Abstract
Comment on: Gavathiotis E, et al. Mol Cell 2010; 40:481-92.
Cells are hard-wired to undergo programmed cell death or apoptosis to literally shape the organism during development and maintain tissue homeostasis throughout the normal lifespan. BCL-2 family proteins participate in a web of pro-life and pro-death interactions that adjudicate cell fate in response to apoptotic stimuli. When a death sentence is rendered, one of the key executioner proteins named BAX is activated to form toxic mitochondrial pores that effectively lance the cell's power plants. Deciphering the explicit mechanism of BAX activation is fundamental to advancing our understanding of the mitochondrial death pathway and how the apoptotic machinery can be manipulated for therapeutic benefit in diseases of uncontrolled cell survival or premature cell death.
It is widely reported that BAX, in its “innocent” form, lies inactive in the cytosol until provoked by cellular stress to translocate to the mitochondria and homo-oligomerize into a deadly membrane channel. Ever vigilant anti-apoptotic proteins deploy a groove on their surface to bind and capture the exposed BCL-2 homology 3 (BH3) α-helical death domain of activated BAX.Citation1 This inhibitory interaction is overcome by overwhelming the anti-apoptotic protein reserve with activated forms of BAX or through competitive displacement of BAX BH3 from the anti-apoptotic groove by the BH3-only subclass of BCL-2 family death proteins, a heterogeneous group of stress sensors whose only sequence commonality lies within a single BH3 α-helix. How BAX becomes conformationally unmasked in response to stress stimuli, such that it can translocate from the cytosol to the mitochondria, expose its BH3 interaction surface and transform into a self-propagating homo-oligomer, has remained an intriguing structural and mechanistic mystery.
To investigate the mechanism of BAX activation, we tested the hypothesis that a subset of BH3-only proteins could directly bind and activate BAX.Citation2,Citation3 We generated Stabilized Alpha-Helix of BCL-2 domains (SAHBs) that recapitulate the natural α-helical structure of BH3 domains to screen for BAX-interacting ligands. Among our initial trio of BID, BIM and BAD SAHBs, only BID and BIM BH3 α-helices exhibited direct and functional binding interactions with BAX.Citation4 We then conducted a series of NMR analyses of the BIM SAHB/BAX interaction and located the BH3-binding site at the junction of α-helices 1 and 6 on the N-terminal side of the BAX protein,Citation5 a geographically distinct region from the canonical BH3-binding pocket of anti-apoptotic proteins.Citation1 Having identified a trigger site for BAX activation, we wondered how “pulling the trigger” actually transformed BAX from its quiescent to killer state?
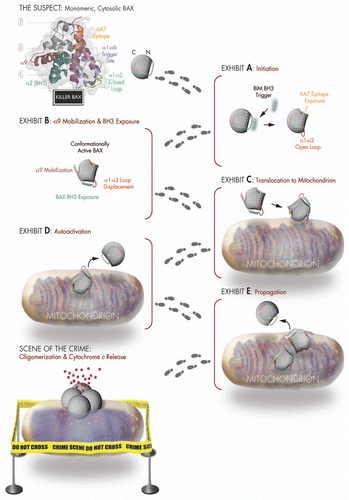
Our first clue came from a comparison between the unbound and bound calculated model structures, which revealed that BAX optimally accommodates BIM SAHB at the trigger site by converting the loop between α-helices 1 and 2 from a closed to an open position. To test the mechanistic importance of loop displacement, we installed a disulfide tether to reversibly lock the α1–α2 loop to the base of the trigger site. Whereas BIM SAHB could still bind to loop-tethered BAX, ligand-triggered BAX activation could only ensue if the covalent lock was eliminated by disulfide reduction, implicating “opening” of the α1–α2 loop as the very first conformational change of the BAX activation pathway (, Exhibit A).Citation6
A major hurdle in studying BAX activation has been the inability to capture any structural information beyond that of the much-celebrated “mug shot” of the inactive monomerCitation7 (, The Suspect) due to the catch 22 of trying to study a moving target using structural methods. By examining dose-responsive chemical shift changes in 15N-BAX induced by BIM SAHB at relatively short time points, we were able to detect allosteric sensing of BH3 triggering at the BAX C-terminus.Citation6 Indeed, the C-terminal α-helix 9 of BAX has been implicated as a membrane insertion helix that is released from its binding pocket for translocation to the mitochondria.Citation7 By tethering α9 to its binding pocket using an installed disulfide, we demonstrated that BIM SAHB triggering could only induce BAX mitochondrial translocation and cytochrome c release upon reduction of the C-terminal covalent lock.Citation6 By trapping BAX in a semi-activated state in which N-terminal triggering is arrested by α9 restraint, we were also able to detect, both by NMR and anti-BAX BH3 pull down, BIM SAHB-induced exposure of BAX BH3, revealing another fingerprint of the major structural reorganization that ensues upon initiation of direct BAX activationCitation6 (, Exhibits B and C).
The exposed BAX BH3 domain is essential to the lethality of BAX but its explicit role in the downstream plot to destroy the mitochondria has remained elusive. Close inspection of the BAX BH3 sequence revealed that it shares remarkable sequence identity to BIM BH3, suggesting that, once exposed, it too may participate in luring inactive BAX monomers into the crime. By generating a stapled BAX BH3 helix for use in NMR and complementary mutagenesis studies, we demonstrated that BAX SAHB directly and functionally engaged the BAX trigger site, implicating BAX BH3 in auto-activation and propagation of the death signalCitation6 (, Exhibits D and E). These key pieces of structural and biochemical evidence, combined with a compelling body of scientific testimony,Citation8–Citation11 helped solve the mechanistic mystery of the infamous “hit and run” behavior of the BAX activation process.
Each step along the BAX activation continuum provides a pharmacologic opportunity to aid and abet BAX's criminal exploits to eliminate renegade cells or intercept BAX to keep cells safe. By cloaking its pore-forming interactions within the confines of the outer mitochondrial membrane (, Scene of the Crime), BAX continues to elude and taunt investigators who are eager to root out its mitochondrial evil doings at the source and, at long last, write the final chapter—“BAX's Last Stand”—in riveting structural and biochemical detail.
Figures and Tables
Figure 1 BH3-triggered structural reorganization drives the activation of pro-apoptotic BAX. Killer BAX is disguised as an inactive cytosolic protein (see mug shot of “The Suspect”) and is coerced to undergo a series of conformational changes upon engagement of its α1/α6 binding site (purple) by a triggering BH3 α-helix. BAX's transformation from an innocent to a guilty protein begins with displacement of its α1–α2 loop from a closed (green) to an open (red) position (Exhibit A), which reveals the 6A7 epitope (orange) and leads to mobilization of the C-terminal α9 helix (maroon) for mitochondrial translocation and exposure of the BAX BH3 death domain (cyan) (Exhibits B and C). BAX propagates its own activation through triggering interactions between the exposed BAX BH3 α-helix of fully activated monomers and the α1/α6 binding site of inactive monomers (Exhibits D and E), ultimately assembling into an elusive oligomeric pore that promotes apoptosis by releasing mitochondrial accomplices such as cytochrome c at the “Scene of the Crime.”
Comment on: Gavathiotis E, et al. Mol Cell 2010; 40:481 - 492
References
- Sattler M, et al. Science 1997; 275:983 - 986
- Kuwana T, et al. Cell 2002; 111:331 - 342
- Wang K, et al. Genes Dev 1996; 10:2859 - 2869
- Walensky LD, et al. Mol Cell 2006; 24:199 - 210
- Gavathiotis E, et al. Nature 2008; 455:1076 - 1081
- Gavathiotis E, et al. Mol Cell 2010; 40:481 - 492
- Suzuki M, et al. Cell 2000; 103:645 - 654
- Tan C, et al. J Biol Chem 2006; 281:14764 - 14775
- Lovell JF, et al. Cell 2008; 135:1074 - 1084
- Kim H, et al. Mol Cell 2009; 36:487 - 499
- Cartron PF, et al. Mol Cell 2004; 16:807 - 818