Abstract
The DNA damage response (DDR) is a complex signaling network that leads to damage repair while modulating numerous cellular processes. DNA double-strand breaks (DSBs)—a highly cytotoxic DNA lesion—activate this system most vigorously. The DSB response network is orchestrated by the ATM protein kinase, which phosphorylates key players in its various branches. Proteasome-mediated protein degradation plays an important role in the proteome dynamics following DNA damage induction. Here, we identify the nuclear proteasome activator PA28γ (REGγ; PSME3) as a novel DDR player. PA28γ depletion leads to cellular radiomimetic sensitivity and a marked delay in DSB repair. Specifically, PA28γ deficiency abrogates the balance between the two major DSB repair pathways—nonhomologous end-joining and homologous recombination repair. Furthermore, PA28γ is found to be an ATM target, being recruited to the DNA damage sites and required for rapid accumulation of proteasomes at these sites. Our data reveal a novel ATM-PA28γ-proteasome axis of the DDR that is required for timely coordination of DSB repair.
Introduction
DNA damage caused by internal or external damaging agents is a major threat to the integrity of the cellular genome. The cellular defense system against this threat is the DNA damage response (DDR)—an elaborate signaling network that repairs the damage while swiftly modulating many physiological processes.Citation1 One of the most powerful triggers of the DDR is the DNA double-strand break (DSB).Citation2 The major DSB repair pathways in eukaryotic cells are error-prone nonhomologous end-joining (NHEJ) Citation2–Citation4 and the high-fidelity process, homologous recombination repair (HRR).Citation2,Citation5 The overall cellular response to DSBs is a powerful signaling network that swiftly and vigorously affects a large number of cellular systems.Citation1,Citation6 Its initial stage is performed by the sensor/mediator proteins that are recruited to the damaged sites,Citation1,Citation7–Citation9 where they are involved in damage recognition and processing, chromatin reorganization and activation of the transducers of the DNA damage alarm: protein kinases that phosphorylate numerous downstream effectors.Citation10,Citation11 The DSB response involves extensive protein post-translational modifications, most notably phosphorylation and modification by the ubiquitin and ubiquitin-like proteins.Citation8,Citation9,Citation12,Citation13
The primary transducer of the DSB alarm is the ATM protein kinase. In response to DSBs, ATM is rapidly activated and phosphorylates a plethora of key players in various damage response pathways.Citation11,Citation14 Null mutations in the ATM gene lead to the severe genomic instability syndrome, ataxiatelangiectasia (A-T).Citation15,Citation16 ATM is a member of the PI3-kinase-like protein kinase (PIKK) family, which includes several protein kinases that regulate a variety of cellular stress responses.Citation10 Among them are the DNA-dependent protein kinase (DNA-PK) Citation17 and the A-T- and RAD3-related protein (ATR),Citation18 which maintain complex collaborative relationships with ATM in response to different genotoxic stresses.
We recently identified a DDR branch mediated by the KAP-1 protein, whose phosphorylation by ATM allows it to induce chromatin decondensation.Citation19 This pathway has been specifically implicated in facilitating DSB repair in the vicinity of heterochromatin.Citation20 While investigating the mechanism of KAP-1 action, we identified new KAP-1-interacting proteins, one of which turned out to be PA28γ (PSME3; REGγ). PA28γ is a 28 kDa component of the 11S REG/PA28 regulatory particle that activates the 20S proteasome in an ATP- and ubiquitin-independent manner.Citation21,Citation22 The proteasome is a large, multi-subunit proteolytic complex composed of a cylindrical 20S core and two regulatory (“activator”) subunits. The three types of activators are PA700 (19S proteasomal activator), PA28 (11S proteasomal activator, REG), and PA200. The PA28 activator can be composed of the PA28α and PA28β proteins, which are expressed in the cytoplasm and assembled as a heteroheptamer, or be a homoheptamer of the PA28γ protein, which is nuclear.Citation23,Citation24
Recent biochemical studies revealed that PA28γ specifically directs ubiquitin- and ATP-independent degradation of proteins such as steroid receptor co-activator 3,Citation25 ubiquitin ligase Smurf1,Citation26 HCV core proteinCitation27–Citation29 and the cell cycle regulators PTTG1,Citation30 p21Cip1, p16INK4a and p19ARF.Citation31,Citation32 On the other hand, it enhances the MDM2-mediated ubiquitylation and subsequent proteasomal degradation of the p53 protein.Citation33 Notably, PA28γ has been implicated in the maintenance of centrosome and chromosomal stabilityCitation34 and was found to interact with the damage checkpoint kinase Chk2 and be involved in regulation of the number of nuclear PML bodies.Citation35 The suggested role for PA28γ in maintenance of genomic stability prompted us to explore its involvement in the DDR. Here, we report that PA28γ is an ATM target and plays a role in a pathway that is required for timely coordination of DSB repair, which involves recruitment of proteasome particles to sites of DNA damage.
Results
PA28γ is required for timely DSB repair.
Initial indication that PA28γ plays a role in the cellular DSB response came from the observation that cells depleted of PA28γ exhibit hypersensitivity to the radiomimetic drug neocarzinostatin (NCS), as demonstrated by a clonogenic survival assay. The sensitivity of PA28γ-depleted cells to NCS was intermediate between that of wild-type and ATM-depleted cells (). Such sensitivity is suggestive of interference with DSB repair. Further evidence of such a defect may come from the altered dynamics of the clearance of damage-induced nuclear foci of phosphorylated histone H2AX (γH2AX) Citation36 or foci formed by damage response proteins such as MDC1, 53BP1 and BRCA1.Citation8 Importantly, PA28γ depletion increased the duration of such foci compared with PA28γ-proficient cells (). It was also important to distinguish between the possible involvement of PA28γ in the initial recruitment of the DDR players to damage sites and its effect on their retention at these sites. When we followed the recruitment of these proteins to sites of DNA damage induced by a focused laser beam, PA28γ depletion did not seem to affect the initial formation of γH2AX along the damage tracks or the early accumulation of MDC1, 53BP1, RNF8 and BRCA1 (Fig. S1). Collectively, these results suggest that PA28γ is required for timely disappearance of the damage hallmarks, presumably by affecting DSB repair or the mechanism leading to dismantling of the nuclear foci at sites of DNA damage.
In order to directly examine the effect of PA28γ depletion on DSB sealing, we used the sensitive neutral comet assay.Citation37–Citation39 Two hr after treatment with an NCS dose of 200 ng/ml, significant differences in comet tail moment were observed between PA28γ-proficient and -deficient cells, indicating a marked retardation in DSB closure in the absence of PA28γ ().
PA28γ is involved in coordinating DSB repair pathways.
In order to dissect the effect of PA28γ depletion on the two major DSB repair routes, NHEJ and HRR, we examined the performance of each route separately in PA28γ-depleted cells. Interestingly, reduction in PA28γ amounts led to opposite effects on the two processes: moderate reduction in NHEJ but marked elevation in HRR ( and B). In view of these results, we monitored the accumulation of a major HRR player, RAD51, at DSBs formed by focused irradiation with α particles.Citation40 Interestingly, enhanced accumulation of RAD51 at DSB tracks was observed in cells depleted of PA28γ, particularly at early time points following DNA damage induction ().
Because HRR functions in the late S and G2 phases of the cell cycle, prolonged arrest at S/G2 can potentially affect the NHEJ:HRR ratio. Flow cytometric analysis showed that PA28γ depletion had minor effects on cell cycle distribution (Fig. S2). The most noticeable effect was an increased proportion of cells in S phase following NCS treatment, most evident 12 h after the treatment (Fig. S2), but overall, reduction of PA28γ cellular levels did not have a marked effect on the activation of the damage-induced cell cycle checkpoints. Collectively, the results suggested that the markedly elevated HRR that followed the loss of PA28γ could not be attributed to a major change in cell cycle distribution compared with PA28γ-proficient cells, but rather to interference with the repair pathways following the loss of this protein.
A fraction of PA28γ is recruited to DNA damage sites.
A protein's involvement in the early stage of the DDR often entails relocalization to the damaged sites of at least a fraction of its cellular portion. Following NCS treatment, we observed rapid recruitment of PA28γ to the chromatin fraction after damage induction, which subsequently subsided after 2 h (). Following induction of localized DNA damage and immunostaining with an antibody against PA28γ, we noticed that a fraction of this protein was, indeed, recruited to the damage sites () (for a test of the antibody's specificity see Fig. S3). Live cell imaging used to follow the relocalization of ectopic, GFP-tagged PA28γ to laser-induced DNA damage again showed recruitment of a fraction of PA28γ to the damage sites as early as few minutes after damage induction (). These observations suggest that PA28γ functions at the damage sites during the early stage of the DDR.
PA28γ is an ATM target.
Players in the cellular DSB response often undergo a variety of DNA damage-induced post-translational modifications,Citation12 with ATM-mediated phosphorylation being a prominent one.Citation11 Protein phosphorylations often lead to changes in gel migration of the targeted proteins. We noticed a slower migrating form of PA28γ that appeared following NCS treatment, and this band-shift could be augmented using the Phos-tag reagentCitation41 (). Importantly, this mobility shift was dose-dependent () and was not observed in cellular extracts treated with lambda protein phosphatase (λPPase), suggesting that it represented a phosphorylated form of PA28γ. PA28γ phosphorylation was ATM-dependent and DNA-PK-independent (), and its kinetics () roughly correlated with that of chromatin recruitment of PA28γ (). Notably, it was clear that only a fraction of the total cellular content of PA28γ underwent damage-induced phosphorylation. Collectively, the results suggest that a fraction of PA28γ is involved in the DSB response, being recruited to DNA damage sites and tagged by ATM-mediated phosphorylation.
Relocalization of proteasomes to DNA damage sites is PA28γ- and ATM-dependent.
Since PA28γ is usually part of a large protein complex, we explored its interactions with other proteins in unprovoked cells and following DNA damage induction. We immunoprecipitated PA28γ from untreated and NCS-treated HEK293 cells and identified co-precipitating proteins. Most of the proteins identified in this manner were the expected proteasome subunits, and no change was observed in the interaction of PA28γ with other proteins following DNA damage induction (Fig. S4). Notably, subunits of the 19S proteasome lid and subunits of the 20S proteasome core were identified. This result suggested that PA28γ could be found in mixed complexes (“hybrid proteasomes”), in which 20S particles are bound at one end by the 19S lid and at the other end by the PA28γ homoheptamer.
A natural corollary of this observation was that PA28γ-containing proteasome particles might be recruited to the damage sites. Using immunostaining, we followed the subcellular localization of endogenous PSMA6, a subunit of the 20S core proteasome, following NCS treatment ( and S5). We observed massive damage-induced relocalization of PSMA6 into the nucleus, where it exhibited punctuate staining reminiscent of damage-induced foci. Indeed, PSMA6 foci co-localized with γH2AX foci that are thought to mark DSB sites (). Further evidence for PSMA6 recruitment to DNA damage sites was obtained when localized damage was induced with a focused laser beam (). Importantly, this relocalization was largely PA28γ- and ATM-dependent (). Further indication that PSMA6 recruitment represented relocalization of proteasomes to the DNA damage sites was obtained using antibodies against two other proteasome subunits, PSMA1 and PSMA4 (Fig. S6).
Discussion
We describe here a novel ATM- and PA28γ-dependent DNA damage response pathway that involves recruitment of proteasomes to DNA damage sites and is required for optimal DSB repair. The function of PA28γ, a novel DDR player and an ATM target, is unique compared with those of many previously described ATM targets, most of them enzymes that mediate specific reactions, or regulators of nucleic acids transactions or chromatin organization. The PA28γ heptamer is one of the proteasome regulators and is involved in ubiquitin- and ATP-independent degradation of specific proteins.Citation25,Citation28,Citation31,Citation32 Its ATM-dependent phosphorylation and recruitment to DNA damage sites place it at the early stage of the DDR, together with many other proteins that are called to action at these sites following damage induction.Citation1,Citation7–Citation9 Our data further suggest that PA28γ is recruited by the ATM-mediated DDR as a proteasome component. Presumably, the aim of this pathway is to degrade proteins in situ at the damage sites. A cardinal question is whether the substrates of this degradation are chromatin components whose rapid degradation is required for chromatin reorganization at damage sites, or if they are DDR players that are removed from the scene during DDR recovery. PA28γ's ATM-dependent phosphorylation may affect the substrate specificity of the corresponding proteasome.Citation23 Previously documented post-translational modifications of PA28γ are MEKK3-mediated phosphorylation,Citation42 whose functional significance is still unclear, and SUMOylation, which affects its interaction with the p21 protein and leads to its cytosolic translocation.Citation43
It is interesting to compare our results with those of Blickwedehl et al.,Citation44,Citation45 who observed DNA damage-induced recruitment to chromatin of the proteasome activator PA200 and proteasome subunits and damage-induced formation of hybrid proteasomes containing the PA200 activator. However, the PA200-associated proteasome recruitment occurred at much later time points compared with the time course that we report and was ATM-independent but DNA-PK-dependent. Of note, PA200-deficient cells were radiosensitive. Collectively, the data indicate involvement of proteasomes of different compositions in different stages of the DDR.
Proteasome involvement in the DDR has previously been associated with the ubiquitin-proteasome pathway of protein degradation.Citation12,Citation13,Citation44–Citation61 Direct involvement of the proteasome in DSB repair was suggested based on observations in yeastCitation48,Citation51,Citation53,Citation56 and human cells.Citation44,Citation45,Citation49 Ben Aroya and colleaguesCitation48 recently suggested that in yeast, proteasome-mediated disassembly of the damage-associated foci is required for the recruitment and retention of repair proteins. In their study, proteasome-mediated degradation of the yeast protein Mms22 was necessary and sufficient for cell cycle progression following a damage-induced G2/M arrest. Shi et al.Citation62 reported that disassembly of MDC1 foci was dependent on the ubiquitin-proteasome pathway of protein degradation.Citation62
The mechanisms that maintain the balance between the NHEJ and HRR pathways at the late S and G2 phases of the cell cycle are a subject of intensive research. Our data may reflect a primary reduction in NHEJ efficiency, which, in turn, leads to elevation of HRR. Murakawa et al.Citation63 observed suppression of HRR following treatment with proteasome inhibitors. It should be noted that this drastic treatment reduces the cellular ubiquitin pools and markedly affects many processes compared with PA28γ reduction. Gudmundsdottir et al.Citation49 noticed that proteasome inhibition affects the relative extent of different DSB repair pathways in human cells. More recently, Ju et al.Citation51 observed abrogation of the NHEJ pathway in yeast upon interference with a feedback loop that involves degradation of Rpn4, a transcription factor that enhances the expression of proteasome unit genes. Evidence is thus emerging for involvement of proteasome-mediated protein degradation in maintaining the balance between DSB repair pathways.
It should be noted that even without the induction of acute DNA damage, reduction in the cellular amounts of proteasome subunits,Citation48 including PA28γ,Citation34 was found to lead to ongoing genomic instability. In view of the marked elevation in HRR that we noticed in PA28γ-depleted cells, a possible explanation for this chromosomal instability is abortive HR in the absence of sister chromatids. These observations indicate that proteasome-mediated processes function continuously in maintenance of genomic stability, probably in the face of ongoing, low-level DNA damage inflicted by metabolic by-products.
The interface between the arenas of the DDR and the ubiquitin-like (UBL) proteins is becoming thicker, as additional UBL-driven DDR pathways are discovered.Citation13,Citation60,Citation64,Citation65 Ours and previous observations highlight the proteasome in its various compositions as an important player in the complex cascade of processes taking place at DNA damage sites at different stages of the DDR. These observations call for further elucidation of proteasome-mediated pathways in the DDR and identification of their targets and regulators.
Materials and Methods
Cell lines.
HEK293, U2OS, HeLa and CAL51 cells were grown in DMEM with 10% fetal bovine serum at 37°C in 5% CO2 atmosphere.
Chemicals, antibodies and vectors.
DharmaFECT 1 transfection reagent from Dharmacon (T-2001-03). Neocarzinostatin (NCS) was obtained from Kayaku Chemicals. Phos-tag was purchased from the NARD Institute (ALL-107). The anti-ATM monoclonal antibody MAT3 was produced in our laboratory in collaboration with N.I. Smorodinsky; monoclonal anti-53BP1 antibody for the immunostaining assay and polyclonal antibody against RNF8 were generous gifts from T. Halazonetis (Geneva University) and N. Mailand (University of Copenhagen), respectively; polyclonal anti-γH2AX were from Bethyl Laboratories, Inc. (A300-081A); monoclonal anti-HA (SC-7392), anti-HSC70 (SC-7298) antibodies were obtained from Santa Cruz Biotechnology, Inc.; monoclonal anti PSMA1 (ab3325) and PSMA4 (ab55625) Abcam, Inc.; monoclonal anti-γH2AX from Millipore, Inc., (05-636); monoclonal anti-MDC1 from Sigma-Aldrich (M2444); monoclonal anti-PA28γ from BD Transduction, Inc. (611180); polyclonal anti-RAD51 was obtained from BD Biosciences, Inc. (551922); polyclonal anti pS824-KAP-1 from Bethyl Laboratories, Inc. (BL1067); poly-clonal anti-PSMA6 from Cell Signaling, Inc. (2459S); polyclonal anti-BRCA1 from Millipore, Inc. (07-434); polyclonal anti-H3 Abcam (ab1791); Secondary antibodies: anti-mouse and anti-rabbit IgG-Alexa 488 (A11001, A11008) and 568 (A11004 and A110011) were purchased from Molecular Probes, and HRP-conjugated anti-rabbit IgG (111-035-144) or anti-mouse IgG (115-035-062) were obtained from Jackson Immunoresearch Laboratories, Inc. The vector pEGFP-PA28γ (human cDNA) was a generous gift from Y. Masuura (Osaka University).Citation66
RNA interference.
OnTarget Plus SMARTpool siRNAs targeting PA28γ were obtained from Dharmacon. The following siRNA sequences were designed and subsequently synthesized by Dharmacon with the OnTarget Plus modifications:
Luciferase: CGU ACG CGG AAU ACU UCG ATT,
RAD51: GAG CUU GAC AAA CUA CUU CUU,
GFP: GGA GCG CAC CAT CTT CTT C,
ATM: NNG ACU UUG GCU GUC AAC UUU CG.
U2OS cells were grown to 20–50% confluence and transfected with siRNA using DharmaFECT1.
DDR assays.
Clonogenic survival of cell lines and immunostaining procedures were performed as previously described in reference Citation19. Detection of 53BP1, γH2AX, MDC1 and BRCA1 nuclear foci was performed as previously described in references Citation39 and Citation62. Induction of localized DNA damage and monitoring of protein recruitment were performed according to previous protocols.Citation39,Citation40,Citation67 DSB were introduced by irradiating cells with α-particles as previously described in references Citation40 and Citation68.
DSB repair.
NHEJ and HRR assays were performed as previously described in references Citation39 and Citation69–Citation71. siRNAs against the RAD51 and Ku70 served as controls. The comet assay was performed as previously described in references Citation38 and Citation39.
Immunoblotting, immunoprecipitation, protein identification and chromatin fractionation.
Immunoblotting and immunoprecipitation were performed as previously described in reference Citation67. Identification of PA28γ was done by the Smoler Proteomics Center at the Technion. PA28γ interactors were identified by the Biological Mass Spectrometry Facility at the Weizmann Institute of Science. To detect chromatin-bound proteins, cells were washed with ice-cold PBS and then lysed in a buffer containing 0.5% NP40, 100 mM NaCl, 50 mM Tris pH = 7.5 and 2 mM EDTA, supplemented with protease and phosphatase inhibitors, at 4°C for 10 min. The insoluble chromatin fractions were separated by centrifugation at 4,000 rpm at 4°C for 10 min. The chromatin-enriched pellet was then resuspended in nuclease incubation buffer containing 0.5% NP40, 100 mM NaCl, 50 mM Tris pH = 7.5, 1.5 mM MgCl2 and 5 units/µl benzonase (Novagen) to digest DNA and RNA, supplemented with protease and phosphatase inhibitors, at 4°C for 60 min. The sample was cleared by centrifugation at 20,000x g for 10 min, and the supernatant containing the solubilized native chromatin proteins was collected.
Cell cycle analysis using flow cytometry.
The cells were harvested and prepared for flow cytometry as previously described in reference Citation72. Sorting was performed using the FACSort flow cytometer (Becton Dickinson) at 10,000 events/sample. Cell cycle analysis was performed using ModFit LT software.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| AT | = | ataxia-telangiectasia |
| ATR | = | ataxiatelangiectasia and RAD3-related |
| DDR | = | DNA damage response |
| DNA-PK | = | DNA-dependent protein kinase |
| DSB | = | double-strand breaks |
| HRR | = | homologous recombination repair |
| NHEJ | = | nonhomologous end-joining |
| PIKK | = | PI3 kinase-related protein kinase |
Figures and Tables
Figure 1 Depletion of PA28γ enhances cellular sensitivity to radiomimetic treatment. Left part: clonogenic survival curves of CAL51 cells transfected with siRNA against PA28γ for 96 h and subsequently treated with various concentrations of the radiomimetic drug NCS. Cells transfected with siRNAs against ATM or GFP served as controls. The experiment was performed in triplicates. Right part: protein gel blotting analysis showing the extent of protein knockdown. Total cellular extracts of CAL51 cells transfected with the various siRNAs for 96 h were blotted with the indicated antibodies.
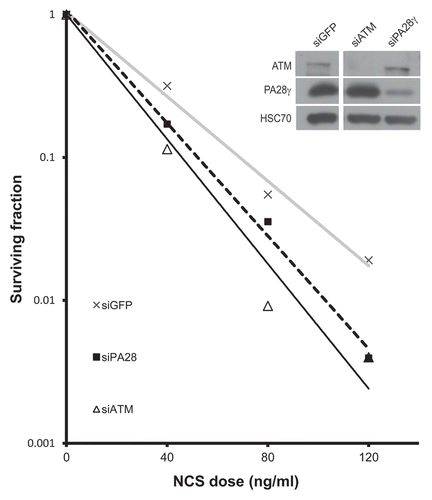
Figure 2 Depletion of PA28γ affects the disappearance of DNA damage-induced nuclear foci of γH2AX, 53BP1, MDC1 and BRCA1. CAL51 cells were treated with 10 ng/ml NCS 72 h after transfection with siRNAs against PA28γ or GFP, fixed and stained with antibodies against γH2AX (A), 53BP1 (B), MDC1 (C) and BRCA1 (D). (A, C and D) The number of cells in which more than 10 foci were counted. Mean of three independent experiments is presented, and error bars represent SD (*p-value < 0.05; **p-value < 0.01; ***p-value <0.0005, by χ2 analysis). (B) The mean of 53BP1 foci per cell is presented, and error bars represent standard error (*p-value < 0.05; ***p-value < 0.0005; by student's t-test). Experiments were performed in triplicates. (E) Protein gel blotting analysis of total cellular extract of CAL51 cells 72 h after siRNA transfection, showing the extents of protein knockdown in this experiment.
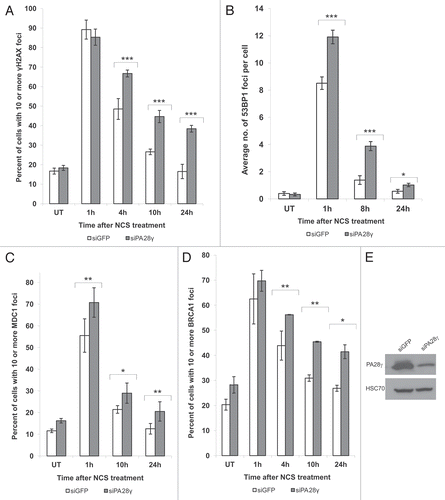
Figure 3 Deficiency of DSB repair in PA28γ-depeleted cells. (A) Direct observation of DNA damage 2 h after treatment of U2OS cells with 200 ng/ml of NCS using a neutral comet assay. (B) Quantitation of the comet data. The length and intensity of SYBR green-stained DNA tails relative to heads is shown as the relative comet tail moment (n = 100). p-values indicate the statistical significance of the difference between samples (student's t-test). Bars represent standard error of the mean based on two independent experiments. (C) Protein gel blotting analysis showing the extent of PA28γ knockdown.
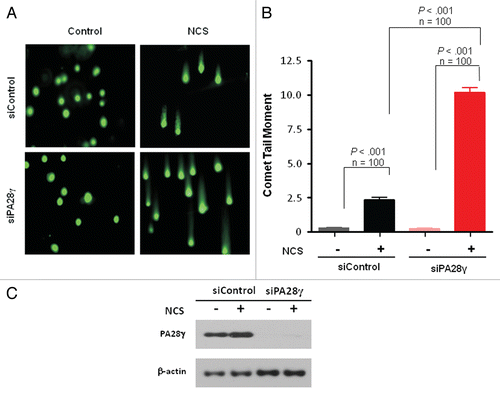
Figure 4 Effect of PA28γ depletion on the NHEJ and HRR pathways of DSB repair. The experimental systems are based on cells in which interrupted GFP-encoding sequences containing recognition sites of the rare cutter restriction endonuclease I-SceI were incorporated into the cellular genome. In one system, the repair of I-SceI-induced DSB via HRR regenerates an active GFP-encoding sequence,Citation69,Citation71 and in the other, this effect is obtained following NHEJ-mediated repair of the break.Citation70 In both cases, the GFP signal is monitored using FACS. (A) Effect of PA28γ depletion on NHEJ. HeLa cells containing the reporter sequences for NHEJCitation39,Citation70 were transfected with an I-SceI-encoding plasmid along with the indicated siRNA oligonucleotides and analyzed 72 h later by flow cytometry. GFP-positive cells are gated, and the percentage of GFP-positive cells in PA28γ-depleted cells is normalized against that of cells transfected with irrelevant siRNA (siLuciferase). Cells depleted of the KU70 protein, a major NHEJ player, served as a positive control. Shown is the mean of the NHEJ ratio (average of triplicates). Error bars represent standard error. Results of one of three independent experiments are shown. (B) Similar analysis in U2OS cells containing the HRR reporter.Citation69 Cells depleted of the RAD51 protein, a major HRR player, served as positive control. Results of one of four independent experiments are shown. Error bars represent standard deviation. (C) Quantification of the accumulation of RAD51 at DSBs in the presence or absence of PA28γ. U2OS cells were transfected with siRNAs against luciferase (siControl) or PA28γ, cultured for 48 h and irradiated with α-particles as described in references Citation40 and Citation68 to produce linear tracks of DSBs. The effectiveness of the downregulation was analyzed by immunoblotting, as displayed on the right. At the indicated time points after irradiation, the cells were stained for DNA (DAPI), γH2AX and RAD51. The γH2AX staining marked the tracks of DSBs, and the percentage of DSB tracks that co-localized with RAD51 was determined for 100 DSB track positive cells per data point. Error bars represent the SEM of three independent experiments.
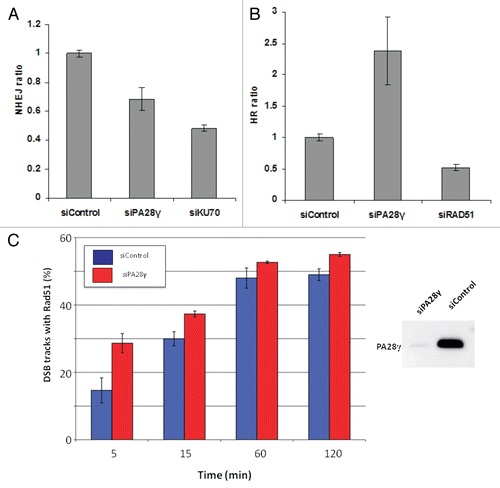
Figure 5 A fraction of PA28γ is recruited to DNA damage sites. (A) U2OS cells were treated with 400 ng/ml of NCS. At various time points following damage induction, the chromatin fraction was isolated and blotted with the indicated antibodies. (B) DNA damage was induced in U2OS cells using a focused laser microbeam. Twenty min later, the cells were treated with 0.25% NP40, fixed and co-stained with antibodies against PA28γ and γH2AX. (C) U2OS cells stably expressing GFP-PA28γ were transfected with DsRed-PCNA, and localized DNA damage was induced using a focused laser micro-beam. The cells were monitored using time-lapse imaging. Note the fraction of PA28γ that was recruited to the sites of DNA damage and co-localized with DsRed-PCNA, lasting up to 1 h following damage induction.
Figure 6 A fraction of PA28γ is phosphorylated by ATM in response to DNA damage. HEK293 cells were treated with NCS and cellular extracts separated on SDS-PAGE using 30 µM Phos-tag. (A) Immunoblots of PA28γ from cells treated with 400 ng/ml NCS for 30 min. The cellular extracts were or were not treated with λPPase. (B) Immunoblots of PA28γ from cells treated with various doses of NCS for 30 min. (C) Effect of the ATM inhibitor KU-55933 (ATMi) and DNA-PKs inhibitor NU-7441 (DNA-PKcs i) at 10 µM concentration on PA28γ phosphorylation, demonstrated using Phos-tag or regular gel. One hr after addition of the inhibitor, the cells were treated with 400 ng/ml of NCS for 30 min. HSC70 served as loading control, and phosphorylation of KAP-1 Citation19 represented the activation of the ATM-dependent DNA damage response. (D) Kinetics of PA28γ phosphorylation demonstrated using the Phos-tag or regular gel. The cells were treated with 400 ng/ml of NCS.
Figure 7 PA28γ- and ATM-dependent recruitment of the 20S proteasome to DNA damage sites. (A) CAL51 cells were treated with 200 ng/ml NCS for 30 min and co-stained with antibodies against the 20S core proteasome subunit PSMA6 and γH2AX. (B) U2OS cells were transfected with siRNAs against GFP or PA28γ or ATM, and localized DNA damage was induced 72 h later by a laser microbeam. Twenty min later, the cells were co-stained with antibodies against PSMA6 and γH2AX.
Additional material
Download Zip (7.8 MB)Acknowledgments
We are grateful to Olivier Coux and Michael Glickman for useful discussions, Amir Oryan for comments on the manuscript, Thanos Halazonetis for the anti-53BP1 antibody, Niels Mailand for the anti-RNF8 antibody, Jochem Dahm-Daphi for the NHEJ assay system and the Smoler Proteomics Center at the Technion and the Proteomic Center of the Weizmann Institute of Science for protein identification. This work was supported by research grants from the Israel Science Foundation, the A-T Medical Research Foundation, the Israel Cancer Research Fund, and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to YS), the National Institutes of Health (NIH CA 50519) to DJC, and the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement No. HEALTH-F2-2010-259893 and the Netherlands Genomics Initiative / Netherlands Organization for Scientific Research (to RK). Yosef Shiloh is a Research Professor of the Israel Cancer Research Fund.
References
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179 - 204; PMID: 20965415; http://dx.doi.org/10.1016/j.molcel.2010.09.019
- Hiom K. Coping with DNA double strand breaks. DNA Repair (Amst) 2010; 9:1256 - 1263; PMID: 21115283; http://dx.doi.org/10.1016/j.dnarep.2010.09.018
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 2010; 79:181 - 211; PMID: 20192759; http://dx.doi.org/10.1146/annurev.biochem.052308.093131
- Mladenov E, Iliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat Res 2011; 711:61 - 72; PMID: 21329706; http://dx.doi.org/10.1016/j.mrfmmm.2011.02.005
- Holthausen JT, Wyman C, Kanaar R. Regulation of DNA strand exchange in homologous recombination. DNA Repair (Amst) 2010; 9:1264 - 1272; PMID: 20971042; http://dx.doi.org/10.1016/j.dnarep.2010.09.014
- Warmerdam DO, Kanaar R. Dealing with DNA damage: relationships between checkpoint and repair pathways. Mutat Res 2010; 704:2 - 11; PMID: 20006736; http://dx.doi.org/10.1016/j.mrrev.2009.12.001
- Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol 2006; 173:195 - 206; PMID: 16618811; http://dx.doi.org/10.1083/jcb.200510130
- Bekker-Jensen S, Rendtlew Danielsen J, Fugger K, Gromova I, Nerstedt A, Lukas C, et al. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat Cell Biol 2010; 12:80 - 86
- Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol 2011; 13:1161 - 1169; PMID: 21968989; http://dx.doi.org/10.1038/ncb2344
- Lovejoy CA, Cortez D. Common mechanisms of PIKK regulation. DNA Repair (Amst) 2009; 8:1004 - 1008; PMID: 19464237; http://dx.doi.org/10.1016/j.dnarep.2009.04.006
- Bhatti S, Kozlov S, Farooqi AA, Naqi A, Lavin M, Khanna KK. ATM protein kinase: the linchpin of cellular defenses to stress. Cell Mol Life Sci 2011; 68:2977 - 3006; PMID: 21533982; http://dx.doi.org/10.1007/s00018-011-0683-9
- Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011; 25:409 - 433; PMID: 21363960; http://dx.doi.org/10.1101/gad.2021311
- Ramaekers CH, Wouters BG. Regulatory functions of ubiquitin in diverse DNA damage responses. Curr Mol Med 2011; 11:152 - 169; PMID: 21342128; http://dx.doi.org/10.2174/156652411794859269
- Derheimer FA, Kastan MB. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett 2010; 584:3675 - 3681; PMID: 20580718; http://dx.doi.org/10.1016/j.febslet.2010.05.031
- Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 2008; 9:759 - 769; PMID: 18813293; http://dx.doi.org/10.1038/nrm2514
- Perlman SL, Boder Deceased E, Sedgewick RP, Gatti RA. Ataxia-telangiectasia. Handb Clin Neurol 2011; 103:307 - 332; PMID: 21827897; http://dx.doi.org/10.1016/B978-0-444-51892-7.00019-X
- Neal JA, Meek K. Choosing the right path: Does DNAPK help make the decision?. Mutat Res 2011; 711:73 - 86; PMID: 21376743; http://dx.doi.org/10.1016/j.mrfmmm.2011.02.010
- Nam EA, Cortez D. ATR signalling: more than meeting at the fork. Biochem J 2011; 436:527 - 536; PMID: 21615334; http://dx.doi.org/10.1042/BJ20102162
- Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol 2006; 8:870 - 876; PMID: 16862143; http://dx.doi.org/10.1038/ncb1446
- Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell 2008; 31:167 - 177; PMID: 18657500; http://dx.doi.org/10.1016/j.molcel.2008.05.017
- Rechsteiner M, Hill CP. Mobilizing the proteolytic machine: cell biological roles of proteasome activators and inhibitors. Trends Cell Biol 2005; 15:27 - 33; PMID: 15653075; http://dx.doi.org/10.1016/j.tcb.2004.11.003
- Hill CP, Masters EI, Whitby FG. The 11S regulators of 20S proteasome activity. Curr Top Microbiol Immunol 2002; 268:73 - 89; PMID: 12083009; http://dx.doi.org/10.1007/978-3-642-59414-4_4
- Mao I, Liu J, Li X, Luo H. REGgamma, a proteasome activator and beyond?. Cell Mol Life Sci 2008; 65:3971 - 3980; PMID: 18679578; http://dx.doi.org/10.1007/s00018-008-8291-z
- Stadtmueller BM, Hill CP. Proteasome activators. Mol Cell 2011; 41:8 - 19; PMID: 21211719; http://dx.doi.org/10.1016/j.molcel.2010.12.020
- Li X, Lonard DM, Jung SY, Malovannaya A, Feng Q, Qin J, et al. The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell 2006; 124:381 - 392; PMID: 16439211; http://dx.doi.org/10.1016/j.cell.2005.11.037
- Nie J, Wu M, Wang J, Xing G, He F, Zhang L. REGgamma proteasome mediates degradation of the ubiquitin ligase Smurf1. FEBS Lett 2010; 584:3021 - 3027; PMID: 20580715; http://dx.doi.org/10.1016/j.febslet.2010.05.034
- Moriishi K, Shoji I, Mori Y, Suzuki R, Suzuki T, Kataoka C, et al. Involvement of PA28gamma in the propagation of hepatitis C virus. Hepatology 2010; 52:411 - 420; PMID: 20683941; http://dx.doi.org/10.1002/hep.23680
- Suzuki R, Moriishi K, Fukuda K, Shirakura M, Ishii K, Shoji I, et al. Proteasomal turnover of hepatitis C virus core protein is regulated by two distinct mechanisms: a ubiquitin-dependent mechanism and a ubiquitin-independent but PA28gamma-dependent mechanism. J Virol 2009; 83:2389 - 2392; PMID: 19091860; http://dx.doi.org/10.1128/JVI.01690-08
- Mori Y, Moriishi K, Matsuura Y. Hepatitis C virus core protein: its coordinate roles with PA28gamma in metabolic abnormality and carcinogenicity in the liver. Int J Biochem Cell Biol 2008; 40:1437 - 1442; PMID: 18321762; http://dx.doi.org/10.1016/j.biocel.2008.01.027
- Ying H, Furuya F, Zhao L, Araki O, West BL, Hanover JA, et al. Aberrant accumulation of PTTG1 induced by a mutated thyroid hormone beta receptor inhibits mitotic progression. J Clin Invest 2006; 116:2972 - 2984; PMID: 17039256; http://dx.doi.org/10.1172/JCI28598
- Chen X, Barton LF, Chi Y, Clurman BE, Roberts JM. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGgamma proteasome. Mol Cell 2007; 26:843 - 852; PMID: 17588519; http://dx.doi.org/10.1016/j.molcel.2007.05.022
- Li X, Amazit L, Long W, Lonard DM, Monaco JJ, O'Malley BW. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGgamma-proteasome pathway. Mol Cell 2007; 26:831 - 842; PMID: 17588518; http://dx.doi.org/10.1016/j.molcel.2007.05.028
- Liu J, Yu G, Zhao Y, Zhao D, Wang Y, Wang L, et al. REGgamma modulates p53 activity by regulating its cellular localization. J Cell Sci 2010; 123:4076 - 4084; PMID: 21084564; http://dx.doi.org/10.1242/jcs.067405
- Zannini L, Lecis D, Buscemi G, Carlessi L, Gasparini P, Fontanella E, et al. REGgamma proteasome activator is involved in the maintenance of chromosomal stability. Cell Cycle 2008; 7:504 - 512; PMID: 18235248; http://dx.doi.org/10.4161/cc.7.4.5355
- Zannini L, Buscemi G, Fontanella E, Lisanti S, Delia D. REGgamma/PA28gamma proteasome activator interacts with PML and Chk2 and affects PML nuclear bodies number. Cell Cycle 2009; 8:2399 - 2407; PMID: 19556897; http://dx.doi.org/10.4161/cc.8.15.9084
- Xie A, Odate S, Chandramouly G, Scully R. H2AX post-translational modifications in the ionizing radiation response and homologous recombination. Cell Cycle 2010; 9:3602 - 3610; PMID: 20703100; http://dx.doi.org/10.4161/cc.9.17.12884
- Liao W, McNutt MA, Zhu WG. The comet assay: a sensitive method for detecting DNA damage in individual cells. Methods 2009; 48:46 - 53; PMID: 19269328; http://dx.doi.org/10.1016/j.ymeth.2009.02.016
- Tsai WB, Chung YM, Takahashi Y, Xu Z, Hu MC. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nat Cell Biol 2008; 10:460 - 467; PMID: 18344987; http://dx.doi.org/10.1038/ncb1709
- Moyal L, Lerenthal Y, Gana-Weisz M, Mass G, So S, Wang SY, et al. Requirement of ATM-Dependent Monoubiquitylation of Histone H2B for Timely Repair of DNA Double-Strand Breaks. Mol Cell 2011; 41:529 - 542; PMID: 21362549; http://dx.doi.org/10.1016/j.molcel.2011.02.015
- Stap J, Krawczyk PM, Van Oven CH, Barendsen GW, Essers J, Kanaar R, et al. Induction of linear tracks of DNA double-strand breaks by alpha-particle irradiation of cells. Nat Methods 2008; 5:261 - 266; PMID: 18309310; http://dx.doi.org/10.1038/nmeth.f.206
- Kinoshita E, Kinoshita-Kikuta E, Ujihara H, Koike T. Mobility shift detection of phosphorylation on large proteins using a Phos-tag SDS-PAGE gel strengthened with agarose. Proteomics 2009; 9:4098 - 4101; PMID: 19658103; http://dx.doi.org/10.1002/pmic.200900020
- Hagemann C, Patel R, Blank JL. MEKK3 interacts with the PA28 gamma regulatory subunit of the proteasome. Biochem J 2003; 373:71 - 79; PMID: 12650640; http://dx.doi.org/10.1042/BJ20021758
- Wu Y, Wang L, Zhou P, Wang G, Zeng Y, Wang Y, et al. Regulation of REGgamma cellular distribution and function by SUMO modification. Cell Res 2011; 21:807 - 816; PMID: 21445096; http://dx.doi.org/10.1038/cr.2011.57
- Blickwedehl J, Agarwal M, Seong C, Pandita RK, Melendy T, Sung P, et al. Role for proteasome activator PA200 and postglutamyl proteasome activity in genomic stability. Proc Natl Acad Sci USA 2008; 105:16165 - 16170; PMID: 18845680; http://dx.doi.org/10.1073/pnas.0803145105
- Blickwedehl J, McEvoy S, Wong I, Kousis P, Clements J, Elliott R, et al. Proteasomes and proteasome activator 200 kDa (PA200) accumulate on chromatin in response to ionizing radiation. Radiat Res 2007; 167:663 - 674; PMID: 17523843; http://dx.doi.org/10.1667/RR0690.1
- Russell SJ, Reed SH, Huang W, Friedberg EC, Johnston SA. The 19S regulatory complex of the proteasome functions independently of proteolysis in nucleotide excision repair. Mol Cell 1999; 3:687 - 695; PMID: 10394357; http://dx.doi.org/10.1016/S1097-2765(01)80001-0
- Jacquemont C, Taniguchi T. Proteasome function is required for DNA damage response and fanconi anemia pathway activation. Cancer Res 2007; 67:7395 - 7405; PMID: 17671210; http://dx.doi.org/10.1158/0008-5472.CAN-07-1015
- Ben-Aroya S, Agmon N, Yuen K, Kwok T, McManus K, Kupiec M, et al. Proteasome nuclear activity affects chromosome stability by controlling the turnover of Mms22, a protein important for DNA repair. PLoS Genet 2010; 6:1000852; PMID: 20174551; http://dx.doi.org/10.1371/journal.pgen.1000852
- Gudmundsdottir K, Lord CJ, Ashworth A. The proteasome is involved in determining differential utilization of double-strand break repair pathways. Oncogene 2007; 26:7601 - 7606; PMID: 17563742; http://dx.doi.org/10.1038/sj.onc.1210579
- Lin CP, Ban Y, Lyu YL, Desai SD, Liu LF. A ubiquitin-proteasome pathway for the repair of topoisomerase I-DNA covalent complexes. J Biol Chem 2008; 283:21074 - 21083; PMID: 18515798; http://dx.doi.org/10.1074/jbc.M803493200
- Ju D, Wang X, Ha SW, Fu J, Xie Y. Inhibition of proteasomal degradation of rpn4 impairs nonhomologous end-joining repair of DNA double-strand breaks. PLoS ONE 2010; 5:9877; PMID: 20376190; http://dx.doi.org/10.1371/journal.pone.0009877
- Schauber C, Chen L, Tongaonkar P, Vega I, Lambertson D, Potts W, et al. Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature 1998; 391:715 - 718; PMID: 9490418; http://dx.doi.org/10.1038/35661
- Krogan NJ, Lam MH, Fillingham J, Keogh MC, Gebbia M, Li J, et al. Proteasome involvement in the repair of DNA double-strand breaks. Mol Cell 2004; 16:1027 - 1034; PMID: 15610744; http://dx.doi.org/10.1016/j.molcel.2004.11.033
- Raman M, Havens CG, Walter JC, Harper JW. A Genome-wide Screen Identifies p97 as an Essential Regulator of DNA Damage-Dependent CDT1 Destruction. Mol Cell 2011; 44:72 - 84; PMID: 21981919; http://dx.doi.org/10.1016/j.molcel.2011.06.036
- Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C A. Proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics 2011; 10:111.013284; PMID: 21890473; http://dx.doi.org/10.1074/mcp.M111.013284
- Cabrera R, Sha Z, Vadakkan TJ, Otero J, Kriegenburg F, Hartmann-Petersen R, et al. Proteasome nuclear import mediated by Arc3 can influence efficient DNA damage repair and mitosis in Schizosaccharomyces pombe. Mol Biol Cell 2010; 21:3125 - 3136; PMID: 20668161; http://dx.doi.org/10.1091/mbc.E10-06-0506
- Chen S, Blank JL, Peters T, Liu XJ, Rappoli DM, Pickard MD, et al. Genome-wide siRNA screen for modulators of cell death induced by proteasome inhibitor bortezomib. Cancer Res 2010; 70:4318 - 4326; PMID: 20460535; http://dx.doi.org/10.1158/00085472.CAN-09-4428
- Zecevic A, Hagan E, Reynolds M, Poage G, Johnston T, Zhitkovich A. XPA impacts formation but not proteasome-sensitive repair of DNA-protein cross-links induced by chromate. Mutagenesis 2010; 25:381 - 388; PMID: 20410141; http://dx.doi.org/10.1093/mutage/geq017
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 2011; 44:325 - 340; PMID: 21906983; http://dx.doi.org/10.1016/j.molcel.2011.08.025
- Ramadan K, Meerang M. Degradation-linked ubiquitin signal and proteasome are integral components of DNA double strand break repair: New perspectives for anti-cancer therapy. FEBS Lett 2011; 585:2868 - 2875; PMID: 21536036; http://dx.doi.org/10.1016/j.febslet.2011.04.046
- Shi W, Ma Z, Willers H, Akhtar K, Scott SP, Zhang J, et al. Disassembly of MDC1 foci is controlled by ubiquitin-proteasome-dependent degradation. J Biol Chem 2008; 283:31608 - 31616; PMID: 18757370; http://dx.doi.org/10.1074/jbc.M801082200
- Shi W, Ma Z, Willers H, Akhtar K, Scott SP, Zhang J, et al. Disassembly of MDC1 foci is controlled by ubiquitin-proteasome-dependent degradation. J Biol Chem 2008; 283:31608 - 31616; PMID: 18757370; http://dx.doi.org/10.1074/jbc.M801082200
- Murakawa Y, Sonoda E, Barber LJ, Zeng W, Yokomori K, Kimura H, et al. Inhibitors of the proteasome suppress homologous DNA recombination in mammalian cells. Cancer Res 2007; 67:8536 - 8543; PMID: 17875693; http://dx.doi.org/10.1158/0008-5472.CAN-07-1166
- Al-Hakim A, Escribano-Diaz C, Landry MC, O'Donnell L, Panier S, Szilard RK, et al. The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair (Amst) 2010; 9:1229 - 1240; PMID: 21056014; http://dx.doi.org/10.1016/j.dnarep.2010.09.011
- Lukas J. The interface between the ubiquitin family and the DNA damage response. EMBO Rep 2010; 11:907 - 909; PMID: 21072060; http://dx.doi.org/10.1038/embor.2010.180
- Moriishi K, Okabayashi T, Nakai K, Moriya K, Koike K, Murata S, et al. Proteasome activator PA28gamma-dependent nuclear retention and degradation of hepatitis C virus core protein. J Virol 2003; 77:10237 - 10249; PMID: 12970408; http://dx.doi.org/10.1128/JVI.77.19.10237-49.2003
- Salton M, Lerenthal Y, Wang SY, Chen DJ, Shiloh Y. Involvement of matrin 3 and SFPQ/NONO in the DNA damage response. Cell Cycle 2010; 9:1568 - 1576; PMID: 20421735; http://dx.doi.org/10.4161/cc.9.8.11298
- Agarwal S, van Cappellen WA, Guenole A, Eppink B, Linsen SE, Meijering E, et al. ATP-dependent and independent functions of Rad54 in genome maintenance. J Cell Biol 2011; 192:735 - 750; PMID: 21357745; http://dx.doi.org/10.1083/jcb.201011025
- Puget N, Knowlton M, Scully R. Molecular analysis of sister chromatid recombination in mammalian cells. DNA Repair (Amst) 2005; 4:149 - 161; PMID: 15590323; http://dx.doi.org/10.1016/j.dnarep.2004.08.010
- Mansour WY, Schumacher S, Rosskopf R, Rhein T, Schmidt-Petersen F, Gatzemeier F, et al. Hierarchy of nonhomologous end-joining, single-strand annealing and gene conversion at site-directed DNA double-strand breaks. Nucleic Acids Res 2008; 36:4088 - 4098; PMID: 18539610; http://dx.doi.org/10.1093/nar/gkn347
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 1999; 13:2633 - 2638; PMID: 10541549; http://dx.doi.org/10.1101/gad.13.20.2633
- Vindeløv LL, Christensen IJ. A review of techniques and results obtained in one laboratory by an integrated system of methods designed for routine clinical flow cytometric DNA analysis. Cytometry 1990; 11:753 - 770; PMID: 2272241; http://dx.doi.org/10.1002/cyto.990110702