with anti-Swi6. Immunoprecipitation assays carried out with the FLAG peptide competitor or without anti-FLAG (non-conjugated sepharose beads were included as a negative control).
Dimerization of Swi6 mutants was determined by co-immuniprecipitation of GFP-swi6 and Flag-swi6. GFP-swi6 integrated cells were transformed with either pREP81X-Flag (#1604) or pREP81X Flag-Swi6 plasmids (#1605, 1606, 1607) carried the same swi6 mutation, and grown in selected medium. 2 µg of soluble protein in B88 lysis buffer, with protease and phosphatase inhibitors, was immunoprecipitated with 10 µl of FLAG M2-agarose conjugated beads (Sigma). Co-immunoprecipitation was detected by western blotting with anti-Swi6 antibodies. Two transformed colonies were tested for each mutant.
In vitro co-precipitation assay.
Pull-down assay were carried out with purified 6 his-FLAG-Swi6 and MAL-Myc-tagged Cdc18 proteins. 0.3 µg 6 his-FLAG-Swi6 was added to 50 µl anti-FLAG (M2) agarose beads and incubated for 1 h at 4°C with gentle agitation. Beads were washed 5 times with 1 ml of pull-down buffer (PB) (50 mM Hepes pH 7.5, 150 mM NaCl, 2.5 mM MgCl2, 1 mM β-mercaptoethanol, 10% glycerol, 2.5 µg/ml soybean tryspsin inhibitor, 4 µg/ml aprotinin, 2 mM pefabloc, 20 µg/ml leupeptin, 20 µg/ml bestatin, 40 µg/ml TLCK, 2 µg/ml pepstatin, 1% triton X-100). 0.3 µg MAL-Myc-tagged Cdc18 proteins was added to anti-FLAG (M2) agarose beads and incubated for 1 h at 4°C with gentle agitation. Beads were washed 5 times with 1 ml of pull-down buffer PB. The beads were finally resuspended in PB and analysed on gel. Cdc18 detected (Santa Cruz) and Swi6 with (Sigma). The beads were finally resuspended in 1x LB and the bound proteins were analysed by SDS-PAGE. Cdc18 was detected by immunoblotting with anti-Myc monoclonal antibody (9E10). FLAG-Swi6 was detected with anti-FLAG monoclonal antibody (M2) and the endogenous Swi6 with anti-Swi6. Pull-down assays carried out without pre-incubation with 6 his-FLAG-Swi6 was included as a negative control. The anti-HA monoclonal antibody (F7) and anti-Myc monoclonal antibody (9E10) were purchased from Santa Cruz Biotechnology. The anti-FLAG monoclonal antibody (M2) was purchased from Sigma. The anti-Swi6 polyclonal antibody was raised by injecting a rabbit with purified 6 his-FLAG-Swi6.
Silencing assay.
Silencing assays were performed described previously in reference Citation48. with the following modifications. Cells were grown to mid-log phase to 5–10 × 106 cells/ml and spotted on selective medium with 1:5 or 1:10 serial dilutions. For ura4+ expression, cells were spotted on PMG lacking uracil, non-selective medium or non-selective with 1 g/L of 5′ FOA. For ade6+ expression, cells were spotted on EMM lacking adenine, 10 mg/L of adenine or non-selective medium. Cells were grown at 32°C for 3–4 days.
RT-PCR.
Cells were grown to mid-log phase and broken in RNA lysis buffer (50 mM NaOAC, 10 mM EDTA, 1% SDS) by acid-washed glass beads. Total RNA was extracted by phenol-chloroform and phenol:chloroform:isoamyl extraction followed by ethanol precipitation. Total RNA was resuspended in TE and checked for integrity by formaldehyde/agarose electrophoresis. The cDNA strand was synthesized with equal amount of total RNA using First-strand cDNA synthesis kit (Amersham) following the manufacturer's instructions. The amount of ura4+ RNA expression was determined by PCR and analyzed by BioRad FX machine and Quantity One software. Results were plotted as fold expression relative to the mini-gene ura4DS/E, located at the endogenous ura4+ locus.
Chromatin immunoprecipitation.
Cells were grown to mid-exponential phase and cross-linked with 1% formaldehyde at room temp for 15 min. The reaction was stopped by 125 mM Glycine at room temp for 5 min. Cells were washed once with TBS and transferred to a 2 ml screw-cap tube. After suspending in 500 µl of ice-cold ChIP lysis buffer (50 mM HEPES/KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1%) Na-Deoxycholate, protease inhibitors cocktail (P8215, Sigma) and 500 µl acid-washed glass-beads (Sigma), cells were broken by Fast-Prep (power 5.5, 45 sec repeated 3 times) or vortex (5 min for 3 times). The genomic DNA was sheared by sonication to between 500–1,000 bp. 2 mg of the cell lysate was pre-cleared by incubation with 30 µl of protein A beads (ThermoScientific) at 4°C for 1 hour. Swi6 antibody,Citation37 GFP antibody (ab-290, Abcam) or H3K9me3 antibody (ab-8898, Abcam) was used for immunoprecipitation at 4°C for 2–3 hours. After incubation with 30 µl of protein A beads at 4°C for 1 hour, the beads were washed by the following buffers on the rotator for 5 min each: 1 ml of ChIP lysis buffer for twice, 1 ml of ChIP lysis buffer/500 mM NaCl, 1 ml of Wash buffer (10 mM Tris/HCl pH 8, 250 mM LiCl, 0.5% NP-40, 0.5% Na-Deoxycholate, 1 mM EDTA) and 1 ml of TE. After aspirating the buffer, the beads were suspended in 100 µl TES (TE with 1% SDS) and incubated at 65°C for 30 min. The supernatant was transferred to a new tube and cross-linking reversed for 16 hours at 65°C. After incubation with 50 µg/µl of protease K at 37°C for 2 hours, the DNA was purified by PCR Purification Kit (Qiagen). The PCR products were visualized by SYBR Green stained agarose gel and quantified by Quantity One software (Bio-rad). The experiment was repeated three times independently. Results were determined as fold expression relative to the mini-gene allele ura4DS/E at the endogenous ura4+ locus.
BrdU ChIP.
Strains competent to take up exogenous thymidine were used.Citation44 Cells were grown to OD600 0.3–0.5 and nitrogen starved for 16 hours at 25°C. Cells were released by refeeding with nitrogen-containing medium at 25°C. After 1.5 hours release, 100 µg/ml of BrdU (Sigma) and 10 mM hydroxyurea (Sigma) were added and indicated time points were collected. Harvested cells were fixed by 0.2% of sodium azide and washed by ice-cold TBS and transfered a to 2 ml screw-cap tube. Cells were suspended in TES (TE with 1% SDS) and broken by glass-beads lysis. The DNA was sheared by sonication to an average size of ∼500 bp. The BrdU-incorporated genomic DNA was extracted by phenol/chloroform and precipitated by ethanol. The samples were treated with RNase and protease K and purified by PCR purification Kit (Qiagen). The BrdU immunoprecipitation was performed as follows: The mixture of 40 µl of extracted DNA and 20 µg of salmon sperm single strand DNA (Invitrogen) was denatured at 95°C for 5 min and snap-cooled on ice-water. 0.5 µl of BrdU antibody (GE Healthcare) was added and incubated for 1 hour at 4°C followed by G-Sepharose beads (Pierce). Reverse cross-linking was performed at 65°C for 15 min followed by PCR purification kit (Qiagen). The immunoprecipitated material and a 1:100 dilution input control were monitored by PCR as follows: 20 cycles were used for ars2004, non-ars, dg and dh PCR; 23 cycles were used for imr and cnt. The PCR products were monitored by SYBR Green stained agarose gel, scanned on an FX machine (BioRad) and quantified by Quantity One software (BioRad). The sequences of primers is listed on the Supplemental Table 2. The experiment was repeated three times independently.
Microscopy.
Whole fixed cells were stained as described for DAPI.Citation61 Live cells were patched on positively charged slides (Fisher Scientific). Cells were visualized with a Leica DMR microscope. Objectives used were Leica 63X/1.32, PLApo, NA = 1.32. Images were captured with a Hamamatsu digital camera and Improvision Openlab software (Improvision, Lexington, MA). Images were assembled using Canvas (ACD/Deneba) and adjusted for contrast.
Abbreviations
| BrdU | = | bromodeoxyuridine |
| ChIP | = | chromatin immunoprecipitation |
| CSD | = | chromoshadow domain |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| EDTA | = | ethylenediaminetetraacetic acid |
| GFP | = | Green fluorescent protein |
| H3K9me | = | histone H3K9 methylated |
| HU | = | hydroxyurea |
| preRC | = | prereplication complex |
| RT-PCR | = | reverse transcriptase polymerase chain reaction |
| SDS | = | sodium dodecyl sulfate |
| TBZ | = | thiabendazole |
Figures and Tables
Figure 1 In vivo co-precipitation of Cdc18 and Swi6. Cell extracts were prepared from transformants expressing pREP4X cdc18 3HA and pREP81X FlagSwi6. In vivo binding studies were performed by immunoprecipitating cells extracts on FLAG M2-agarose conjugate beads in the absence (F1, B1) or presence (F2, B2) of FLAG peptide or by incubating the extracts on Sepharose beads (F3, B3). Immunoblotting was done with F7 anti-HA antibodies for cdc18 and anti-FLAG or polyclonal antibody for Swi6. I input, F flow through; B beads.

Figure 2 Mapping of the Swi6 interaction domain by in vitro binding. (A) Schematic representation of the Swi6 protein and mutational analysis. The chromo domain (CD) is linked to the chromo shadow domain (CSD) through the hinge (H) region summarizes the binding results. (B) In vitro binding studies were performed by precipitation of bacterial recombinant His6FLAG-Swi6 wild-type or fragments or mutants with FLAG M2-agarose conjugate beads. Recombinant MBP-Myc Cdc18 wild type was added to the beads and its binding capacity was measured by western blotting. Immunoblotting was performed with anti-Myc antibodies for Cdc18 or anti-FLAG antibodies for Swi6.
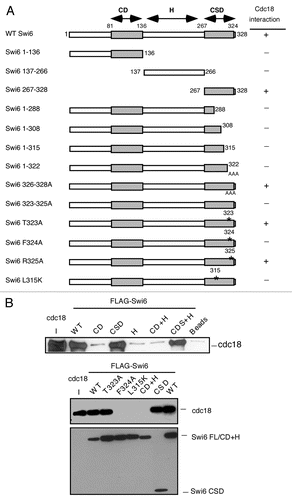
Figure 3 Characterization of Swi6 mutations in vivo. (A) Visualization of Swi6-GFP foci in live cells. Quantification of GFP foci results of wild type (FY3386), swi6-L315K (FY3388), swi6-T323A (FY3389), swi6-F324A (FY 3390) and swi6-R325A (FY3391), showing fraction of cells with 0, 1 or ≥2 foci. 100 cells were counted and repeated three times independently. (B) Dimerization in vivo of mutant forms of Swi6. GFP-swi6 integrated cells were transformed with either pREP81X-Flag or pREP81X Flag-Swi6 plasmids containing the same version of Swi6, and grown in selected medium. Soluble protein was immunoprecipitated with FLAG M2-agarose conjugated beads. Coimmunoprecipitation was detected by western blotting with anti-Swi6 antibodies. Two transformed colonies were tested in each mutant. (C) Silencing of the ura4+ or ade6+ marker in the cen1 dh, imr or dg repeats (indicated by the schematic). Expression in different swi6 mutants was determined by sensitivity to FOA or viability on adenine-minus medium. N/S, non selective (Uracil+) medium. Swi6 localization in the cen1 dh repat was measured by chromatin immunoprecipitation (ChIP) using primers to detect an integrated ura4+ marker compared to a ura4-DS/E minigene that contains an internal deletion, which is located at the normal (euchromatic) locus. The same primers detect both ura4-DS/E and ura4+. W: Input; I: IP. Swi6-F324A localization at cen1(dh) is significantly lower than wild type, with a p value of 0.048. (D) Expression of ura4+ marker in cen2 or cen3 dg repeat in different swi6 mutants was determined by sensitivity to FOA. Swi6 localization at cen2 dg or cen3 dg was performed as in (C), with the indicated significance values. (E) Swi6 localization in different mutants at global dg was measured by ChIP using total dg primers compared to act1. Swi6-F324A is lower but not significant than wild type. Statistical analysis compared three independent experimental samples using the Student's t-test.
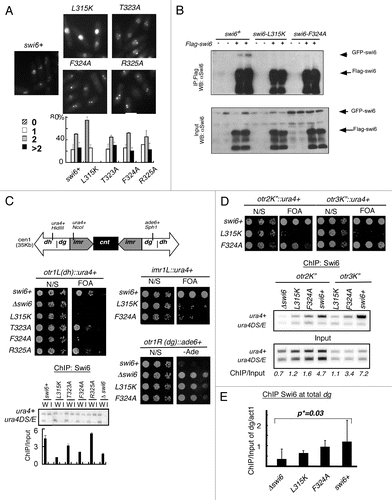
Figure 4 Mapping of Cdc18 interaction domain by in vitro binding. (A) Schematic representation of the Cdc18 protein and mutations. The N-terminal portion includes 7 Cdc2 phosphorylation sites (*). The protein also contains the ATP Walker A and B motifs. +, interaction between Cdc18 and Swi6; −, no interaction. (B) In vitro binding studies were performed by immunoprecipitation of recombinant MBP-Myc Cdc18 wild-type or fragments or mutants in absence (Fo, Bo) or presence (F, B) of bacterially produced recombinant His6FLAG-Swi6 wild-type on FLAG M2-agarose conjugate beads. Cdc18 binding was measured by western blotting. Immunoblotting was performed with anti-Myc antibodies for cdc18 or anti-FLAG antibodies for Swi6. I, input; Fo/F, flow through; Bo/B, beads.
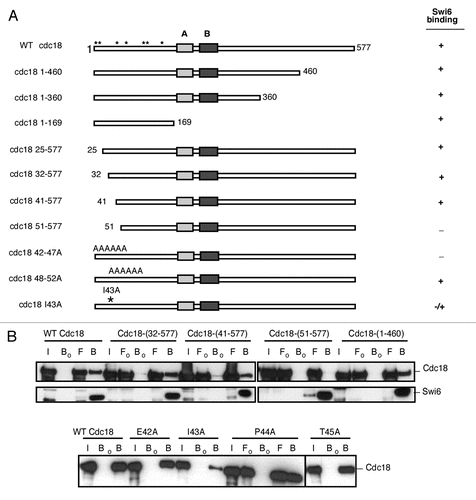
Figure 5 Region specific silencing defects in cdc18-I43A. (A) Expression of the ura4+ or ade6+ marker at the cen1 dh, imr or dg repeat in cdc18-I43A mutant was determined by senstitivity to FOA or viability on minus adenine medium. N/S, non selective (Ura+) medium. (B) Expression of ura4+ marker in cen2 or cen3 dg repeat was determined by sensitivity to FOA. (C) Swi6 localization at transgenes in cen1 dg repeat (ade6+) in cdc18-I43A was determined by ChIP. As an internal control, the same primers detect an ade6ΔN/N minigene at their normal euchromatic loci. Swi6 localization at global dg (D) or dh (E) repeat in cdc18-I43A mutant was determined by ChIP compared to act1. Swi6 in cdc18-I43A is significant higher at cen1(dg) and total dg regions, with p value of 0.027 and 0.002, but lower at dh regions. Statistical analysis was performed on 3 independent experimental samples using the Student's t-test.
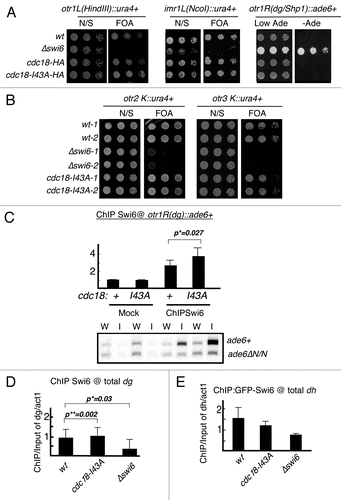
Figure 6 Timing of centromere replication in cdc18-I43A and Δswi6. Wild-type (FY2318), Δswi6 (FY3812), cdc18-I43A (FY3813) and swi6-F324A (FY5048) cells were synchronized at G1 by nitrogen starvation and released into 10 mM of hydroxyurea and 100 µg/ml of BrdU. DNA replication was determined by BrdU ChIP. (A) Replication timing at euchromatin ars2004. (B) Centromeric replication timing in wt, cdc18-I43A, Δswi6 or swi6-F324A. Schematic representation of the structure of the three centromeres and the location of the primers used for ChIP. The dh and dg primers recognize sequences in all three centromere. The primers for imr1 are cen1 specific. The primers for cnt are able to detect cen1 and 3. All experiments were repeated three times and representative data are shown.
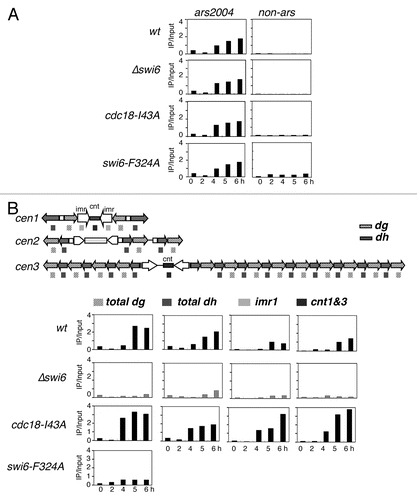
Figure 7 Model for Swi6 regulation of centromere replication. (A) In normal cells, Swi6 bound at the methylated histone enriches the local concentration of DDK which promotes origin activation (i). This is antagonized by Swi6 binding Cdc18 to prevent premature replication (ii). (B) In the absence of Swi6, origins are late-firing because they fail to recruit DDK effectively, and because there is nothing to counter fully repressive methylated chromatin. Similar results are observed for DDK mutants specifically defective in Swi6 binding.Citation38 (C) In the absence of Swi6 binding to Cdc18 (cdc18-I43A), the inhibitory effect of Swi6 on origin firing is relieved. Swi6 is still able to bind methylated histone and recruit DDK to promote accelerated replication.
Acknowledgements
We thank Ji-Ping Yuan for help in strain construction, and Marc Green for help with microscopy. We thank Robin Allshire, Junichi Nakayama, Danesh Moazed and Janet Partridge for strains. Angel Tabancay and Chris Viggiani advised on ChIP protocols. We thank members of the Forsburg and Kelly labs for critical reading of the manuscript. This work was supported by NIH GM059321 (S.L.F.) and NSF MCB 0743448 (S.L.F.).
References
- Kelly TJ, Brown GW. Regulation of chromosome replication. Annu Rev Biochem 2000; 69:829 - 880
- Forsburg SL. Eukaryotic MCM proteins: Beyond replication initiation. Microbiol Mol Biol Rev 2004; 68:109 - 131
- Smith JG, Caddle MS, Bulboaca GH, Wohlgemuth JG, Baum M, Clarke L, et al. Replication of centromere II of Schizosaccharomyces pombe. Mol Cell Biol 1995; 15:5165 - 5172
- Kim SM, Dubey DD, Huberman JA. Early-replicating heterochromatin. Genes Dev 2003; 17:330 - 335
- Heichinger C, Penkett CJ, Bahler J, Nurse P. Genome-wide characterization of fission yeast DNA replication origins. EMBO J 2006; 25:5171 - 5179
- Hayashi M, Katou Y, Itoh T, Tazumi A, Yamada Y, Takahashi T, et al. Genome-wide localization of pre-RC sites and identification of replication origins in fission yeast. EMBO J 2007; 26:1327 - 1339 Epub 2007 Feb 15
- Allshire RC, Karpen GH. Epigenetic regulation of centromeric chromatin: Old dogs, new tricks?. Nat Rev Genet 2008; 9:923 - 937
- Grewal SI, Jia S. Heterochromatin revisited. Nat Rev Genet 2007; 8:35 - 46
- Eissenberg JC, Elgin SC. The HP1 protein family: getting a grip on chromatin. Curr Opin Genet Dev 2000; 10:204 - 210
- Lechner MS, Schultz DC, Negorev D, Maul GG, Rauscher FJ 3rd. The mammalian heterochromatin protein 1 binds diverse nuclear proteins through a common motif that targets the chromoshadow domain. Biochem Biophys Res Commun 2005; 331:929 - 937
- Thiru A, Nietlispach D, Mott HR, Okuwaki M, Lyon D, Nielsen PR, et al. Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin. EMBO J 2004; 23:489 - 499
- Cowieson NP, Partridge JF, Allshire RC, McLaughlin PJ. Dimerisation of a chromo shadow domain and distinctions from the chromodomain as revealed by structural analysis. Curr Biol 2000; 10:517 - 525
- Brasher SV, Smith BO, Fogh RH, Nietlispach D, Thiru A, Nielsen PR, et al. The structure of mouse HP1 suggests a unique mode of single peptide recognition by the shadow chromo domain dimer. EMBO J 2000; 19:1587 - 1597
- Bernard P, Maure JF, Partridge JF, Genier S, Javerzat JP, Allshire RC. Requirement of heterochromatin for cohesion at centromeres. Science (Washington DC) 2001; 294:2539 - 2542
- Nonaka N, Kitajima T, Shihori Y, Xiao G, Yamamoto M, Grewal SI, et al. Recruitment of cohesin to heterochromatic regions by Swi6/HP1 in fission yeast. Nat Cell Biol 2002; 4:89 - 93
- Ekwall K, Javerzat JP, Lorentz A, Schmidt H, Cranston G, Allshire R. The chromodomain protein SWi6: A key component at Fission yeast centromeres. Science (Washington DC) 1995; 269:1429 - 1431
- Pidoux AL, Allshire RC. Kinetochore and heterochromatin domains of the fission yeast centromere. Chromosome Research 2004; 12:521 - 534
- Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, et al. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 2005; 438:1116 - 1122
- Hirota T, Lipp JJ, Toh BH, Peters JM. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 2005; 438:1176 - 1180
- Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, Grewal SI. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature 2008; 451:734 - 737
- Kloc A, Zaratiegui M, Nora E, Martienssen R. RNA interference guides histone modification during the S phase of chromosomal replication. Curr Biol 2008; 18:490 - 495
- Pak DTS, Pflumm M, Chesnokov I, Huang DW, Kellum R, Marr J, et al. Association of the origin recognition complex with heterochromatin and HP1 in higher eukaryotes. Cell 1997; 91:311 - 323
- Shareef MM, King C, Damaj M, Badagu R, Huang DW, Kellum R. Drosophila heterochromatin protein 1 (HP1)/origin recognition complex (ORC) protein is associated with HP1 and ORC and functions in heterochromatin-induced silencing. Mol Biol Cell 2001; 12:1671 - 1685
- Badugu R, Shareef MM, Kellum R. Novel Drosophila heterochromatin protein 1 (HP1)/origin recognition complex-associated protein (HOAP) repeat motif in HP1/HOAP interactions and chromocenter associations. J Biol Chem 2003; 278:34491 - 34498
- Huang DW, Fanti L, Pak DTS, Botchan MR, Pimpinelli S, Kellum R. Distinct cytoplasmic and nuclear fractions of Drosophila heterochromatin protein 1: Their phosphorylation levels and associations with origin recognition complex proteins. J Cell Biol 1998; 142:307 - 318
- Auth T, Kunkel E, Grummt F. Interaction between HP1alpha and replication proteins in mammalian cells. Exp Cell Res 2006; 312:3349 - 3359
- Quivy JP, Roche D, Kirschner D, Tagami H, Nakatani Y, Almouzni G. A CAF-1 dependent pool of HP1 during heterochromatin duplication. EMBO J 2004; 23:3516 - 3526
- Murzina N, Verreault A, Laue E, Stillman B. Heterochromatin dynamics in mouse cells: interaction between chromatin assembly factor 1 and HP1 proteins. Mol Cell 1999; 4:529 - 540
- Dohke K, Miyazaki S, Tanaka K, Urano T, Grewal SI, Murakami Y. Fission yeast chromatin assembly factor 1 assists in the replication-coupled maintenance of heterochromatin. Genes Cells 2008; 13:1027 - 1043
- Maison C, Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol 2004; 5:296 - 304
- Wallace JA, Orr-Weaver TL. Replication of heterochromatin: insights into mechanisms of epigenetic inheritance. Chromosoma 2005; 114:389 - 402
- Lejeune E, Bortfeld M, White SA, Pidoux AL, Ekwall K, Allshire RC, et al. The chromatin-remodeling factor FACT contributes to centromeric heterochromatin independently of RNAi. Curr Biol 2007; 17:1219 - 1224
- Gambus A, Jones RC, Sanchez-Diaz A, Kanemaki M, van Deursen F, Edmondson RD, et al. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol 2006; 8:358 - 366
- Nakayama Ji, Allshire RC, Klar AJS, Grewal SIS. A role for DNA polymerase alpha in epigenetic control of transcriptional silencing in fission yeast. EMBO J 2001; 20:2857 - 2866
- Ahmed S, Saini S, Arora S, Singh J. Chromodomain protein Swi6-mediated role of DNA polymerase alpha in establishment of silencing in fission Yeast. J Biol Chem 2001; 276:47814 - 47821
- Natsume T, Tsutsui Y, Sutani T, Dunleavy EM, Pidou AL, Iwasaki H, et al. A DNA polymerase alpha accessory protein, Mcl1, is required for propagation of centromere structures in fission yeast. PLoS One 2008; 3:2221
- Bailis JM, Bernard P, Antonelli R, Allshire RC, Forsburg SL. Hsk1-Dfp1 is required for heterochromatin-mediated cohesion at centromeres. Nat Cell Biol 2003; 5:1111 - 1116
- Hayashi MT, Takahashi TS, Nakagawa T, Nakayama J, Masukata H. The heterochromatin protein Swi6/HP1 activates replication origins at the pericentromeric region and silent mating-type locus. Nat Cell Biol 2009; 11:357 - 362
- Leatherwood J, Lopez-Girona A, Russell P. Interaction of Cdc2 and Cdc18 with a fission yeast ORC2-like protein. Nature 1996; 379:360 - 363
- Lorentz A, Ostermann K, Fleck O, Schmidt H. Switching gene swi6, involved in repression of silent mating-type loci in fission yeast, encodes a homologue of chromatid-associated proteins from Drosophila and mammals. Gene (Amsterdam) 1994; 143:139 - 143
- Ekwall K, Nimmo ER, Javerzat JP, Borgstrom B, Egel R, Cranston G, et al. Mutations in the fission yeast silencing factors clr4+ and rik1+ disrupt the localisation of the chromo domain protein Swi6p and impair centromere function. Journal of Cell Science 1996; 109:2637 - 2648
- Liu J, Smith CL, DeRyckere D, DeAngelis K, Martin GS, Berger JM. Structure and function of Cdc6/Cdc18: implications for origin recognition and checkpoint control. Mol Cell 2000; 6:637 - 648
- Sivakumar S, Porter-Goff M, Patel PK, Benoit K, Rhind N. In vivo labeling of fission yeast DNA with thymidine and thymidine analogs. Methods 2004; 33:213 - 219
- Hodson JA, Bailis JM, Forsburg SL. Efficient labeling of fission yeast Schizosaccharomyces pombe with thymidine and BUdR. Nucleic Acids Res 2003; 31:134
- Feng W, Collingwood D, Boeck ME, Fox LA, Alvino GM, Fangman WL, et al. Genomic mapping of single-stranded DNA in hydroxyurea-challenged yeasts identifies origins of replication. Nat Cell Biol 2006; 8:148 - 155
- Smothers JF, Henikoff S. The HP1 chromo shadow domain binds a consensus peptide pentamer. Curr Biol 2000; 10:27 - 30
- Shankaranarayana GD, Motamedi MR, Moazed D, Grewal SIS. Sir2 regulates histone H3 lysine 9 methylation and heterochromatin assembly in fission yeast. Curr Biol 2003; 13:1240 - 1246
- Freeman-Cook LL, Gomez EB, Spedale EJ, Marlett J, Forsburg SL, Pillus L, et al. Conserved locus-specific silencing functions of Schizosaccharomyces pombe sir2+. Genetics 2005; 169:1243 - 1260
- Li H, Motamedi MR, Yip CK, Wang Z, Walz T, Patel DJ, et al. An alpha motif at tas3 c terminus mediates rits cis spreading and promotes heterochromatic gene silencing. Mol Cell 2009; 34:155 - 167
- Seeler JS, Marchio A, Sitterlin D, Transy C, Dejean A. Interaction of SP100 with HP1 proteins: a link between the promyelocytic leukemia-associated nuclear bodies and the chromatin compartment. Proc Natl Acad Sci USA 1998; 95:7316 - 7321
- Prasanth SG, Prasanth KV, Siddiqui K, Spector DL, Stillman B. Human Orc2 localizes to centrosomes, centromeres and heterochromatin during chromosome inheritance. EMBO J 2004; 23:2651 - 2663
- Tabancay AP Jr, Forsburg SL. Eukaryotic DNA replication in a chromatin context. Curr Top Dev Biol 2006; 76:129 - 184
- Bell SP, Kobayashi R, Stillman B. Yeast origin recognition complex functions in transcription silencing and DNA replication. Science 1993; 262:1844 - 1849
- Foss M, McNally FJ, Laurenson P, Rine J. Origin recognition complex (ORC) in transcriptional silencing and DNA replication in S. cerevisiae. Science 1993; 262:1838 - 1844
- Dillin A, Rine J. Separable functions of ORC5 in replication initiation and silencing in Saccharomyces cerevisiae. Genetics 1997; 147:1053 - 1062
- Bachant J, Jessen SR, Kavanaugh SE, Fielding CS. The yeast S phase checkpoint enables replicating chromosomes to bi-orient and restrain spindle extension during S phase distress. J Cell Biol 2005; 168:999 - 1012
- Forsburg SL, Rhind N. Basic methods for fission yeast. Yeast 2006; 23:173 - 183
- Forsburg SL, Sherman DA. General purpose tagging vectors for fission yeast. Gene 1997; 191:191 - 195
- Forsburg SL. Comparison of Schizosaccharomyces pombe expression systems. Nucleic Acids Res 1993; 21:2955 - 2956
- Siam R, Dolan WP, Forsburg SL. Choosing and using Schizosaccharomyces pombe plasmids. Methods 2004; 33:189 - 198
- Gomez EB, Forsburg SL. Analysis of the fission yeast Schizosaccharomyces pombe cell cycle. Methods Mol Biol 2004; 241:93 - 111