Abstract
Aurora kinase A (Aur-A), a mitotic kinase, regulates initiation of mitosis through centrosome separation and proper assembly of bipolar spindles. LIM kinase 1 (LIMK1), a modulator of actin and microtubule dynamics, is involved in the mitotic process through inactivating phosphorylation of cofilin. Phosphorylated LIMK1 is recruited to the centrosomes during early prophase, where it colocalizes with γ-tubulin. Here, we report a novel functional cooperativity between Aur-A and LIMK1 through mutual phosphorylation. LIMK1 is recruited to the centrosomes during early prophase and then to the spindle poles, where it colocalizes with Aur-A. Aur-A physically associates with LIMK1 and activates it through phosphorylation, which is important for its centrosomal and spindle pole localization. Aur-A also acts as a substrate of LIMK1, and the function of LIMK1 is important for its specific localization and regulation of spindle morphology. Taken together, the novel molecular interaction between these two kinases and their regulatory roles on one another’s function may provide new insight on the role of Aur-A in manipulation of actin and microtubular structures during spindle formation.
Introduction
Aurora A kinase (Aur-A) is a serine/threonine kinase and a member of the Aurora kinase family, which plays important roles in various but distinct mitotic processes.Citation1,Citation2 Although not a bona fide oncogene, Aur-A is overexpressed in a variety of adenocarcinomas, including cancers of the breast, skin and prostate.Citation3–Citation5 Therefore, much attention has been focused on identification of Aur-A inhibitors as anticancer agents,Citation6–Citation8 some of which showed success in clinical trialsCitation9–Citation11 singly or in combination with EGFR inhibitor for drug-resistant cancer or with actinomycin D in p53-based cyclotherapy.Citation12,Citation13 At the same time, an increased interest in understanding the mechanism of activation of Aur-A, and identification of interacting partners and substrates that are phosphorylated by Aur-A led to a multitude of published reports in recent years.Citation14–Citation18
Aur-A activity increases in G2, with its targeting to the centrosomes by activated Plk1,Citation19 which then allows initiation of early mitotic events, such as centrosome maturation and separation and spindle assembly.Citation1,Citation2,Citation20–Citation22 Aur-A plays a role in centrosome maturation through recruitment of γ-tubulin,Citation23 ChToh,Citation24 NDEL1,Citation25 TACCCitation25 and LATS2 Citation26 to the centrosomes and bipolar spindle assembly through interaction with microtubule (MT)-associated proteins TPX2, XMAP and HURP, forming a complex.Citation27–Citation29 Aur-A phosphorylates LATS2 at a specific site (S380), which allows its colocalization with the other family member Aur-B at the central spindle.Citation30 Recent studies showed that MT-binding protein TPX2 targets Aur-A to the spindle microtubules and induces autophosphorylation of Aur-A at T288 through a conformational change.Citation17,Citation31,Citation32 Binding of TPX2 prevents dephosphorylation of Aur-AT288 and promotes accumulation of activated Aur-A. Phosphorylated Aur-A (T288), the active form of Aur-A,Citation33 then phosphorylates a variety of substrates,Citation34 including LIM domain containing AjubaCitation35 and Plk1,Citation36 which allows spindle assembly and bipolarity.Citation26
Ajuba interacts with centrosomal Aur-A through its LIM domain with the non-catalytic N-terminal region of Aur-A, which then induces its phosphorylation by Aur-A. This interaction and subsequent phosphorylation promotes autophosphorylation of Aur-A and its complete activation.Citation35 Although a number of studies indicated involvement of a variety of interacting partners of Aur-A,Citation37,Citation38 some of which are responsible for inhibition of Aur-A catalytic function,Citation33,Citation39 the understanding of Aur-A regulation and protein functions regulated by Aur-A during mitotic phases is far from complete.
Recent studies on LIM domain containing protein LIM kinase 1 (LIMK1) showed its localization to the centrosomes and association with γ-tubulin.Citation40 Furthermore, LIMK1 was shown to be involved in positioning of mitotic spindles during metaphase through modulation of cortical actin through phosphorylation of cofilin.Citation41 LIMK1 is a LIM domain containing serine/threonine kinase, which modulates actin and microtubule dynamics and participates in a variety of cellular processes (reviewed in refs. Citation42 and Citation43). Function of LIMK1 on actin cytoskeleton is mediated through inactivating phosphorylation of actin depolymerizing family protein cofilinCitation44,Citation45 and microtubule-binding protein p25alpha.Citation46 LIMK1 has two N-terminal LIM domains, a PDZ domain and a C-terminal kinase domain. LIMK1 interacts with a variety of proteins through its LIM and PDZ domains.Citation47–Citation49 LIMK1 also has a nuclear localization signal and two nuclear exit signal sequences. LIMK1 is activated by phosphorylation at T508 by ROCK, PAK1 or PAK4 and phosphorylates cofilin at S3, rendering it inactive. LIMK1 is activated in early mitotic phases, but its inactivation is required for cytokinesis.Citation50,Citation51 Overexpression of LIMK1 leads to cytokinesis defectsCitation51 and formation of multipolar spindles,Citation52 which are noted in a variety of cancers.Citation52,Citation53 Although LIMK1 needs to be phosphorylated during early prophaseCitation50 for its targeting to the centrosomes,Citation40 it is not known which kinase(s) phosphorylates LIMK1 during mitosis. Amano et al. showed that LIMK1 is not phosphorylated by ROCK or PAK during mitosis.Citation51 In this study, we show that LIMK1 is phosphorylated by the centrosomal kinase Aur-A and also participates in phosphorylating Aur-A. We further show that functions of both LIMK1 and Aur-A are important for integrity and bipolarity of mitotic spindles.
Results
LIMK1 and Aur-A colocalize to the centrosomes during mitosis.
Previously, we showed that phosphoLIMK1 (T508)(pLIMK1) colocalized with γ-tubulin to the centrosomes during early prophase till early telophase, while nonphosphorylated LIMK1 did not localize to the centrosomes.Citation40 To further elucidate the involvement of LIMK1 during early mitotic phases, we examined the correlative expression pattern of LIMK1 and Aur-A, which is involved in recruitment of γ-tubulin to the centrosomes during early prophase,Citation23 in PC-3 cells. Immunofluorescence analysis (IFA) showed strong colocalization of pLIMK1 with Aur-A at the centrosomes during early prophase (), as determined by analysis of Pearson's colocalization coefficient (0.70) (). Aur-A and pLIMK1 remained colocalized to the centrosomes at the spindle poles during metaphase and anaphase (). We used anti-LIMK1 rabbit polyclonal antibodies developed against the N-terminal region, which recognized both phosphorylated and nonphosphorylated LIMK1. Although some Aur-A remained localized to the spindle microtubules from pro-metaphase through anaphase, no colocalization of LIMK1 with microtubule-associated Aur-A could be seen.
LIMK1 acts as a substrate of Aur-A in vitro.
To study if LIMK1 is phosphorylated by Aur-A, we performed in vitro kinase assays using recombinant GST-tagged inactive LIMK1 and active GST-Aur-A. The activity of recombinant LIMK1 and Aur-A was tested by their ability to phosphorylate cofilin and MBP, respectively, which showed that LIMK1 was not active, while Aur-A retained a high level of activity. Kinase assays showed a radioactive polypeptide band corresponding to LIMK1 in the presence of GST-Aur-A ( and B). Because GST-LIMK1 was inactive, LIMK1 phosphorylation was mediated by Aur-A. We also noted autophosphorylation of Aur-A during in vitro assays.
Because Aur-A interacts with the LIM domains while phosphorylating Ajuba,Citation35 we studied if LIM domains are required for Aur-A mediated phosphorylation of LIMK1. We used recombinant inactive His-tagged kinase domain of LIMK1 (LIMK1K) (Fig. S1A), which has the known phosphorylation site T508, as the substrate for kinase assays. Indeed, the kinase domain of LIMK1 was phosphorylated by Aur-A independently of the LIM domains. Since the autophosphorylated His-Aur-A was similar in size to that of His-LIMK1K (∼50 kD) ( and D and S1B), the specificity of phosphorylation was confirmed using increasing amounts of His-LIMK1K as the substrate, which showed a corresponding increase in phosphorylation ( and D). No phosphorylation was detected when His-LIMK1K was incubated with the inactive Aur-A (Aur-AK162M) (Fig. S2).
Aurora A interacts with the LIM and kinase domains of LIMK1.
The physical association between Aur-A and LIMK1 was determined by co-immunoprecipitation (co-IP) and pull-down assays using both BPH-1 (benign prostatic hyperplasia) stable subline expressing FLAG-tagged LIMK1 (F-LIMK1) (BPHLCA) and transiently transfected RWPE-1 cells expressing different domains of LIMK1. We chose BPH-1 cells for ectopic expression of LIMK1, as these cells express low amounts of LIMK1 (Fig. S3). We used a construct of constitutively active phospho-mimic (CA) mutant of LIMK1 (T508EE) for stable expression to have fully active LIMK1 (Fig. S1A). In the unphosphorylated form of LIMK1, the N-terminal LIM domains associate with the kinase domain, preventing its full activation. Phosphorylation at T508 disrupts this association, making the protein optimally active. Co-IP assays followed by western blotting with anti-Aur-A antibodies showed that Aur-A was pulled down with LIMK1 in BPHLCA cells but not in the vector-only controls (). There was no increase in overall expression of Aur-A in cells expressing F-LIMK1CA(Fig. S3). In a reverse experiment, LIMK1 was detected when Aur-A was immunoprecipitated from BPHLCA extracts ().
Experiments using extracts of PC-3 cells, which naturally overexpress LIMK1, and immobilized His-Aur-A or His-Aur-AK162M (kinase dead) followed by western blotting showed that LIMK1 was pulled down with His-Aur-A and His-Aur-AK162M but not the bead-only control (). Densitometric analysis revealed that Aur-AK162M was ∼50% less efficient than Aur-A at pulling down LIMK1 (). Lysates from RWPE-1 cells transiently transfected with pCMVLIMK1-FLAG (RWPE-1L) (Fig. S1A and B) also showed pull down of LIMK1 with His-Aur-A (, top part). Next, we examined if it interacts with the LIM domains of LIMK1. We used pCMVLIMK1LD-FLAG and pCMVLIMK1K-FLAG constructs containing the LIM domains or the kinase domain of LIMK1 (LIMK1LD or LIMK1K) (Fig. S1A and B) and repeated the pull-down assays with extracts from RWPE-1 cells expressing these domains (RWPE-1LD or RWPE-1K). Both LIMK1LD and LIMK1K were pulled down with His-Aur-A and not with the bead-only control (, bottom and middle parts).
Aurora A phosphorylates LIMK1 at S307.
Phosphopeptide analysis by mass spectrometry was used next to identify all sites of phosphorylation of inactive LIMK1 by Aur-A. In vitro kinase assays were performed with His-Aur-A, His-Aurora AK162M and GST-LIMK1, and gel extracted LIMK1 bands were used for mass spectrometry. Phosphopeptide analysis showed that LIMK1 was phosphorylated at S307 by Aur-A but not at T508 (). LIMK1 was not phosphorylated at either site by inactive Aur-A (His-Aur-AK162M) (data not shown). Phosphorylation of LIMK1 at S307 by Aur-A was confirmed by in vitro kinase assays using wild type (His-LIMK1) and LIMK1 with mutated serine to alanine at position 307 (His-LIMK1S307A). Wild-type LIMK1 was strongly phosphorylated by Aur-A, but phosphorylation of HisLIMK1S307A was barely detectable ( and C). To elucidate the phosphorylation site further, nonradioactive kinase assays were performed, and phosphorylation of LIMK1 was detected using phosphospecific LIMK1 (T508) antibodies. Although a strong phosphorylated band of LIMK1 was noted for wild-type LIMK1, only a weak phosphorylation of T508 was detected for His-LIMK1S307A (, lane 5).
Serine307 lies within the gap region between the PDZ domain and kinase domain, an area that contains many sites of serine phosphorylation (). Published studies showed phosphorylation of LIMK1 at S307,Citation54–Citation56 specifically during mitosis as a site of phosphorylationCitation54,Citation55 but functional implication of phosphorylation at this residue is unknown. Motif analysis indicated a partial homology of the S307 phosphorylation site to one of the motifs that are phosphorylated by Aur-A (pS/T with a bias of L at the +1 position) (). Interestingly, the motif at the T508 phosphorylation site (KRYpTV) shows a perfect match to the motif of Aur-A phosphorylation site ([K/N/R]-R-X-[pS/pT]-V) (Fig. S1A).
To confirm Aur-A phosphorylation of LIMK1 in vivo, we performed in vitro kinase assays using whole-cell extracts and immunoprecipitated protein complex. Extracts of RWPE-1 cells expressing LIMK1 (RWPE-1L) were phosphatase treated to remove any phosphorylated residues and used for kinase assays with His-Aur-A with addition of phosphatase inhibitor. We detected phosphorylation of LIMK1/2 by Aur-A in western blotting using anti-pLIMK1/2 (pT508/pT505) antibodies (). Phosphorylation of LIMK1 at T508 was further confirmed using immunoprecipitated LIMK1 from phosphatase-treated PC-3 extracts and from RWPE-1L cells using anti-FLAG antibodies ( and C). Strong phosphorylation at T508 in both assays was noted after incubation with recombinant His-Aur-A ( and C). Phosphorylated LIMK1 was not detected upon incubating with inactive Aur-A (Aur-AK162M) (), confirming that the phosphorylation was due to Aur-A activity rather than LIMK1 autophosphorylation. These experiments show that Aur-A also phosphorylates LIMK1 at T508 but requires an intact S307 phosphorylation site. Western blot analysis of total extract of RWPE-1 cells expressing FLAG-tagged LIMK1, LIMK1S307A and LIMK1T508A showed phosphorylation of LIMK1, LIMK1S307A but not LIMK1T508A (Fig. S4) at T508. This data confirms that expressed LIMK1S307A could be phosphorylated by kinases other than Aur-A.
The importance of phosphorylation of LIMK1 at S307 was further assessed by immunolocalization analysis of LIMK1S307A and pAur-A in mitotic cells. P69 prostate cells were transiently transfected with pCMVLIMK1-FLAG or pCMVLIMK1S307A-FLAG, and colocalization of FLAG-tagged LIMK1 with pAur-A was analyzed using antibodies against FLAG and pAur-A. Although LIMK1 was colocalized with pAur-A at the centrosomes at the spindle poles (), no obvious colocalization between pAur-A and LIMK1S307A was noted. Analysis of Pearson's correlation coefficient confirmed the loss of colocalization between these two proteins (). Furthermore, α-tubulin staining showed aster formation, but proper spindle structure was rarely seen in cells expressing LIMK1S307A mutants. We have also noted loss of colocalization between LIMK1S307A and γ-tubulin (data not shown).
Functional inactivation of Aurora A kinase was associated with pLIMK1 mislocalization.
Next, we studied the implication of Aur-A mediated phosphorylation on intracellular localization of LIMK1. We treated PC-3 and RWPE-1 cells with a specific Aur-A inhibitor, MLN8237,Citation10,Citation57 and studied the spindle morphology and targeting of pLIMK1 to the centrosomes. MLN8237 treatment (0.01 µM) showed distinct defects in spindle morphology, multipolarity and diffused staining of pAur-A, including distinct speckles of pAur-A (). Inhibition of Aur-A activity showed appearance of a stretched spindle, possibly due to defects in nuclear membrane dissolution (). This observation supports studies showing a role of Aur-A in nuclear membrane breakdown.Citation58,Citation59 In RWPE-1 cells, spindles were formed but not as tightly organized as the vehicle-treated cells. Inhibition of Aur-A activity also severely disrupted localization of pLIMK1 in mitotic PC-3 and RWPE-1 cells ( and D). In MLN8237 treated PC-3 cells, pLIMK1 was not localized to the centrosomes, but located toward the cell periphery. Inhibition of Aur-A activity did not affect centrosomal localization of centrin, as two distinct spots of centrin staining were observed in PC-3 and RWPE-1 cells. MLN8237 treated RWPE-1 cells, showed similar results, with pLIMK1 localized to the edge of the cell, rather than the centrosomes, as noted in the vehicle-treated cells. Centrin staining was largely localized to the cell periphery in these cells ( and D).
Aur-A also acts as a substrate of LIMK1 in vitro.
To determine any potential reciprocal catalytic relationship between LIMK1 and Aur-A, we examined the ability of endogenous LIMK1 from PC-3 cells to phosphorylate inactive His-Aur-AK162M in vitro using immunocomplex kinase assays. We noted that Aur-A-K162M was phosphorylated by LIMK1 ( and B). Immunoprecipitated LIMK1 also phosphorylated His-tagged cofilin as its bona fide substrate. Importantly, LIMK1-mediated phosphorylation was not at T288, Aur-A autophosphorylation site, as anti-Aur-A phosphospecific (pT288) antibodies failed to recognize the phosphorylated Aur-A polypeptide band in western blots (Fig. S5). Phosphospecific Aur-ApT288 antibodies were able to recognize autophosphorylation of Aur-A in kinase assays, which was not seen for inactive Aur-A (Fig. S5). This observation suggests that LIMK1 phosphorylates Aur-A at a site different than the activating autophosphorylation site.
Knockdown of LIMK1 was associated with decreased pAur-A (pT288) levels, mislocalized pAur-A and abnormal spindle structures.
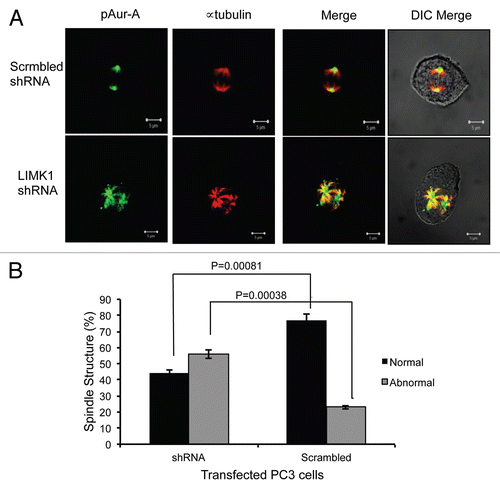
To elucidate the implication of Aur-A phosphorylation by LIMK1, we examined the effect of knockdown of LIMK1 on the levels of pAur-A (pT288). PC-3 cells were transfected with LIMK1 shRNA constructs, and 72 h post-transfection, total pAur-A levels were examined by western blotting (). A substantial decrease in the overall pT288 Aur-A levels in LIMK1 shRNA transfected cells compared with nonspecific shRNA transfected cells was noted (). Densitometric quantification showed that inhibition of expression of LIMK1 resulted in a ∼40–50% decrease in pAur-A levels compared with the control cells (), while total Aur-A levels were unaltered. Knockdown of LIMK1 also interfered with localization of pAur-A in mitotic PC-3 cells, which was more diffused compared with control cells (). The majority of pAur-A remained associated with α-tubulin, which appears to be organized in astral microtubules. The overall spindle structure was disorganized and not as tight and uniform as noted in nonspecific shRNA expressing cells. Knockdown of LIMK1 expression interfered with centrosome separation and spindle bipolarity, although it did not inhibit centrosomal localization of pAur-A. To verify the effect of LIMK1 knockdown on spindle structure, we quantified the number of transfected cells containing mitotic spindle abnormalities. There was a significant increase (2.4-fold) in the number of cells with abnormal spindles for LIMK1 shRNA expressing cells compared with the scrambled shRNA expressing cells (). Taken together, these results suggest that LIMK1 may regulate mitotic spindle organization and bipolarity through localization of pAur-A.
Discussion
The findings presented in this study suggest that a functional cooperation between Aur-A and LIMK1 is important in the early mitotic phase, specifically during mitotic spindle formation. This study also partly explains our recent observation showing localization of pLIMK1T508 to the centrosomes during prophase through telophase.Citation40 In this study, we noted that pLIMK1T508 colocalizes with Aur-A to the centrosomes during mitosis. At the centrosomes, upon activation through autophosphorylation at T288, Aur-A phosphorylates a number of proteins, including LATS2,Citation60 NDELCitation25 for centrosome maturation, kinesin motor protein Eg5,Citation61 MCAKCitation62 for spindle bipolarity and ASAPCitation63 for spindle formation. It is speculated that activated Aur-A maintains continued activation of centrosomal LIMK1 throughout its localization to the spindle poles. The requirement of sustained activation of LIMK1 at the spindle poles is supported by studies showing that LIMK1-induced cofilin phosphorylation is essential for accurate spindle orientation during metaphase through stabilization of cortical actin network.Citation41
The observation that pLIMK1T508 colocalized with Aur-A (this study) and γ-tubulinCitation40 to the centrosomes during prophase suggests that recruitment of LIMK1 to the centrosomes is necessary for proper spindle formation through modulation of actin filaments. We noted that Aur-A binds to the LIM domains and the kinase domain of LIMK1 independently and phosphorylates LIMK1 in vitro. Published studies showing similar interaction of Aur-A with the LIM domains of Ajuba and subsequent phosphorylation of Ajuba and autophosphorylation suggest that Aur-A exhibits preference for binding to LIM domain containing proteins.Citation35,Citation64
Our data further show that Aur-A phosphorylates LIMK1 primarily at S307, which lies outside the kinase domain of LIMK1, and that interaction between LIMK1 and Aur-A results in phosphorylation of LIMK1 at T508. We speculate that once S307 is phosphorylated, a possible change in conformation makes the T508 residue accessible for phosphorylation as the secondary site. Results from in vitro kinase assays and immunoprecipitation followed by immunoblot analysis suggest that Aur-A-mediated phosphorylation at S307 is essential for its phosphorylation at T508 by Aur-A. Active Aur-A was unable to phosphorylate inactive recombinant LIMK1S307A at T508. It is possible that the conformational change induced by S307 phosphorylation could either (1) allow Aurora A to directly phosphorylate at T508 or (2) allow LIMK1 to autophosphorylate at T508. Earlier, it was shown that LIMK1 becomes hyperphosphorylated upon initiation of mitosis at a site other than T508, but the site was not identified.Citation51 Our data show phosphorylation of LIMK1 at an additional site S307 by the mitotic kinase Aur-A and colocalization of these two proteins to the centrosomes. Treatment with Aur-A inhibitor MLN8237 showed a diffused accumulation of pLIMK1 (T508) in the cytoplasm. It could be speculated that LIMK1 is phosphorylated at T508 by other kinases in the absence of functional Aur-A, but pLIMK1 was not recruited to the centrosomes. This suggests that targeting of LIMK1 to the centrosomes requires Aur-A-mediated phosphorylation at S307. This speculation is further supported by our result showing that LIMK1S307A does not colocalize with Aur-A in mitotic cells.
Unlike interaction of Aur-A with Ajuba, association of Aur-A with LIMK1 also induces phosphorylation of Aur-A but not at the autophosphorylation site (T288). LIMK1-mediated phosphorylation of Aur-A was at a site other than T288, as phospho-specific (T288) Aur-A antibodies did not recognize the resulting phosphopeptide. Catalytic activation of Aur-A is through T-loop phosphorylation at T288 directly by PAK1 38 or mainly through autophosphorylation by Aur-A. Nevertheless, the possibility of a kinase that phosphorylates Aur-A in vivo and sensitizes it for autophosphorylation cannot be ruled out. Immunoprecipitated LIMK1 effectively phosphorylated kinase-dead His-Aur-A, suggesting that active LIMK1 was capable of such phosphorylation. This observation is in support of the report showing that cell extracts immunodepleted of pT288 Aur-A retained the ability of T288 phosphorylation of GST-fused Aur-A activation loop peptide.Citation65 Depletion of LIMK1 resulted in 40–50% reduced levels of pT288 Aur-A, which suggests an indirect regulatory role of LIMK1 in Aur-A phosphorylation at T288. Furthermore, knockdown of LIMK1 indicated a physiological consequence in centrosome separation and spindle bipolarity. Inhibition of LIMK1 did not inhibit Aur-A targeting to the centrosomes and actually favored microtubular localization of pAur-A. However, knockdown of LIMK1 expression interfered separation of asters needed for spindle bipolarity. Additionally, a 2.4-fold increase in the number of abnormal mitotic spindles was noted in PC-3 cells following knockdown of LIMK1. Based on our observation, we speculate that decreased phosphorylation of Aur-A at T288 could occur by two different mechanisms. First, upon binding to LIMK1, Aur-A may autophosphorylate at T288, as it does upon interaction with Ajuba. Second, LIMK1 knockdown disrupts proper Aur-A sub-cellular localization, whereby it may prevent interaction of Aur-A with interacting proteins that stimulate direct phosphorylation or autophosphorylation at T288. Nonetheless, LIMK1 induced phosphorylation of Aur-A may be important for optimal activation of Aur-A at the microtubule organization center (MTOC), and regulation of spindle bipolarity. It is known that activated Aur-A mediates formation of bipolar spindles through regulation of microtubule dynamics by inactivating phosphorylation of MCAK at the center of the aster.Citation62
In this study, we presented a novel functional relationship between Aur-A and LIMK1. This functional relationship seems to be mediated through reciprocal phosphorylation of one another. Our data show that small-molecule inhibitors alter mitotic progression not only through direct inhibition of Aur-A, but also through altered LIMK1 localization and function. Although Aur-A regulates functions of a variety of proteins, not many kinases that regulate Aur-A function are known to date. Our study provides evidence of a new mechanism whereby the function of Aur-A is regulated and that Aur-A has an additional regulatory function during mitosis. Additionally, our data suggests that development of small-molecule inhibitors targeted toward LIMK1 may have the added benefit of disrupting Aur-A function.
Material and Methods
Cell culture and transfection.
RWPE-1 cells were cultured following ATCC guidelines. PC-3 cells were maintained in 50% DMEM (InvitrogenA) and 50% F12 Ham (Sigma) containing 10% FBS (Atlanta Biologicals). BPHV and BPHLCA cells were cultured as described in reference Citation54. P69 cells (a gift from Dr. Ware) were maintained in RPMI containing EGF (10 ng/ml), dexamethasone (0.1 µM), Gentamycin (50 µg/ml), ITS mix (Insulin 5 µg/ml, Transferrin 5 µg/ml, Selenium 5 ng/ml).Citation66 RWPE-1 cells were transfected with Fugene HD (Roche). PC-3 cells were transfected with either Fugene HD or LipofectAMINE Plus and Lipofectamine LTX (Invitrogen). P69 cells were transfected with X-tremeGENE HP (Roche).
Reagents and antibodies.
GST-LIMK1 (catalytically inactive) (Abnova) and GST-Aur-A (Cell Signaling,) were used for kinase assays. Aur-A inhibitor MLN8237 was a gift from Selleck. Recombinant myelin basic protein (MBP) was from Sigma. The following antibodies were used: rabbit anti-humanphospho-Aur-A (pT288) (Cell Signaling), rabbit anti-humanphospho-LIMK1 (pT508) (Abcam), mouse anti-human-GAPDH (Sigma), mouse anti-FLAG (Sigma), rabbit anti-human-Aur-A (Bethyl), rabbit anti-human-LIMK1 (Santa Cruz), mouse antihuman LIMK1 (BD PharMingen) rabbit anti-human-α-tubulin (Sigma), anti-mouse Cy3 (Jackson Laboratory), rat anti-human-α-tubulin (Abcam), anti-rabbit Cy3 (Jackson Laboratory), anti-mouse Alexafluor-647 (Invitrogen), anti-rabbit Alexafluor-488 (Invitrogen).
Plasmid DNA and shRNA constructs.
BPHV and BPHLCA were previously described in reference Citation54. The coding sequence of LIMK1 was cloned into pCMV14-3xFLAG mammalian expression vector to generate pCMVLIMK1-FLAG. pCMV-LIMK1LD-FLAG and pCMVLIMK1K-FLAG were generated by cloning the 1–137 amino acids fragment containing LIM1 and LIM2 domains and the 258–647 fragment containing the kinase domain and the flanking sequences, respectively. LIMK1 shRNA constructs and scrambled shRNA were described previously in reference Citation54. pCMVLIMK1S307A-FLAG and pCMVLIMK1T508A-FLAG were generated by site directed mutagenesis.
Expression of recombinant protein.
The coding sequence of human Aur-A was cloned into pET30Ek/LIC vector (Novagen). Aur-AK162M kinase-dead mutant sequence was generated by site directed mutagenesis. Proteins were expressed by transforming BL21-CodonPlus (DE3) RIPL cells using our published method.Citation67 Transformed cells were induced with 1 mM IPTG at 20°C overnight. Recombinant proteins were purified using a Talon Beads cobalt affinity column (Clontech). Recombinant His-LIMK1, the kinase domain of LIMK1 (His-LIMK1K) and the His-LIMK1 S307A mutant fusion proteins were expressed in bacteria. The coding sequence of LIMK1 was cloned into pET50b(+) expression vector, and LIMK1S307A mutant sequence was generated by site directed mutagenesis. The kinase domain of LIMK1 was cloned into pET30Ek/LIC expression vector. These constructs were used for transformation of BL21-codon plus (DE3) RIPL cells and expression was induced as described above. Protein was isolated using a Protein Refolding Kit (Novagen). Briefly, inclusion bodies were solubilized in 1x solubilization Buffer (50 mM CAPS, pH 11.0, 0.3% N-lauroylsarcosine, 1 mM DTT) at room temperature for 15 min. Proteins were clarified by centrifugation at 10,000x g for 10 min at room temperature. Proteins were refolded in dialysis buffer (1 M TRIS-HCl, pH 8.5) with 0.1 mM DTT for 3 hrs at 4°C. DTT was removed by additional dialysis in dialysis buffer without DTT for 3 h at 4°C. Next, the protein was dialyzed in dialysis buffer with 1 mM reduced glutathione and 0.2 mM oxidized glutathione, overnight at 4°C. Purity of the expressed proteins was determined by SDS-PAGE followed by Coomassie staining. Recombinant Hiscofilin was purified as previously described in reference Citation68. Wild-type Aur-A fusion protein was catalytically active, but all LIMK1 and Aur-AK162M fusion proteins were inactive.
Kinase assays.
For in vitro kinase assays, GST-LIMK1, His-LIMK1, His-LIMK1K, His-LIMK1S307A, GST-Aur-A, His-Aur-A or His-Aurora AK162M recombinant proteins were incubated with either cofilin or MBP as the substrates in the either kinase assay buffer containing 50 mM HEPES, 150 mM NaCl, 5 mM MgCl2, 5 mM MnCl2, 250 µM ATP for LIMK1; or the assay buffer containing 50 mM MOPS pH 7.2, 25 mM β-glycerophosphate, 10 mM EGTA, 4 mM EDTA, 50 mM MgCl2, 0.5 mM DTT, 250 µM ATP for Aur-A and 5 nM γ-32P-ATP. The reaction mix was incubated for 30 min at room temperature (Aur-A as the kinase) or 30 min at 30°C (LIMK1 as the kinase). Reaction was stopped by the addition of Lammeli sample buffer. Phosphorylated bands were determined by SDS-PAGE followed by autoradiography of the dried gels. Non-radioactive kinase assays were performed as above in the absence of [γ-32P] ATP. In some experiments, total extracts of PC-3 or transfected RWPE-1 cells were prepared without phosphatase inhibitors and incubated with calf intestinal phosphatase (100 units for IP, 5 units for whole cell extracts) at 37°C for 30 min (NEB) to remove existing phosphorylation. Next, either total extracts or immunoprecipitated LIMK1 were used for kinase assays in the presence or absence of phosphatase inhibitor (sodium orthovanadate, sodium fluoride and β-glycerophosphate). LIMK1 phosphorylation was detected by western blotting with anti-pT508-LIMK1 antibodies. Immunocomplex kinase assays were performed using the method as described earlier.Citation68 Briefly, PC-3 and pCMV-LIMK1-FLAG transfected RWPE-1 cells were harvested using RIPA lysis buffer (50 mM Tris pH 7.5, 2 mM EDTA, 150 mM NaCl, 1% Nonidet P-40, 1 mM phenylmethylsulfonylfluoride, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 40 mM β-glycerophosphate, 1 µg/mL aprotinin, 1 µg/mL leupeptin). Cell extracts were incubated with 2 µg anti-LIMK1 (PC-3) or anti-FLAG (RWPE-1) antibodies and immunocomplex were precipitated using Sepharose A/G beads. Bead-bound LIMK1 was suspended in kinase assay buffer and used for radioactive or nonradioactive kinase assays.
Immunoprecipitation, immunoblotting and His-pull-down assays.
Immunoprecipitation experiments were performed as described before in reference Citation40, using 500 µg whole cell extract in RIPA lysis buffer and 2 µg of anti-FLAG antibodies or antiAur-A antibodies or anti-rabbit or anti-mouse IgG as the control antibodies, for 6 hr at 4°C. Sepharose A/G beads (Santa Cruz) were added next and incubation continued at 4°C overnight. The beads were washed with RIPA buffer and resuspended in Lammeli sample buffer. Immunoprecipitated proteins were detected by western blotting using anti-FLAG or anti-Aur-A antibodies. For immunoblots, 30–50 µg of total extracts were directly mixed with Lammeli sample buffer and separated on SDS-PAGE. Immunoblots were performed using appropriate primary and horseradish peroxidase conjugated respective secondary antibodies. Positive signals were detected using a chemiluminiscence ECl kit (Pierce). His-tag affinity precipitation was determined using PC-3 and RWPE-1 cell extracts. RWPE-1 cells were transfected with LIMK1 constructs and harvested at 24 h post-transfection. For the assay, 30 µg of recombinant HisAur-A or His-Aur-AK162M was incubated with MagNi-His beads (Promega) for 45 min at room temperature. Beads were washed with 100 mM HEPES, pH 7.5 and incubated with 500 µg whole cell lysates at room temperature for 1 hr. Beads were washed with a buffer (100 mM HEPES, pH 7.5) containing 20 mM imidazole and proteins were eluted in the same buffer containing 500 mM imidazole. The presence of LIMK1 in the eluates was determined by western blotting using anti-LIMK1 or anti-FLAG antibodies.
Phosphopeptide analysis.
GST-LIMK1 was incubated with His-Aur-A or His-Aur-AK162M in Aur-A kinase assay buffer at room temperature for 30 min and separated on a 4–20% SDS gradient gel. Proteins were stained, and LIMK1 bands were excised from the gel for LC MS/MS analysis performed at W.M. Keck Foundation Biotechnology Resource Laboratory (Yale Cancer Center Mass Spectrometry Resources). Samples were digested with trypsin, and phosphopeptides were enriched with TiO2. Enriched fractions and flow-through were analyzed on a LTQ Orbitrap mass spectrometer. The enriched fraction contained phosphopeptides, and the flow-through contained all the other peptides that did not bind to TiO2 column. All MS/MS spectra were searched using the automated Mascot algorithm with a confidence level set at 95% against the NCBInr database taxonomy human.
Double-label immunofluorescence.
PC-3, RWPE-1 and P69 cells were seeded onto culture dishes or poly-L-lysine coated coverslips in multi-well plates in complete growth medium and incubated for 16 hrs. When ready for processing, media was removed, and cells were washed with 100 mM sodium phosphate buffer. Cells were fixed with 4% paraformaldehyde for 5 min and then fixed/permeabilized in methanol for 10 min at −20°C. Cells were blocked for 90 min with blocking solution (10% goat serum, 2% BSA, 0.1% Tween-20 in 100 mM sodium phosphate buffer) at room temperature. Coverslips were incubated with primary antibodies for one hour at room temperature in 2% BSA and 0.1% Tween in 100 mM sodium phosphate buffer. Cells then were washed and incubated with secondary antibodies for 30 min at room temperature. Cells were post-fixed with 4% paraformaldehyde in 100 mM sodium phosphate buffer and coverslips were mounted using DAPI Fluoromount G (Southern Biotech). Cells were visualized either in a Zeiss 710 or a Leica TCS SP5II confocal microscope.
For LIMK1 expression experiments, P69 cells were seeded onto poly-L-lysine-coated covereslips placed in a 24-well plate containing complete growth medium and transfected with pCMVLIMK1-FLAG constructs. After 16 hrs and 24 hrs post transfection, cells in coverslips were processed as described above.
For LIMK1-knockdown experiments, PC-3 cells were transfected with LIMK shRNA or non-targeting shRNA, as described earlier in references Citation54. At 36 hrs post transfection, PC-3 cells were treated with 100 nM nocodazole (Sigma). Following 20 hrs of treatment, mitotic cells were isolated by shake off and released into complete media for 40 min.
For experiments with Aur-A inhibitor MLN8237, cells (2 × 104) were seeded onto poly-L-lysine-coated coverslips. 24 hrs after seeding, cells were treated with nocodazole (PC-3 100 nM, RWPE-1 50 nM), as described above, and MLN8237 (0.01 µM) or DMSO. After 24 h, cells were washed with PBS and released for 15 min in media containing MLN8237 (0.01 µM). Cells were then fixed/permeabilized, blocked, probed and mounted as described above. Spindle abnormalities were quantified by counting the number of transfected cells containing normal and abnormal spindles. A total of 200 cells were counted. Colocalization was quantified by selecting the region of interest and threshold values were set using single labels. The Pearson's correlation was quantified using Leica LAS AF Software. Pearson's correlation: [∑ (Ch1i − Ch1avg) (Ch2i − Ch2avg)]/(√(sum [(Ch1i − Ch1avg)2 (Ch2i − Ch2avg)2])].
Statistical analysis.
Quantitative results from densitometry are presented as mean ± SD of the number of independent experiments performed. Statistical differences were calculated using Student's t-test in GraphPad/Prism 4.0a. A p value of < 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
L.R. conducted all kinase assays, mutagenesis experiments, pull down assays and involved in writing the manuscript. R.O. performed all microscopy and knockdown experiments. M.R. performed coimmunoprecipitation experiments. R.C. conceived the idea, analyzed data and wrote the manuscript.
Acknowledgements
We are thankful to the National Cancer Institute of National Institute of Health and the Prostate Cancer Research Program of the Department of Defense for funding this research.
Abbreviations
| LIMK1 | = | LIM kinase1 |
| Aur-A | = | Aurora kinaseA |
| GST | = | glutathione-S-transferase |
| MBP | = | myelin basic protein |
| BPH | = | benign prostatic hyperplasia |
| IFA | = | immunofluorescence analysis |
| IP | = | immunoprecipitation |
Figures and Tables
Figure 1 LIMK1 and Aur-A colocalized to the centrosomes in mitotic cells. (A) Upper and lower parts: immunofluorescence analysis of pLIMK-pT508 (red) and Aur-A (green) in PC-3 cells. DNA was counterstained with DAPI. Colocalization of pLIMK1 with Aur-A in early prophase was mainly to the centrosomes (white arrows). (B) Pearson's correlation of colocalization between LIMK1 and Aur-A from (A). (C) Immunofluorescence of LIMK1 (red) and Aur-A (green) during mitosis. A distinct colocalization of pLIMK1 (only pLIMK1 localizes to the centrosomes) and Aur-A at the centrosomes during prophase was evident. Strong association of Aur-A with the microtubules also could be noted. At prometaphase, continued localization of pLIMK1 at the separating centrosomes along with Aur-A could be noted (white arrows). In metaphase, redistribution of pLIMK1 at the spindle poles but not at the spindle microtubules and its sustained colocalization with Aur-A was seen (white arrows). In anaphase, pLIMK1 remained colocalized with Aur-A to the poles of the elongated spindle. All pictures are representatives of at least three independent experiments. Scale bar 5 µm.
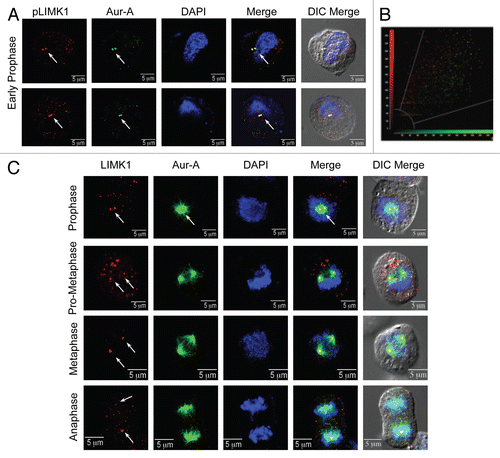
Figure 2 LIMK1 acts as a substrate of Aur-A. (A and B) Kinase assays with inactive GST-LIMK1 (500 ng) and GST-Aur-A (50 ng) kinases and His-cofilin (1 µg) or MBP (0.5 µg) as respective substrates. (A) Coomassie stained SDS-PAGE showing location of the peptide bands. MW, molecular weight marker. (B) Autoradiogram showing no phosphorylation of His-cofilin by GST-LIMK1 (lane 2), which confirms its inactivity. Strong phosphorylation of MBP and LIMK1 by Aur-A (lanes 4 and 5) and autophosphorylation of Aur-A (lane 4 and 5) could be seen. Aur-A autophosphorylation seemed to be enhanced in the presence of MBP and LIMK1. (C and D) Phosphorylation of His-LIMK1K by active His-Aur-A. (C) Coomassie stained SDS-PAGE. (D) Autoradiogram showing increased phosphorylation intensity with increasing amounts of His-LIMK1K (0.25 µg, 0.5 µg and 1 µg) by active His-Aur-A (0.22 µg) (lanes 4–6) but not by inactive His-Aur-AK162M kinase (0.22 µg) (lanes 7–9). His-Aur-AK162M was also unable to phosphorylate MBP (1 µg) (lane 2), which confirms catalytic inactivity of Aur-AK162M mutant. SDS-PAGE images are representatives of three to five independent experimental repeats.
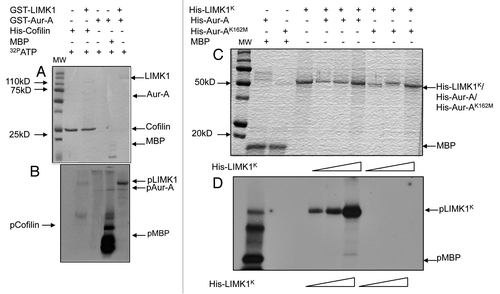
Figure 3 Aur-A physically associates with LIMK1. (A) Immunoprecipitation and reverse immunoprecipitation of FLAG-LIMK1 or Aur-A from BPHV and BPHLCA cell extracts. 500 µg total extracts were immunoprecipitated using either anti-FLAG or anti-Aur-A antibodies. Rabbit IgG was used as the negative control. Data shows strong immunoprecipitated FLAG-LIMK1 or Aur-A bands from cells expressing FLAG-LIMK1 (lanes 2 and 4). No bands could be seen in lanes for BPHV control cell extracts (lanes 3, 5, 7 and 8). No nonspecific bands were noted with rabbit antibodies (lanes 6 and 7). (B and C) Interaction between His-Aur-A and endogenous LIMK1 affinity precipitated from PC-3 extracts. (B) Immunoblots showing binding of LIMK1 with both His-Aur-A and His-Aur-AK162M (lanes 3 and 4). No nonspecific binding was noted with the beads (lane 2). (C) Densitometric analysis of the binding affinity of Aur-A and Aur-AK162M to LIMK1 from equal amounts of extracts. Data shows a 50% reduction in the affinity of binding of Aur-AK162M with LIMK1. Data represents mean ± SD of three independent experiments. (D) Interaction of Aur-A with different domains of LIMK1. Total extracts of RWPE-1 cells transiently transfected with pCMVLIMK1-FLAG, pCMVLIMK1LD-FLAG or pCMVLIMK1K-FLAG were used for affinity precipitation with His Aur-A. Data shows that in addition to wild-type LIMK1, Aur-A was capable of binding both LIM domains and kinase domain independently. The lane for bead control (lane 1) shows some nonspecific binding, but the intensity was much lower than the beads with bound Aur-A. Data shows representative images from three independent experiments.
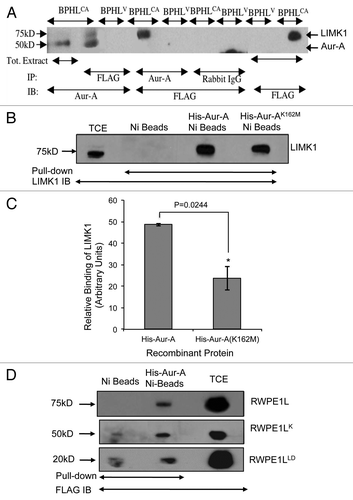
Figure 4 Aur-A phosphorylated LIMK1 at a site different than its activating phosphorylation site. (A) Phosphopeptide analysis of GST-LIMK1 after incubation with GST-Aur-A in kinase assay buffer showing phosphorylation of S307 at the linking region between the PDZ, and the kinase domain, which shows a partial motif for Aur-A phosphorylation (L at +1 position after phosphorylating residue). (B and C) Kinase assay showing loss of phosphorylation by His-Aur-A of His-LIMK1 with S307A mutation. (B) Coomassie stained SDS-PAGE. (C) Autoradiogram of the kinase assay with His-LIMK1 (0.5 µg), His-LIMK1S307A (0.5 µg), His-Aur-A (0.22 µg) and His-cofilin (1 µg). Both His-LIMK1 and His-LIMK1S307A were inactive, as no His-cofilin phosphorylated polypeptide was detected (lanes 4 and 7). While strong phosphorylation of the wild-type LIMK1 by His-Aur-A was noted (lane 4), very weak to no phosphorylation could be seen with LIMK1S307A mutant protein (lane 8), which further confirms the unique Aur-A phosphorylation site on LIMK1. (D) Phosphorylation of His-LIMK1 at S307 by Aur-A was essential for phosphorylation at T508. Immunoblot analysis of nonradioactive kinase assays using phosphospecific antibodies (pT508) showing strong phosphorylation of wild-type His-LIMK1 at T508 by His-Aur-A (lane 2), which was not noted with Aur-AK162M (lane 3). No phosphorylation at T508 could be seen when His-LIMK1S307A was incubated with His-Aur-A (lane 5). Some nonspecific signals were noted in lanes 1, 3 and 5. Data presented are representatives of at least three separate experiments.
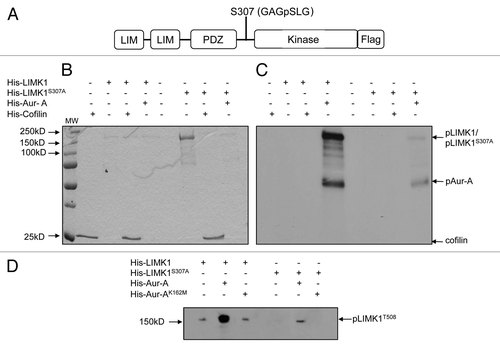
Figure 5 Aur-A allows T508 phosphorylation on endogenously expressed LIMK1. (A) Nonradioactive kinase assays using calf intestinal phosphatase (CIP) (5 units) treated extracts (50 µg) of RWPE-1 cells transfected with pCMVLIMK1-FLAG. CIP-treated extracts were incubated with His-Aur-A (0.22 µg) with phosphatase inhibitor (PPI) (lane 3), and phosphopeptide band was detected by western blotting using anti-p508-LIMK1 antibodies. Data show strong phosphorylation at T508 by His-Aur-A but not without Aur-A. No phosphorylated LIMK1 (T508) could be seen in the absence of PPI (lane 1). GAPDH was used as the loading control. (B) and (C) Nonradioactive kinase assays using immunoprecipitated FLAG-tagged LIMK1 from CIP treated (100 units) transfected RWPE-1 cell extracts (500 µg) or LIMK1 from CIP treated (100 units) PC-3 cell extracts (500 µg) and His-Aur-A (0.22 µg). Phosphorylated LIMK1 at T508 was detected by immunoblotting using anti-pT508-LIMK1 antibodies. (B) A strong phosphorylated band of FLAG-LIMK1 at T508 was evident upon incubation with His-Aur-A but not in the lane without Aur-A. (C) A similar phosphorylation at T508 of immunoprecipitated LIMK1 by His-Aur-A was noted (lane 3) which was not seen upon incubation with Aur-AK162M, which confirms the requirement of active Aur-A to achieve phosphorylation at T508 of LIMK1. Data show a representative image of at least three independent experiments.
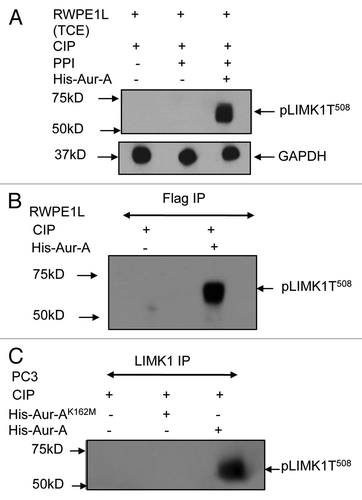
Figure 6 LIMK1 mutant did not colocalize with pAur-A. (A) Upper two parts: immunofluorescence analysis of LIMK1 (red) using anti-FLAG antibodies and pAur-A (green) in P69 cells transfected with LIMK1-FLAG constructs. Lower two parts: localization of LIMK1S307A using anti-FLAG antibodies and pAur-A in transfected P69 cells. Mitotic spindle and asters are shown by α-tubulin staining (magenta). DNA was counterstained with DAPI. Scale bar: 10 µm. (B) Relative correlation coefficient of the colocalized proteins as determined using the average value of two centrosomes per sample/cell. Results presented as mean ± SD of 10 individual cells from at least three independent experiments.
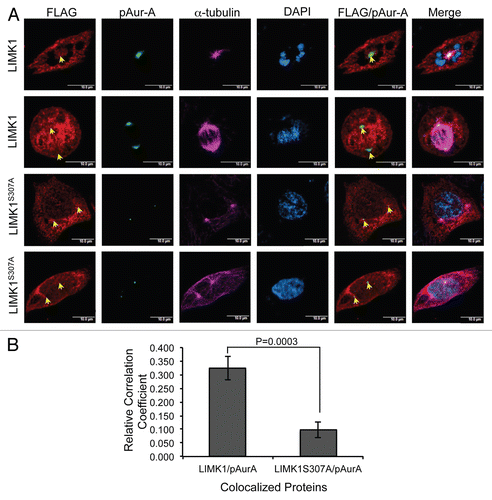
Figure 7 Aur-A inhibition led to mislocalization of pLIMK1. (A and B) Immunofluorescence analysis of nocodazole synchronized mitotic shake-off PC-3 and RWPE-1 cells following treatment with MLN8237 or vehicle. (A and B) Mitotic spindles are shown by α-tubulin staining in red, and spindle poles localization of pAur-A-pT288 is shown in green. Treatment with MLN8237 showed a distinct deformation of the spindles and altered localization of pAur-A in the mitotic cells (lower parts) compared with vehicle-treated cells. (C and D) Centrosomes are shown by staining of centrosomal protein centrin in red and pLIMK1 (T508) in green. Upper parts: in vehicle-treated cells, pLIMK1 was colocalized with centrin at the centrosomes. Inhibitor treatment (lower parts) showed a diffused staining of pLIMK1 at the periphery and mislocalization pLIMK1 from the centrosomes (orange arrow). Some diffused staining of centrin at the cell periphery was also noted in MLN8237-treated cells, although some centrosomal localization could be seen (white arrows). Scale bar: 5 µm.
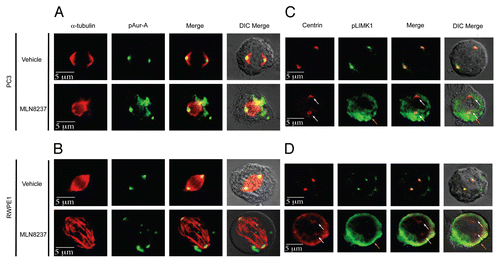
Figure 8 LIMK1 phosphorylates Aur-A. (A and B) Kinase assays using immunoprecipitated LIMK1 and His-Aur-AK162M and His-cofilin as the substrates. (A) Coomassie stained SDS-PAGE of the kinase assays. (B) Autoradiogram of the SDS-PAGE showing phosphorylation of His-Aur-AK162M (lane 3) and His-cofilin (lane 1) by LIMK1. LIMK1 was immunoprecipitated from PC-3 cells using anti-LIMK1 antibodies and incubated with His-Aur-AK162M or His-cofilin in kinase assay buffer with γ-32P-ATP. (C) Knockdown of LIMK1 leads to decreased levels of pAur-A. PC-3 cells were transiently transfected with either scrambled or LIMK1 shRNA vectors. Cells were harvested 48 hr post-transfection and levels of LIMK1, pAur-AT288 and Aur-A were detected by western blotting using anti-LIMK1, anti-pAur-AT288 or anti-Aur-A antibodies. A significant reduction in the levels of pAur-A-pT288 but not total Aur-A was evident in cells with reduced levels of LIMK1. GAPDH was used as the loading control. (D) Quantification of the relative pAur-AT288 levels from (C) by densitometry. Data show mean ± SD of three separate experiments.
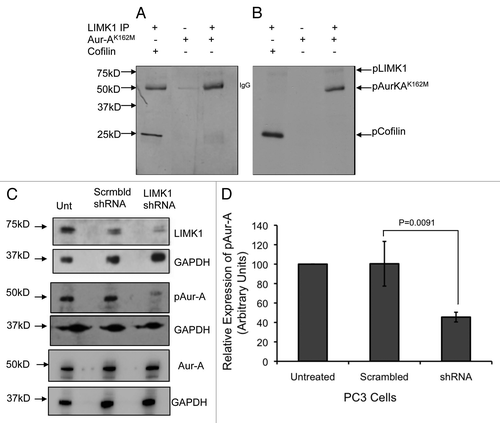
Figure 9 Knockdown of LIMK1 caused altered localization of pAur-AT288 and spindle disorganization. (A) Immunofluorescence analysis of pAur-AT288 (green) localization and α-tubulin staining (red) showing mitotic spindle structures in LIMK1 shRNA transfected PC-3 cells. A diffused staining of pAur-A in the aster microtubules with multiple speckles could be seen in cells with reduced expression of LIMK1 compared with scrambled shRNA-expressing cells. Lack of normal spindle structures was also evident in shRNA-transfected cells. (B) Quantification of spindle abnormalities of scrambled and LIMK1 shRNA transfected PC-3 cells showing a significant increase in the number of cells with abnormal spindles. A total of 200 transfected cells were counted. Data show mean ± SD of the percent change in the spindle structure of three separate experiments.
Additional material
Download Zip (2.3 MB)References
- Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta 2008; 1786:60 - 72; PMID: 18662747
- Barr AR, Gergely F. Aurora-A: the maker and breaker of spindle poles. J Cell Sci 2007; 120:2987 - 2996; PMID: 17715155; http://dx.doi.org/10.1242/jcs.013136
- Das K, Lorena PD, Ng LK, Shen L, Lim D, Siow WY, et al. Aurora-A expression, hormone receptor status and clinical outcome in hormone related cancers. Pathology 2010; 42:540 - 546; PMID: 20854072; http://dx.doi.org/10.3109/00313025.2010.508789
- Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN Jr, Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res 2008; 14:1639 - 1648; PMID: 18347165; http://dx.doi.org/10.1158/1078-0432.CCR-07-2179
- Buschhorn HM, Klein RR, Chambers SM, Hardy MC, Green S, Bearss D, et al. Aurora-A overexpression in high-grade PIN lesions and prostate cancer. Prostate 2005; 64:341 - 346; PMID: 15754349; http://dx.doi.org/10.1002/pros.20247
- Karthigeyan D, Prasad SB, Shandilya J, Agrawal S, Kundu TK. Biology of Aurora A kinase: Implications in cancer manifestation and therapy. Med Res Rev 2010; In press PMID: 20196102; http://dx.doi.org/10.1002/med.20203
- Görgün G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell cycle arrest in multiple myeloma. Blood 2010; 115:5202 - 5213; PMID: 20382844; http://dx.doi.org/10.1182/blood-2009-12-259523
- Tomita M, Mori N. Aurora A selective inhibitor MLN8237 suppresses the growth and survival of HTLV-1-infected T-cells in vitro. Cancer Sci 2010; 101:1204 - 1211; PMID: 20180813; http://dx.doi.org/10.1111/j.1349-7006.2010.01499.x
- Dees EC, Infante JR, Cohen RB, O'Neil BH, Jones S, von Mehren M, et al. Phase 1 study of MLN8054, a selective inhibitor of Aurora A kinase in patients with advanced solid tumors. Cancer Chemother Pharmacol 2011; 67:945 - 954; PMID: 20607239; http://dx.doi.org/10.1007/s00280-010-1377-y
- Macarulla T, Cervantes A, Elez E, Rodriguez-Braun E, Baselga J, Rosello S, et al. Phase I study of the selective Aurora A kinase inhibitor MLN8054 in patients with advanced solid tumors: safety, pharmacokinetics and pharmacodynamics. Mol Cancer Ther 2010; 9:2844 - 2852; PMID: 20724522; http://dx.doi.org/10.1158/1535-7163.MCT-10-0299
- Estevam J, Danaee H, Liu R, Ecsedy J, Trepicchio WL, Wyant T. Validation of a flow cytometry based G(2)M delay cell cycle assay for use in evaluating the pharmacodynamic response to Aurora A inhibition. J Immunol Methods 2011; 363:135 - 142; PMID: 20887727; http://dx.doi.org/10.1016/j.jim.2010.09.021
- Hoellein A, Pickhard A, von Keitz F, Schoeffmann S, Piontek G, Rudelius M, et al. Aurora kinase inhibition overcomes cetuximab resistance in squamous cell cancer of the head and neck. Oncotarget 2011; 2:599 - 609; PMID: 21865609
- Rao B, van Leeuwen IM, Higgins M, Campbel J, Thompson AM, Lane DP, et al. Evaluation of an Actinomycin D/VX-680 aurora kinase inhibitor combination in p53-based cyclotherapy. Oncotarget 2010; 1:639 - 650; PMID: 21317459
- De Luca M, Brunetto L, Asteriti IA, Giubettini M, Lavia P, Guarguaglini G. Aurora-A and ch-TOG act in a common pathway in control of spindle pole integrity. Oncogene 2008; 27:6539 - 6549; PMID: 18663358; http://dx.doi.org/10.1038/onc.2008.252
- Du J, Jablonski S, Yen TJ, Hannon GJ. Astrin regulates Aurora-A localization. Biochem Biophys Res Commun 2008; 370:213 - 219; PMID: 18361916; http://dx.doi.org/10.1016/j.bbrc.2008.03.072
- Van Horn RD, Chu S, Fan L, Yin T, Du J, Beckmann R, et al. Cdk1 activity is required for mitotic activation of aurora A during G2/M transition of human cells. J Biol Chem 2010; 285:21849 - 21857; PMID: 20444701; http://dx.doi.org/10.1074/jbc.M110.141010
- Giubettini M, Asteriti IA, Scrofani J, De Luca M, Lindon C, Lavia P, et al. Control of Aurora-A stability through interaction with TPX2. J Cell Sci 2011; 124:113 - 122; PMID: 21147853; http://dx.doi.org/10.1242/jcs.075457
- Santamaria A, Wang B, Elowe S, Malik R, Zhang F, Bauer M, et al. The Plk1-dependent phosphoproteome of the early mitotic spindle. Mol Cell Proteomics 2011; 10:110.004457; PMID: 20860994; http://dx.doi.org/10.1074/mcp.M110.004457
- De Luca M, Lavia P, Guarguaglini G. A functional interplay between Aurora-A, Plk1 and TPX2 at spindle poles: Plk1 controls centrosomal localization of Aurora-A and TPX2 spindle association. Cell Cycle 2006; 5:296 - 303; PMID: 16418575; http://dx.doi.org/10.4161/cc.5.3.2392
- Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol 2003; 4:842 - 854; PMID: 14625535; http://dx.doi.org/10.1038/nrm1245
- Carmena M, Ruchaud S, Earnshaw WC. Making the Auroras glow: regulation of Aurora A and B kinase function by interacting proteins. Curr Opin Cell Biol 2009; 21:796 - 805; PMID: 19836940; http://dx.doi.org/10.1016/j.ceb.2009.09.008
- Joukov V. Aurora kinases and spindle assembly: variations on a common theme?. Cell Cycle 2011; 10:895 - 903; PMID: 21325891; http://dx.doi.org/10.4161/cc.10.6.14909
- Hannak E, Kirkham M, Hyman AA, Oegema K. Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. J Cell Biol 2001; 155:1109 - 1116; PMID: 11748251; http://dx.doi.org/10.1083/jcb.200108051
- Conte N, Delaval B, Ginestier C, Ferrand A, Isnardon D, Larroque C, et al. TACC1-chTOG-Aurora A protein complex in breast cancer. Oncogene 2003; 22:8102 - 8116; PMID: 14603251; http://dx.doi.org/10.1038/sj.onc.1206972
- Mori D, Yano Y, Toyo-oka K, Yoshida N, Yamada M, Muramatsu M, et al. NDEL1 phosphorylation by Aurora-A kinase is essential for centrosomal maturation, separation and TACC3 recruitment. Mol Cell Biol 2007; 27:352 - 367; PMID: 17060449; http://dx.doi.org/10.1128/MCB.00878-06
- Abe Y, Ohsugi M, Haraguchi K, Fujimoto J, Yamamoto T. LATS2-Ajuba complex regulates gamma-tubulin recruitment to centrosomes and spindle organization during mitosis. FEBS Lett 2006; 580:782 - 788; PMID: 16413547; http://dx.doi.org/10.1016/j.febslet.2005.12.096
- Koffa MD, Casanova CM, Santarella R, Kocher T, Wilm M, Mattaj IW. HURP is part of a Ran-dependent complex involved in spindle formation. Curr Biol 2006; 16:743 - 754; PMID: 16631581; http://dx.doi.org/10.1016/j.cub.2006.03.056
- Kufer TA, Sillje HH, Korner R, Gruss OJ, Meraldi P, Nigg EA. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J Cell Biol 2002; 158:617 - 623; PMID: 12177045; http://dx.doi.org/10.1083/jcb.200204155
- Gruss OJ, Vernos I. The mechanism of spindle assembly: functions of Ran and its target TPX2. J Cell Biol 2004; 166:949 - 955; PMID: 15452138; http://dx.doi.org/10.1083/jcb.200312112
- Yabuta N, Mukai S, Okada N, Aylon Y, Nojima H. The tumor suppressor Lats2 is pivotal in Aurora A and Aurora B signaling during mitosis. Cell Cycle 2011; 10:2724 - 2736; PMID: 21822051; http://dx.doi.org/10.4161/cc.10.16.16873
- Bayliss R, Sardon T, Vernos I, Conti E. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol Cell 2003; 12:851 - 862; PMID: 14580337; http://dx.doi.org/10.1016/S1097-2765(03)00392-7
- Eckerdt F, Pascreau G, Phistry M, Lewellyn AL, DePaoli-Roach AA, Maller JL. Phosphorylation of TPX2 by Plx1 enhances activation of Aurora A. Cell Cycle 2009; 8:2413 - 2419; PMID: 19556869; http://dx.doi.org/10.4161/cc.8.15.9086
- Eyers PA, Erikson E, Chen LG, Maller JL. A novel mechanism for activation of the protein kinase Aurora A. Curr Biol 2003; 13:691 - 697; PMID: 12699628; http://dx.doi.org/10.1016/S0960-9822(03)00166-0
- Sardon T, Pache RA, Stein A, Molina H, Vernos I, Aloy P. Uncovering new substrates for Aurora A kinase. EMBO Rep 2010; 11:977 - 984; PMID: 21072059; http://dx.doi.org/10.1038/embor.2010.171
- Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell 2003; 114:585 - 598; PMID: 13678582; http://dx.doi.org/10.1016/S0092-8674(03)00642-1
- Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008; 320:1655 - 1658; PMID: 18566290; http://dx.doi.org/10.1126/science.1157425
- Satinover DL, Leach CA, Stukenberg PT, Brautigan DL. Activation of Aurora-A kinase by protein phosphatase inhibitor-2, a bifunctional signaling protein. Proc Natl Acad Sci USA 2004; 101:8625 - 8630; PMID: 15173575; http://dx.doi.org/10.1073/pnas.0402966101
- Zhao ZS, Lim JP, Ng YW, Lim L, Manser E. The GIT-associated kinase PAK targets to the centrosome and regulates Aurora-A. Mol Cell 2005; 20:237 - 249; PMID: 16246726; http://dx.doi.org/10.1016/j.molcel.2005.08.035
- Tong Y, Ben-Shlomo A, Zhou C, Wawrowsky K, Melmed S. Pituitary tumor transforming gene 1 regulates Aurora kinase A activity. Oncogene 2008; 27:6385 - 6395; PMID: 18663361; http://dx.doi.org/10.1038/onc.2008.234
- Chakrabarti R, Jones JL, Oelschlager DK, Tapia T, Tousson A, Grizzle WE. Phosphorylated LIM kinases colocalize with gamma-tubulin in centrosomes during early stages of mitosis. Cell Cycle 2007; 6:2944 - 2952; PMID: 18000399; http://dx.doi.org/10.4161/cc.6.23.4957
- Kaji N, Muramoto A, Mizuno K. LIM kinase-mediated cofilin phosphorylation during mitosis is required for precise spindle positioning. J Biol Chem 2008; 283:4983 - 4992; PMID: 18079118; http://dx.doi.org/10.1074/jbc.M708644200
- Scott RW, Olson MF. LIM kinases: function, regulation and association with human disease. J Mol Med 2007; 85:555 - 568; PMID: 17294230; http://dx.doi.org/10.1007/s00109-007-0165-6
- Bernard O. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol 2007; 39:1071 - 1076; PMID: 17188549; http://dx.doi.org/10.1016/j.biocel.2006.11.011
- Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, et al. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 1998; 393:805 - 809; PMID: 9655397; http://dx.doi.org/10.1038/31729
- Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, et al. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 1998; 393:809 - 812; PMID: 9655398; http://dx.doi.org/10.1038/31735
- Acevedo K, Li R, Soo P, Suryadinata R, Sarcevic B, Valova VA, et al. The phosphorylation of p25/TPPP by LIM kinase 1 inhibits its ability to assemble microtubules. Exp Cell Res 2007; 313:4091 - 4106; PMID: 18028908; http://dx.doi.org/10.1016/j.yexcr.2007.08.012
- Lee-Hoeflich ST, Causing CG, Podkowa M, Zhao X, Wrana JL, Attisano L. Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J 2004; 23:4792 - 4801; PMID: 15538389; http://dx.doi.org/10.1038/sj.emboj.7600418
- Sacchetti P, Carpentier R, Segard P, Olive-Cren C, Lefebvre P. Multiple signaling pathways regulate the transcriptional activity of the orphan nuclear receptor NURR1. Nucleic Acids Res 2006; 34:5515 - 5527; PMID: 17020917; http://dx.doi.org/10.1093/nar/gkl712
- Yang X, Yu K, Hao Y, Li DM, Stewart R, Insogna KL, et al. LATS1 tumour suppressor affects cytokinesis by inhibiting LIMK1. Nat Cell Biol 2004; 6:609 - 617; PMID: 15220930; http://dx.doi.org/10.1038/ncb1140
- Sumi T, Matsumoto K, Nakamura T. Mitosis-dependent phosphorylation and activation of LIM-kinase 1. Biochem Biophys Res Commun 2002; 290:1315 - 1320; PMID: 11812007; http://dx.doi.org/10.1006/bbrc.2002.6346
- Amano T, Kaji N, Ohashi K, Mizuno K. Mitosis-specific activation of LIM motif-containing protein kinase and roles of cofilin phosphorylation and dephosphorylation in mitosis. J Biol Chem 2002; 277:22093 - 22102; PMID: 11925442; http://dx.doi.org/10.1074/jbc.M201444200
- Davila M, Jhala D, Ghosh D, Grizzle WE, Chakrabarti R. Expression of LIM kinase 1 is associated with reversible G1/S phase arrest, chromosomal instability and prostate cancer. Mol Cancer 2007; 6:40; PMID: 17559677; http://dx.doi.org/10.1186/1476-4598-6-40
- Chhayi, Saxena M, Singh S, Negi MP, Srivastava AK, Trivedi R, Singh U, et al. Expression profiling of G2/M phase regulatory proteins in normal, premalignant and malignant uterine cervix and their correlation with survival of patients. J Cancer Res Ther 2010; 6:167 - 171; PMID: 20622363; http://dx.doi.org/10.4103/0973-1482.65242
- Tapia T, Ottman R, Chakrabarti R. LIM kinase1 modulates function of membrane type matrix metalloproteinase 1: implication in invasion of prostate cancer cells. Mol Cancer 2011; 10:6; PMID: 21219645; http://dx.doi.org/10.1186/1476-4598-10-6
- Deng X, Bhagat S, Dong Z, Mullins C, Chinni SR, Cher M. Tissue inhibitor of metalloproteinase-3 induces apoptosis in prostate cancer cells and confers increased sensitivity to paclitaxel. Eur J Cancer 2006; 42:3267 - 3273; PMID: 16950615; http://dx.doi.org/10.1016/j.ejca.2006.07.003
- Bonfil RD, Sabbota A, Nabha S, Bernardo MM, Dong Z, Meng H, et al. Inhibition of human prostate cancer growth, osteolysis and angiogenesis in a bone metastasis model by a novel mechanism-based selective gelatinase inhibitor. Int J Cancer 2006; 118:2721 - 2726; PMID: 16381009; http://dx.doi.org/10.1002/ijc.21645
- Sloane DA, Trikic MZ, Chu ML, Lamers MB, Mason CS, Mueller I, et al. Drug-resistant aurora A mutants for cellular target validation of the small molecule kinase inhibitors MLN8054 and MLN8237. ACS Chem Biol 2010; 5:563 - 576; PMID: 20426425; http://dx.doi.org/10.1021/cb100053q
- Portier N, Audhya A, Maddox PS, Green RA, Dammermann A, Desai A, et al. A microtubule-independent role for centrosomes and aurora a in nuclear envelope breakdown. Dev Cell 2007; 12:515 - 529; PMID: 17419991; http://dx.doi.org/10.1016/j.devcel.2007.01.019
- Romé P, Montembault E, Franck N, Pascal A, Glover DM, Giet R. Aurora A contributes to p150(glued) phosphorylation and function during mitosis. J Cell Biol 2010; 189:651 - 659; PMID: 20479466; http://dx.doi.org/10.1083/jcb.201001144
- Toji S, Yabuta N, Hosomi T, Nishihara S, Kobayashi T, Suzuki S, et al. The centrosomal protein Lats2 is a phosphorylation target of Aurora-A kinase. Genes Cells 2004; 9:383 - 397; PMID: 15147269; http://dx.doi.org/10.1111/j.1356-9597.2004.00732.x
- Zhang X, Ladd A, Dragoescu E, Budd WT, Ware JL, Zehner ZE. MicroRNA-17-3p is a prostate tumor suppressor in vitro and in vivo, and is decreased in high grade prostate tumors analyzed by laser capture microdissection. Clin Exp Metastasis 2009; 26:965 - 979; PMID: 19771525; http://dx.doi.org/10.1007/s10585-009-9287-2
- Zhang X, Ems-McClung SC, Walczak CE. Aurora A phosphorylates MCAK to control ran-dependent spindle bipolarity. Mol Biol Cell 2008; 19:2752 - 2765; PMID: 18434591; http://dx.doi.org/10.1091/mbc.E08-02-0198
- Venoux M, Basbous J, Berthenet C, Prigent C, Fernandez A, Lamb NJ, et al. ASAP is a novel substrate of the oncogenic mitotic kinase Aurora-A: phosphorylation on Ser625 is essential to spindle formation and mitosis. Hum Mol Genet 2008; 17:215 - 224; PMID: 17925329; http://dx.doi.org/10.1093/hmg/ddm298
- Prigent C, Giet R. Aurora A and mitotic commitment. Cell 2003; 114:531 - 532; PMID: 13678575; http://dx.doi.org/10.1016/S0092-8674(03)00685-8
- Ohashi S, Sakashita G, Ban R, Nagasawa M, Matsuzaki H, Murata Y, et al. Phospho-regulation of human protein kinase Aurora-A: analysis using anti-phospho-Thr288 monoclonal antibodies. Oncogene 2006; 25:7691 - 7702; PMID: 16785988; http://dx.doi.org/10.1038/sj.onc.1209754
- Plymate SR, Tennant M, Birnbaum RS, Thrasher JB, Chatta G, Ware JL. The effect on the insulin-like growth factor system in human prostate epithelial cells of immortalization and transformation by simian virus-40 T antigen. J Clin Endocrinol Metab 1996; 81:3709 - 3716; PMID: 8855827; http://dx.doi.org/10.1210/jc.81.10.3709
- Ellis JG 4th, Davila M, Chakrabarti R. Potential involvement of extracellular signal-regulated kinase 1 and 2 in encystation of a primitive eukaryote, Giardia lamblia. Stage-specific activation and intracellular localization. J Biol Chem 2003; 278:1936 - 1945; PMID: 12397063; http://dx.doi.org/10.1074/jbc.M209274200
- Davila M, Frost AR, Grizzle WE, Chakrabarti R. LIM kinase 1 is essential for the invasive growth of prostate epithelial cells: implications in prostate cancer. J Biol Chem 2003; 278:36868 - 36875; PMID: 12821664; http://dx.doi.org/10.1074/jbc.M306196200