Abstract
In metazoans, TOR is an essential protein that functions as a master regulator of cellular growth and proliferation. Over the past decade, there has been an explosion of information about this critical master kinase, ranging from the composition of the TOR protein complex to its ability to act as an integrator of numerous extracellular signals. Unfortunately, this plethora of information has also raised numerous questions regarding TOR function. Currently, the prevailing view is that mammalian TOR (mTOR) exists in at least two molecular complexes, mTORC1 and mTORC2, which are largely defined by the presence of either RAPTOR or RICTOR. However, additional co-factors have been identified for each complex, and their importance in mediating mTOR signals has been incompletely elucidated. Similarly, there are differences in mTOR function that reflect the tissue of origin. In this review, we present an alternative view to mTOR complex formation and function, which envisions mTOR regulation and signal propagation as a reflection of cell type- and basal state-dependent conditions. The re-interpretation of mTOR biology in this framework may facilitate the design of therapies most likely to effectively inhibit this central regulator of cell behavior.
Introduction
The mammalian target of rapamycin (mTOR) is one of the most studied signaling complexes, whose deregulated function leads to a variety of human disorders, ranging from cancer and immune dysfunction to autism and epilepsy.Citation1–Citation5 The function of this signaling complex is particularly relevant to nervous system biology. As a regulator of protein translation in nerve cells (neurons), mTOR signaling controls synaptic plasticity by modulating long-term potentiation (LTP) and long-term depression (LTD), important for memory and learning.Citation6–Citation9 Additionally, in the hypothalamus, mTOR regulates food intake and controls hormone production relevant to the onset of puberty.Citation10,Citation11 In neurons, mTOR regulates axonal regeneration in response to nerve injury,Citation12–Citation14 while in glial cells, it functions to limit astrocytic scar formation in the brain and spinal cord.Citation15,Citation16 Similarly, mTOR signaling is affected in a number of neurodegenerative conditions, including Alzheimer disease, Parkinson disease, stroke and Huntington disease, such that pharmacologic inhibition of mTOR activity can reduce cognitive defects associated with these conditions in mice.Citation17–Citation21 In other conditions, abnormal mTOR signaling has been implicated in the genesis of recurrent seizures, where it controls mossy fiber sprouting in the hippocampus.Citation22–Citation24 Finally, recent studies have suggested that deregulated mTOR activation might underlie autistic-like behaviors in rodents.Citation25 Together, these observations highlight the essential role of mTOR in maintaining nervous system homeostasis both in health and in disease.
The critical importance of mTOR to nervous system function derives from studies of inherited cancer predisposition syndromes in which mutations in upstream regulators of mTOR result in mTOR hyperactivation (). These conditions include neurofibromatosis type 1 (NF1) and tuberous sclerosis complex (TSC). NF1 is a common autosomal dominant disorder in which affected children and adults are prone to the development of peripheral nerve sheath tumors (neurofibromas), brain tumors (optic gliomas), learning disabilities and attention deficit.Citation26 The NF1 gene is a classic tumor suppressor gene, such that biallelic NF1 gene inactivation is required for tumorigenesis. The NF1 protein, neurofibromin, functions in part as a negative regulator of the RAS proto-oncogene and serves to accelerate the conversion of active GTP-bound RAS to inactive GDP-bound RAS by virtue of the GTPase-activating protein (GAP) domain.Citation27 Loss of neurofibromin expression in astrocytes or neural stem cells in the brain results in increased RAS activation, and leads to high levels of activated AKT and MEK signaling, which converge on the mTOR complex to regulate cell growth.Citation27–Citation29 In this respect, inhibition of mTOR hyperactivation using the macrolide rapamycin inhibits the growth of malignant peripheral nerve sheath tumors and optic gliomas in genetically engineered mice.Citation30–Citation32 These exciting pre-clinical observations have leveraged the design and execution of new clinical trials for nerve sheath tumors and gliomas in individuals with NF1.
Individuals with TSC are prone to the development of brain tumors (subependymal giant cell astrocytomas, SEGAs), intractable epilepsy, autism and mental retardation.Citation33,Citation34 In addition, affected individuals develop renal, heart and lung tumors, especially pulmonary lymphangioleiomyomatosis (LAM).Citation35 In contrast to NF1, mutations in one of two genetic loci (TSC1 or TSC2) underlie the pathogenesis of TSC.Citation36,Citation37 However, individuals with TSC harbor mutations in either the TSC1 or TSC2 gene, not both. The TSC1 gene encodes hamartin, which forms a regulatory complex with the TSC2 protein tuberin to negatively regulate the activity of a related RAS protein, termed RAS homolog expressed in brain (RHEB).Citation38–Citation43 Bi-allelic inactivation of the TSC1 or TSC2 gene leads to loss of function of the tuberin-hamartin complex and results in increased RHEB activation and mTOR hyperactivation (see below). Similar to NF1, treatment of Tsc genetically engineered mice with rapamycin to inhibit mTOR activity leads to reduced seizure frequency and severity.Citation43 Moreover, clinical trials using rapamycin analogs have demonstrated efficacy in the treatment of SEGAs in individuals with TSC.Citation44–Citation46
Other inherited syndromes that do not affect the nervous system have also provided insights into the role of mTOR signaling in normal growth control. Germline mutations in the PTEN tumor suppressor gene result in a variety of clinical conditions, including Cowden syndrome, Proteus syndrome, autism spectrum disorder (ASD) and Lhermitte-Duclos disease.Citation47–Citation51 In each of these syndromes, mutational inactivation of the PTEN gene leads to deregulated AKT signaling and increased mTOR activation. In genetically engineered mice, Pten inactivation in neuroglial cells results in progressive seizures, which can be dramatically attenuated with rapamycin analog treatment.Citation52–Citation54 Moreover, these Pten conditional knockout mice exhibit abnormal social interactions and exaggerated responses to sensory stimuli, which are also suppressed by pharmacologic mTOR inhibition.Citation25 In addition, mutations in the LKB1 gene are responsible for Peutz-Jeghers syndrome, a disorder of intestinal polyp formation.Citation55 LKB1 is a serine/threonine kinase that activates 5′AMP-activated protein kinase (AMPK) by phosphorylation to inhibit tuberin function.Citation56–Citation58 Mutational inactivation of the LKB1 gene results in mTOR hyperactivation under low energy conditions and underlies the propensity for tumor formation in conditional knockout mouse strains.Citation59,Citation60
In this Extra View, we will review the current understanding of mTOR regulation and signaling, discuss emerging and unresolved controversies in mTOR biology and propose a new conceptual model for mTOR function.
Current Concept of mTOR and its Complexes
Although cell growth, an increase in cell mass and size through macromolecular synthesis, is distinct from cell cycle progression and division, these two processes are generally tightly regulated. In eukaryotic systems, mTOR is a key regulator of cell mass and size, identified through the elucidation of the target of an antifungal agent, rapamycin.Citation61 Using genetic screens, the intracellular rapamycin receptor FKBP12 and TOR were discovered.Citation62–Citation65 Eukaryotic TORs are large proteins (∼280 kDa) that exhibit 40–60% sequence homology at the amino acid level and are members of the phosphatidylinositol-3-kinase-related protein kinase (PIKK) family ().Citation66–Citation68 Unlike PI3K, which possesses lipid kinase activity, TOR functions solely as a protein kinase.Citation69,Citation70 In Saccharomyces cerevisiae, mutations in Fpr1 (gene encoding FKBP12) or Tor1 allow for growth in the presence of rapamycin, firmly establishing the products of these genes as the bona fide rapamycin targets.Citation62 Whereas deletion of Tor1 in yeast results in decreased cell proliferation but not lethality, TOR deletion in metazoans is lethal. Genetic studies in Drosophila melanogaster first revealed the role of dTOR in regulating cell size; dTor-deficient flies exhibit a cell autonomous decrease in both cell size and proliferation.Citation71,Citation72 Mammalian TOR (mTOR) is ubiquitously expressed in tissues, with highest levels of expression in the brain and skeletal muscle.Citation73 Consistent with this pattern of expression, conditional deletion of mTOR in muscle results in decreased muscle mass, muscle dystrophy and decreased oxidative capacity.Citation74
The use of rapamycin has been a valuable tool for identifying molecular targets downstream of mTOR. While many proteins have been elucidated using this strategy, S6K-1 and 4EBP1 are the best-characterized molecules. S6K-1 phosphorylates the ribosomal protein S6 (rpS6).Citation75,Citation76 Rapamycin treatment inhibits S6K-1 activity and, thus, reduces rpS6 phosphorylation in multiple cell types under varying stimuli.Citation77,Citation78 In flies, overexpression of dS6K rescues the growth phenotype of dTOR deficiency, indicating that S6K is a critical downstream effector of dTOR in cell size determination.Citation79 Moreover, S6k1-knockout mice are considerably smaller than their wild-type littermates, further stressing the importance of S6K activity in mediating cell growth and size.Citation80
4EBP1 is a potent translational repressor, binding tightly to the eIF4E mRNA cap-binding protein. Through its interaction with eIF4E, 4EBP1 effectively competes with eIF4G, preventing its access to eIF4E. Stimulation of mTOR signaling results in 4EBP1 phosphorylation and eIF4E release, allowing eIF4E to bind to eIF4G and initiate cap-dependent mRNA translation.Citation81,Citation82 Treatment with rapamycin prevents the dissociation of 4EBP1 from eIF4E, resulting in continued translational repression and securing 4EBP1 as a downstream substrate of mTOR.Citation83
In recent years, a distinction has been made between mTOR complexes based on their sensitivity to rapamycin treatment (). mTOR complex 1 (mTORC1) is sensitive to rapamycin and consists of RAPTOR, PRAS40, mLST8 and DEPTOR proteins.Citation73,Citation84 Rapamycin binds to FKBP12, such that the rapamycin-FKBP12 complex inhibits RAPTOR-bound mTOR. mTOR complex 2 (mTORC2) is relatively insensitive to rapamycin and consists of RICTOR, PROTOR, DEPTOR, mLST8 and mSIN1.Citation85 However, prolonged treatment with rapamycin can inhibit mTORC2, most likely due to the progressive sequestration of pools of mTOR by rapamycin-FKBP12. This model predicts that overproduction of mTOR would restore mTORC2 assembly and activation in the prolonged presence of rapamycin, although this has not formally been tested.
While the individual proteins of the mTOR complexes and their precise functions are hotly debated, several key players have been identified through loss-of-function studies. Depletion of RAPTOR in mammalian cells results in decreased cell size and lower levels of phosphorylated S6K-1, demonstrating a crucial role for RAPTOR in mTOR signaling.Citation73 Moreover, non-ionic detergent treatment of mTORC1 complexes release RAPTOR from mTOR, resulting in markedly attenuated mTOR activity and a lack of 4EBP1 phosphorylation.Citation84 Similarly, depletion of mLST8 results in decreased cell size and lower S6K-1 phosphorylation, establishing mLST8 as an integral component of mTORC1.Citation86 Of note, mLST8 binds to the kinase domain of mTOR and facilitates enhanced binding of RAPTOR to mTOR. These findings further support an integral function of mLST8 in mTOR activation through its ability to bind to mTOR.
RICTOR was discovered in mTOR immunoprecipitates and shares significant regions of homology with the Avo3p rapamycin-insensitive protein from S. cerevisiae. RICTOR is found in distinct mTOR complexes devoid of RAPTOR and is unable to bind FKBP12-rapamycin.Citation85 RICTOR-bound mTOR contains mLST8, but these complexes are unable to phosphorylate S6K-1. Instead, RICTOR-mTOR-mLST8 complexes modulate the phosphorylation of PCKα and AKT.Citation87 Further studies of mTORC2 identified mSIN1 as a necessary protein in the assembly of mTORC2. mSIN1 exists as multiple isoforms, with at least three of these isoforms establishing unique mTORC2 complexes, each being regulated by different upstream stimuli.Citation88 Moreover, mSIN1 mediates the interaction of SGK-1 with mTORC2, suggesting that it might serve as a scaffold for downstream mTOR substrates. PROTOR (1 and 2) was recently added to the list of mTORC2-specific-interacting proteins. However, knockdown of PROTOR-1 does not affect mTORC2 complex formation or activity, suggesting that PROTOR-1 is not an essential mTORC2 component.Citation89
mTOR complexes are controlled by a set of negative regulators, including PRAS40 and DEPTOR. PRAS40 was first identified as an mTOR-interacting protein by mass spectrometry of mTOR complexes.Citation90 PRAS40 binds to the kinase domain of mTOR, although its interaction does not release mLST8. Additionally, PRAS40 is phosphorylated by activated AKT, and this phosphorylation results in the either the full release or lessened affinity of PRAS40 for mTOR through the formation of PRAS40 and 14-3-3 protein complexes.Citation90,Citation91 DEPTOR was discovered following immunoprecipitation of RAPTOR and RICTOR proteins. Knockdown of DEPTOR results in the activation of both mTORC1 and 2, implying that it is a negative regulator of both complexes.Citation92 However, over-expression of DEPTOR only inhibits mTORC1; mTORC2 activity is elevated upon DEPTOR overexpression. While it is unclear how this asymmetrical signaling is achieved, it is clear that mTORC1 and 2 both phosphorylate DEPTOR, signaling for its destruction by the SCF (βTrCP) E3 ligase.Citation93
To date, the evidence for two distinct mTOR complexes is largely based on the identification of independent RAPTOR- and RICTOR-containing immunoprecipitates with mTOR. Clearly, RAPTOR and RICTOR are not found in immunoprecipitates of the other protein, and RICTOR appears to interact with proteins unique to its complex (mSIN1 and PROTOR). However, the finding of multiple mSIN1 isoforms pertaining to multiple mTORC2s suggests that a simple view of two complexes may warrant further scrutiny. Many of these studies were performed in fibroblasts or established cell lines, where components may not be uniformly expressed or functional. Recent studies suggest that mTOR regulation of brain cell function may not follow these rules (see below). These new findings suggest that some of the existing dogma may need to be reassessed.
Upstream Regulation of mTOR Activation
Nutrients, energy, stress and growth factors are the major upstream signaling inputs for the mTORC1 complex (). While the varying flux of signals from each of these pathways cooperates to activate mTOR, we will discuss each component separately. Amino acids are the building blocks of proteins and can be used in the synthesis of nucleic acids and ATP. The RAG family of small GTPases serves as the link between intracellular amino acids and mTORC1.Citation94,Citation95 RAG proteins form heterodimers of RAGA or RAGB with either RAGC or RAGD. In the absence of amino acids, RAG complexes are inactive, and RAGA and RAGB are bound to GDP. The presence of amino acids activates RAGA and RAGB and switches them to GTP-bound proteins that interact with RAPTOR. This interaction causes mTORC1 to re-localize to the surface of endosomes and lysosomes, enabling mTORC1 to interact with another small GTPase, Ras homologous enriched in brain (RHEB).Citation94,Citation96 GDP-loaded RHEB is unable to interact with mTORC1. However, upon growth factor stimulation, AKT phosphorylates and inactivates tuberin (TSC2 protein),Citation97–Citation99 which, together with its binding partner hamartin (TSC1 protein), acts as a GTPase-activating protein (GAP) for RHEB.Citation38,Citation39,Citation41,Citation43 In this manner, TSC1-TSC2 complexes are potent inhibitors of mTORC1 activity. Release of hamartin/tuberin complex negative regulation of RHEB results in increased RHEB GTP-loading, which, in turn, stimulates mTOR kinase activity. Overexpression of RHEB maintains mTORC1 activity even in the absence of nutrients and growth factors,Citation38,Citation39,Citation41,Citation100 demonstrating its central role in activating mTORC1.
Previous studies using established cell lines support a model in which the primary mode of mTOR activation involves TSC-RHEB signaling. In these studies, several signaling intermediates (AMPK, MEK and AKT) regulate mTOR activity by phosphorylating tuberin on a number of residues to result in RHEB-mediated mTOR activation.Citation39,Citation98,Citation101 For example, mTOR hyperactivation and increased tumor cell line growth was inhibited when an interfering tuberin phospho-mutant protein was introduced.Citation97,Citation98 One possible exception to this route of mTOR activation involves PRAS40-mediated mTOR regulation. PRAS40 was initially shown to operate upstream of mTOR, such that AKT phosphorylation of PRAS40 relieved TORC1 suppression.Citation90,Citation91 However, more recent studies suggest that PRAS40 may also be a downstream target of mTORC1 activity.Citation102,Citation103 Knockdown of PRAS40 results in a reduction in RAPTOR bound to mTOR and attenuated mTORC1 activity, suggesting that PRAS40 might positively regulate the assembly or stabilization of mTORC1.Citation102 This would be consistent with dual roles for PRAS40 in negatively regulating the short-term activity of mTORC1 while enhancing the long-term stability of mTORC1. Analysis of the AKT phosphorylation site (Thr-246) and one of the three mTOR phosphorylation sites (Ser-221) revealed their concerted importance in promoting 14-3-3 binding for PRAS40, which helps to remove PRAS40 from the complex, thus establishing that upstream (by AKT) and downstream (by mTORC1) phosphorylation of PRAS40 are fully activated mTORC1.Citation104
Given the importance of mTORC1 in controlling ribosome biogenesis and mRNA translation, both of which require massive amounts of cellular energy, it is appropriate that mTORC1 acts as a sensor of energy and stress. In the absence of nutrients, cellular ATP levels rapidly decline due to altered mitochondrial respiration and glycolysis and trigger the AMP-activated kinase (AMPK). Under increased levels of AMP, AMPK phosphorylates TSC2 to activate its GAP activity toward RHEB and inactivate mTOR.Citation56,Citation105 Additionally, AMPK phosphorylates RAPTOR, leading to RAPTOR-14-3-3 binding and allosteric inhibition of mTORC1.Citation106 AMPK is capable of relaying signals from both energy stress, such as hypoxia,Citation107 as well as other cellular stresses, such as DNA damage.Citation108 The latter is achieved through the p53 transcriptional induction of SESTRIN1 and SESTRIN2, which are both potent activators of AMPK.Citation109 Thus, AMPK is positioned as a central cellular stress relay kinase to mTORC1.
Of note, little is known about the upstream activating signals for mTORC2. While it is appreciated that mTORC2 is largely activated by growth factors,Citation110 its ability to signal in any specific manner seems improbable given the plethora of potential upstream signals. This is further highlighted by the divergence of AKT, SGK and PKCα regulation by mTORC2 in response to various growth factors. Thus, mTORC2 must somehow be capable of distinguishing between growth factor inputs to elicit the proper downstream kinase activation. Our earlier discussion of mSIN1 isoforms might provide some clues to this achieved diversity of mTORC2, but it seems more likely that other, as-yet-unidentified mechanisms must be in place to provide this diversity in mTORC2 signaling.
Downstream mTOR Signaling
After dealing with such a diverse array of input signals, both mTORC1 and mTORC2 must direct a signaling program that leads to the activation of critical cellular processes.Citation111 In this regard, it has been easier to establish consequences of mTORC signaling than to determine the underlying intricate mechanisms of how the mTOR complex performs these decisions. As such, the hallmark of mTORC1 activity is the stimulation of ribosome biogenesis and mRNA translation (). Both processes are inhibited by rapamycin and rescued by proteins downstream of mTORC1. First, active S6K-1 stimulates the translation of mRNAs containing unique 5′-terminal oligopyrimidine tracts (TOPs).Citation112,Citation113 TOP-containing mRNAs encode ribosomal proteins, elongation factors and other critical components of ribosome production.Citation114 However, S6K-1 activity is not obligatory for this important process; cells lacking S6K-1 still actively translate TOP mRNAs in response to growth factor stimulation through a potentially redundant mechanism that does not involve the phosphorylation of rpS6.Citation113,Citation115
Second, phosphorylation of 4EBP1 by mTORC1 signals the onset of cap-dependent translation.Citation116–Citation118 Without 4EBP1 phosphorylation, 40S ribosome subunits (newly produced by S6K-1 signals) would not engage the cap complex of eIF4E, eIF4G and eIF4A to enable the beginning of mRNA scanning. The eIF4G protein is also a direct target of S6K-1. Mitogen signaling induces phosphorylation of Ser422 in a rapamycin-sensitive and S6K-1-dependent manner.Citation119 A Ser422Ala mutant mimicking non-phosphorylated eIF4G is unable to stimulate cap-dependent translation,Citation119 and RNAi knockdown of eIF4G leads to polysome reduction and translational repression of key proteins involved in cell survival and proliferation.Citation120 Additionally, S6K-1 phosphorylates and inactivates the programmed cell death 4 protein (PCD4). Phosphorylation of PCD4 on Ser67 prevents its association with eIF4A and signals the destruction of PCD4 by β-Trap.Citation121 Released eIF4A then interacts with active eIF4G to initiate translation.Citation122 This clearly places S6K-1 as a central kinase in the activation of the cap-dependent pre-initiation complex. Moreover, mTORC1 provides the key signals to both produce ribosomes and to enable those ribosomes to engage in mRNA translation in response to appropriate nutrient, energy and growth signals.
In addition to directly targeting the translational machinery, mTORC1 indirectly enhances ribosome biogenesis.Citation123 Indeed, one of the major targets of mTORC1 signaling is the nucleophosmin (NPM) proto-oncogene, a TOP-containing mRNA. Growth signals, such as those emanating from RAS, stimulate the translation of existing NPM mRNAs in a rapamycin-sensitive mechanismCitation124 through the binding of far upstream binding protein-1 (FUBP-1) to the 3′ untranslated region (UTR) of NPM mRNA.Citation125 In agreement with this finding, astrocytes lacking Nf1 express elevated NPM protein levels and exhibit enhanced protein synthesis.Citation126 NPM proteins engage assembling ribosomes in the nucleolus and aid in their transport into the cytosol, placing NPM as a critical nucleolar sensor of mTOR signalsCitation127,Citation128 (). Moreover, actin cytoskeleton rearrangement and enhanced proliferation of Nf1-deficient astrocytes is dependent on elevated NPM expression and its ability to properly transport ribosomes to the cytosol.Citation129
Only recently have we begun to appreciate the complexity of downstream mTORC2 signaling. Nonetheless, mTORC2 activation of AKT, SGK1 and PKCα appears to place mTORC2 as the effector of numerous and diverse biological processes. AKT itself possesses pleiotropic cellular effects, regulating such events as metabolism, survival and proliferation. While many AKT substrates remain to be uncovered, GSK3β and FOXO1/3A are well-studied targets of AKT, controlling many of the processes previously ascribed to mTORC2. PROTOR-1 serves as an adaptor for the phosphorylation of SGK1 by mTORC2; cells lacking PROTOR fail to activate SGK1.Citation130 SGK1 is also regulated by osmotic stress, and its activation corresponds with increased epithelial sodium channel-dependent Na+ transport.Citation131,Citation132 In glioma cell lines, increased RICTOR levels and higher mTORC2 activity enhanced cellular motility and correlated with increased PKCα activity.Citation133 Although it is unclear how mTORC2 precisely regulates changes in the actin cytoskeleton, PKCα has been implicated in this function.Citation134,Citation135 Additionally, knockdown of mTOR, mLST8 and RICTOR, but not RAPTOR, leads to severe defects in actin reorganization in the presence of serum, implicating mTORC2 in cytoskeleton regulation.Citation85 Moreover, RICTOR knockdown also decreased RAC1 activation, further linking mTORC2 to RHO-type GTPase control of the cytoskeleton and cellular motility.Citation136 Importantly, this process is not inhibited by rapamycin, squarely placing mTORC2 as a central mediator of cell migration and metastasis.Citation136,Citation137
While mTORC1 has been ascribed a primary role in regulating mRNA translation, recent evidence also points to mTORC2 as a key player in this process. mTORC2 has been isolated in polysomes, where it readily associated with individual ribosomal proteins of the 60S large subunit, including rpL23a and rpL26.Citation138,Citation139 Moreover, SIN1 associated with poly(rC) binding protein 2 (PCBP2) and RNA binding protein, which controls mRNA stability and translation in response to various cellular stresses.Citation140 Thus, it appears that mTORC2 is as connected to mRNA translation as mTORC1 (). Indeed, consistent with this notion, cells disrupted for mTORC2 function exhibit more severely attenuated translation compared with those treated with rapamycin,Citation138 forcing a reassessment of what was once thought of as an mTORC1-specific function.
Heterogeneity in mTOR Function
The canonical view of mTOR signaling envisions the presence of two static complexes with a unique set of associated proteins that collectively signal to a distinct set of downstream effectors (). The notion that all cells harbor the same collection of static complexes with limited hard-wired signaling effector pathways has recently been called into question by several new observations in brain cells (astrocytes and neural stem cells). Using a high throughput chemical library screening approach, we identified STAT3 as a downstream effector of Ras/mTOR signaling in Nf1-deficient astrocytes.Citation141 In these astrocytes, STAT3 activation was inhibited by rapamycin (). mTOR-mediated STAT3 regulation involved Rac1 activation,Citation129 which resides upstream of both mTORC1 and mTORC2.Citation142 However, astrocytes express very low levels of RICTOR, and STAT3 regulation was instead RAPTOR-dependent. These findings raise the intriguing possibility that the previously rigid signaling diagrams might not fully represent the true nature of mTOR signaling complexity in primary cells or tissues. Furthermore, it is also possible that different tissues have differing levels of mTOR components, which could determine exactly how the mTOR complex signals in any given tissue (Gutmann DH and Weber JD, unpublished observations).
Compared with other tissues,Citation143–Citation145 the biological effects of mTOR activation resulting from Pten loss, Nf1 loss, Tsc1 loss and RHEB overexpression in astrocytes are distinctCitation28 (). In these experiments, Tsc1 loss and RHEB overexpression did not increase astrocyte proliferation in vitro, whereas Pten and Nf1 loss led to increased cell growth. In all four groups of genetically modified astrocytes, mTOR activation, as measured by ribosomal S6 phosphorylation, was elevated and could be ameliorated by rapamycin treatment. However, neither neurofibromin nor PTEN loss resulted in increased tuberin phosphorylation, suggesting that TSC inactivation was not involved. Consistent with a TSC-independent mechanism of mTOR activation, RHEB silencing using shRNA completely blocked mTOR activation in Tsc1-null astrocytes but had no effect on either mTOR activation or proliferation in Nf1- or Pten-deficient cells. Since previous studies had shown that optic glioma growth in Nf1 genetically engineered mouse models could be inhibited by rapamycin treatment,Citation30 we hypothesized that RAS activation, but not Tsc1 loss or RHEB overexpression, in glial cells in vivo would result in gliomagenesis. In agreement with a model in which mTOR activation due to Tsc1 loss or RHEB over-expression are not equivalent to neurofibromin loss, Nf1 genetically engineered mice with glial Tsc1 loss or RHEB over-expression did not develop optic glioma, whereas those with glial Ras activation did. While the precise mechanism underlying the differential activation of mTOR remains to be completely elucidated, these results support a model in which mTOR function is differentially dictated by the mode of mTOR activation by its upstream regulators.
Further functional support for mTOR heterogeneity dictating cell biology was provided by studies in which neural stem cells (NSCs) from two different regions of the mouse brain were analyzed.Citation146 In these experiments, Nf1-deficient NSCs from the brainstem exhibited a cell autonomous increase in proliferation in vitro and increased gliogenesis in vivo, whereas NSCs from the neocortex did not (). Examination of the signaling pathways deregulated following neurofibromin loss revealed that only brainstem NSCs had increased AKT activation. In contrast, no increase in AKT activity was observed in Nf1-/- cortex NSCs. Interestingly, the AKT hyperactivation and increased NSC proliferation was TORC2-dependent and could be blocked by rapamycin treatment or RICTOR genetic silencing but not by RAPTOR shRNA knockdown. The basis for this striking difference in both mTOR function and NSC biology reflected differential RICTOR expression. In brainstem NSCs, there was significantly higher RICTOR expression relative to cortex NSCs (). These observations establish differential mTOR component expression as a primary determinant of cellular heterogeneity even in nearly identical cell types.
mTOR as a Dynamic Molecular Sensor and Integrator
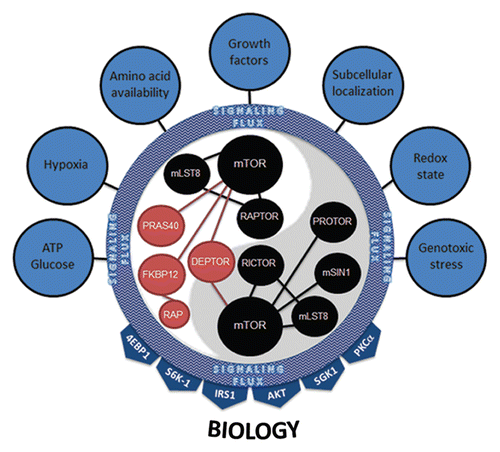
In light of recent experimental observations on the central role of mTOR as a molecular sensor and integrator in a plethora of biological settings, the current models of mTOR function require thoughtful re-examination. We propose a model in which the function of mTOR is dynamically regulated by spatially-, temporally- and signaling network-defined conditions. In this regard, which molecules are physically assembled into the mTOR complex is dictated by their expression levels and activity status. Not only will differing levels of mTOR component expression define the mTOR signaling complex, but also the interplay of the signal transduction pathways activated at any given time within the cell. The activation of these pathways likely do not operate in a linear fashion, such that MEK activation not only affects tuberin phosphorylation to impact on mTOR signaling, but also on the activity and subcellular localization of other proteins that define how mTOR signaling is regulated or the context in which its signals are transmitted. We view this state as the aggregate signaling flux (). In this setting, various mTORC components are individually (on a molecule by molecule basis) modified at the protein level (through expression or modification) to provide an overall landscape of mTOR-associated proteins. In effect, this provides the necessary complexity that allows mTOR to interpret a wide multitude of input signals. Based on this landscape, mTORC1 and mTORC2 act to target select downstream effectors. Many of these targets control cellular processes specific to each mTOR complex but may be maintained by overlapping mTORC1 and mTORC2 functions (e.g., mRNA translation). In any case, the key to understanding mTOR complex function resides in our ability to merge upstream signaling flux into mTOR complex control of downstream substrates and cellular processes. While this may seem a daunting task, recent advances in genetic and proteomic technologies should permit a more detailed view of mTOR complex assembly and how these complexes are dynamically regulated to perform such intricate multitasking within the cell.
Abbreviations
| 4EBP1 | = | eukaryotic translation initiation factor 4E binding protein 1 |
| AKT | = | protein kinase-B |
| AMPK | = | AMP-activated protein kinase |
| DEPTOR | = | DEP domain-containing mTOR-interacting protein |
| FKBP12 | = | FK506 binding protein 12 |
| FKBP38 | = | FK506 binding protein 38 |
| FUBP-1 | = | far upstream binding protein-1 |
| GAP | = | GTPase activating protein |
| MEK | = | mitogen-activated kinase (ERK) kinase |
| mLST8 | = | mammalian lethal with SEC13 protein 8 |
| GβL | = | G protein beta subunit-like |
| mSIN1 | = | mammalian stress-activated protein kinase-interacting protein 1 |
| mTOR | = | mammalian target of rapamycin |
| NF1 | = | neurofibromatosis type 1 |
| NPM | = | nucleophosmin |
| PCD4 | = | programmed cell death 4 |
| PI3K | = | phosphatidylinositol-3-kinase |
| PCBP2 | = | poly(rC) binding protein 2 |
| PRAS40 | = | proline-rich AKT substrate of 40 kDa |
| PROTOR | = | protein observed with RICTOR-1 |
| PTEN | = | phosphatase and tensin homolog |
| RAPTOR | = | regulatory associated protein of TOR |
| RHEB | = | Ras homolog enriched in brain |
| RICTOR | = | Rapamycin-insensitive companion of TOR |
| S6K | = | ribosomal S6 kinase |
| SGK1 | = | serum- and glucocorticoid-induced protein kinase 1 |
| STAT3 | = | signal transducer and activator of transcription 3 |
| TOP | = | terminal oligopyrimidine |
| TORC1 | = | mTOR complex 1 |
| TORC2 | = | mTOR complex 2 |
| TSC | = | tuberous sclerosis complex |
Ethical Statement
The authors have complied with all of the legal requirements governing the use of animals at the Washington University School of Medicine under active and approved Animal Studies Protocols.
Figures and Tables
Figure 1 Current model of mTOR regulation. The mTOR complex is regulated by numerous upstream kinase molecules. Neurofibromin (Nf1 gene product) loss leads to increased RAS activity, leading to AKT-mediated phosphorylation of PRAS40 and release of PRAS40-mediated mTOR inhibition. In addition, the increased RAS activity in Nf1-deficient cells leads to MEK activation, which phosphorylates tuberin (TSC complex), and increased Rheb-mediated mTOR activation. Lastly, LKB1 activates AMPK, which inhibits TSC complex function, such that loss of function mutations in LKB1 lead to increased Rheb-mediated mTOR activation.
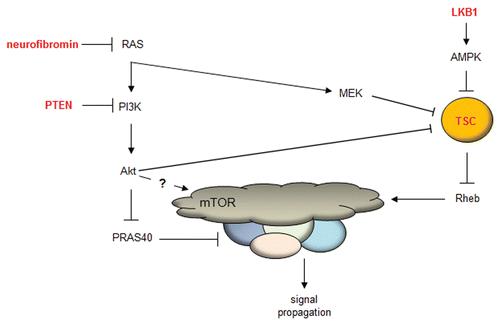
Figure 2 Anatomy of the mTOR protein. The mTOR protein is composed of several domains, including a HEAT (Huntington, EF3A, ATM, TOR), FAT (FRAP, ATM, TTRAP), R (TOR), FATC (FRAP, ATM, TRRAP C-terminal) and kinase domain. The amino acid residues are shown along the bottom.

Figure 3 mTOR complexes. mTOR is composed of two distinct complexes, mTORC1 and mTORC2. TORC1 is composed of mLST8, RAPTOR, DEPTOR and PRAS40, while the mTORC2 complex is composed of mSIN1, mLST8, RICTOR and DEPTOR. mTORC1 signals to S6K1, 4EBP1 and IRS1, whereas mTORC2 activates AKT, SGK1 and PKCα.
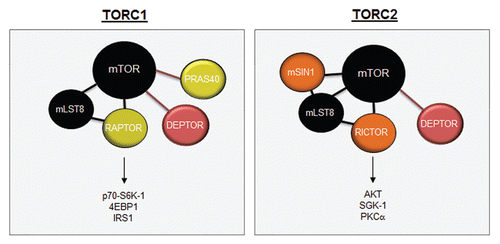
Figure 4 mTOR functions as a molecular integrator. Various inputs to mTOR provide information about amino acid availability, growth factor mitogenic signaling, oxidative damage and oxygen levels. This information must be integrated by mTOR through the use of its binding partners to provide meaningful outputs that dictate mRNA transcription, protein translation, cell growth, cell survival, ribosomal biogenesis and autophagy.
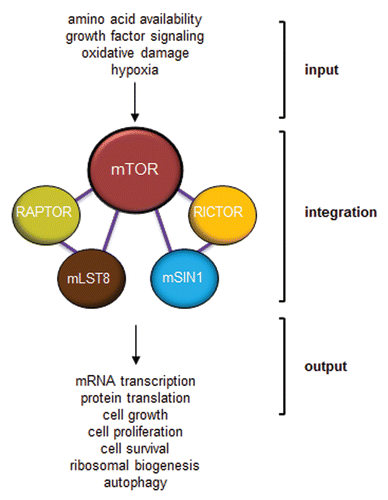
Figure 5 Control of translation by mTORC1 and mTORC2. mTORC1 controls cap-dependent translation through the phosphorylation and inactivation of 4EBP1, freeing eIF4E to bind to the 7-methylguanosine (m7G) cap structure at the 5′ end of mature mRNAs. S6K-1 phosphorylates eIF4G to initiate its interaction with eIF4G at the cap while also inactivating PCD4, the negative regulator of the eIF4A RNA helicase. mTORC1 (in astrocytes) and S6K-1 (in fibroblasts) stimulates the increased expression of nucleophosmin (NPM), which acts in a rate-limiting manner to transport mature 40S and 60S ribosome subunits from the nucleolus/nucleus into the cytosol, where they engage in mRNA translation. mTORC2 interacts with rpL23 and rpL26 of the 60S subunit on polysomes during translational elongation. Inhibition of mTORC1 or mTORC2 dramatically attenuates mRNA translation.
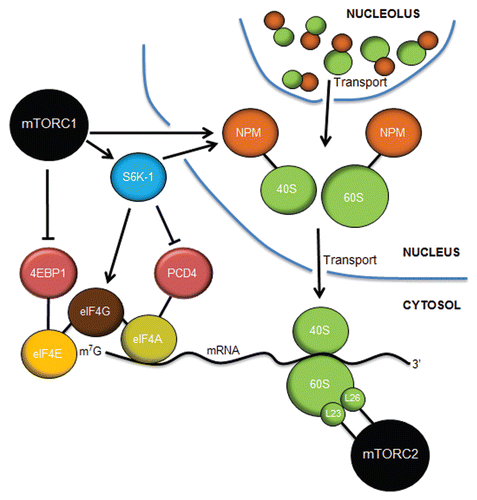
Figure 6 mTOR signaling in the brain does not follow the established canonical rules. (A) In astrocytes, neurofibromin negatively regulates mTOR through RAS/AKT-mediated signaling. Loss of neurofibromin leads to increased mTOR activation, which results in Rac1 and STAT3 activation through RAPTOR. (B) While tuberin phosphorylation results in loss of TSC complex function and increased RHEB-mediated mTOR activation, NF1 protein (neurofibromin) and PTEN loss in astrocytes leads to AKT-dependent mTOR activation, which does not involve TSC complex regulation. Moreover, Tsc1/Tsc2 inactivation or Rheb overexpression in vitro or in vivo does not recapitulate the effects of Nf1 or Pten loss on astrocyte growth or gliomagenesis in genetically engineered mice. (C) mTOR-regulated AKT activation in neural stem cells (NSCs) following Nf1 gene loss is controlled by the levels of RAPTOR and RICTOR. NSCs from the cortex fail to activate AKT and increase their proliferation following Nf1 loss, whereas those from the brainstem activate AKT and exhibit increased proliferation. This region-specific difference reflects the relative abundance of RAPTOR and RICTOR in NSCs from these two brain regions.
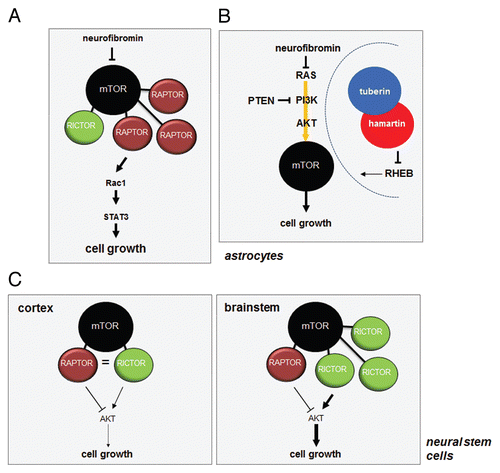
Figure 7 mTORC1 and mTORC2 integrate multiple inputs and stimulate numerous effectors. Seven major categories of inputs signal to mTOR complexes. Their combined inputs are viewed as aggregate signaling flux that is ultimately interpreted by both mTORC1 and mTORC2. The dynamics of this interpretation most likely involve changes in the protein expression level of positive (black) and negative (red) regulators of mTOR kinase activity, post-translational modification of mTORC members, binding of complexes to downstream effectors or, most likely, a combination of all of these. mTOR complexes then activate downstream signaling effectors based on their interpretation of the aggregate signaling flux flowing into mTORC1 and mTORC2. The 4EBP1, S6K-1, IRS1, AKT, SGK1 and PKCαy effectors coalesce to illicit the proper cellular biology (e.g., ribosome biogenesis, translation, growth, proliferation, survival, autophagy, lipid biogenesis, cytoskeletal changes, angiogenesis).
Acknowledgements
We thank the members of our laboratory as well as our colleagues for their helpful comments and suggestions during the preparation of this manuscript. This work was partially supported by grants from the National Institutes of Health (CA127008 to J.D.W. and D.H.G. and NS065547 to D.H.G.).
References
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell 2007; 12:9 - 22; PMID: 17613433; http://dx.doi.org/10.1016/j.ccr.2007.05.008
- Yang Q, Guan KL. Expanding mTOR signaling. Cell Res 2007; 17:666 - 681; PMID: 17680028; http://dx.doi.org/10.1038/cr.2007.64
- Rosner M, Hanneder M, Siegel N, Valli A, Fuchs C, Hengstschlager M. The mTOR pathway and its role in human genetic diseases. Mutat Res 2008; 659:284 - 292; PMID: 18598780; http://dx.doi.org/10.1016/j.mrrev.2008.06.001
- Chong ZZ, Shang YC, Zhang L, Wang S, Maiese K. Mammalian target of rapamycin: hitting the bull'seye for neurological disorders. Oxid Med Cell Longev 2010; 3:374 - 391; PMID: 21307646; http://dx.doi.org/10.4161/oxim.3.6.14787
- Proud CG. mTOR Signalling in Health and Disease. Biochem Soc Trans 2011; 39:431 - 436; PMID: 21428914; http://dx.doi.org/10.1042/BST0390431
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA 2002; 99:467 - 472; PMID: 11756682; http://dx.doi.org/10.1073/pnas.012605299
- Kelleher RJ 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 2004; 116:467 - 479; PMID: 15016380; http://dx.doi.org/10.1016/S0092-8674(04)00115-1
- Slipczuk L, Bekinschtein P, Katche C, Cammarota M, Izquierdo I, Medina JH. BDNF activates mTOR to regulate GluR1 expression required for memory formation. PLoS ONE 2009; 4:6007; PMID: 19547753; http://dx.doi.org/10.1371/journal.pone.0006007
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci 2010; 33:67 - 75; PMID: 19963289; http://dx.doi.org/10.1016/j.tins.2009.11.003
- Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, et al. Hypothalamic mTOR signaling regulates food intake. Science 2006; 312:927 - 930; PMID: 16690869; http://dx.doi.org/10.1126/science.1124147
- Roa J, Garcia-Galiano D, Varela L, Sanchez-Garrido MA, Pineda R, Castellano JM, et al. The mammalian target of rapamycin as novel central regulator of puberty onset via modulation of hypothalamic Kiss1 system. Endocrinology 2009; 150:5016 - 5026; PMID: 19734277; http://dx.doi.org/10.1210/en.2009-0096
- Gao Y, Deng K, Hou J, Bryson JB, Barco A, Nikulina E, et al. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 2004; 44:609 - 621; PMID: 15541310; http://dx.doi.org/10.1016/j.neuron.2004.10.030
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008; 322:963 - 966; PMID: 18988856; http://dx.doi.org/10.1126/science.1161566
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci 2010; 13:1075 - 1081; PMID: 20694004; http://dx.doi.org/10.1038/nn.2603
- Codeluppi S, Svensson CI, Hefferan MP, Valencia F, Silldorff MD, Oshiro M, et al. The Rheb-mTOR pathway is upregulated in reactive astrocytes of the injured spinal cord. J Neurosci 2009; 29:1093 - 1104; PMID: 19176818
- Abe N, Borson SH, Gambello MJ, Wang F, Cavalli V. Mammalian target of rapamycin (mTOR) activation increases axonal growth capacity of injured peripheral nerves. J Biol Chem 2010; 285:28034 - 28043; PMID: 20615870; http://dx.doi.org/10.1074/jbc.M110.125336
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36:585 - 595; PMID: 15146184; http://dx.doi.org/10.1038/ng1362
- Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet 2006; 15:433 - 442; PMID: 16368705; http://dx.doi.org/10.1093/hmg/ddi458
- Santini E, Heiman M, Greengard P, Valjent E, Fisone G. Inhibition of mTOR signaling in Parkinson's disease prevents L-DOPA-induced dyskinesia. Sci Signal 2009; 2:36; PMID: 19622833; http://dx.doi.org/10.1126/scisignal.2000308
- Ma T, Hoeffer CA, Capetillo-Zarate E, Yu F, Wong H, Lin MT, et al. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer's disease. PLoS ONE 2010; 5:12845; PMID: 20862226; http://dx.doi.org/10.1371/journal.pone.0012845
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS ONE 2010; 5:9979; PMID: 20376313; http://dx.doi.org/10.1371/journal.pone.0009979
- Buckmaster PS, Ingram EA, Wen X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci 2009; 29:8259 - 8269; PMID: 19553465
- Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci 2009; 29:6964 - 6972; PMID: 19474323
- Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol 2008; 63:444 - 453; PMID: 18389497; http://dx.doi.org/10.1002/ana.21331
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, et al. Pten regulates neuronal arborization and social interaction in mice. Neuron 2006; 50:377 - 388; PMID: 16675393; http://dx.doi.org/10.1016/j.neuron.2006.03.023
- Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997; 278:51 - 57; PMID: 9207339; http://dx.doi.org/10.1001/jama.1997.03550010065042
- Dasgupta B, Gutmann DH. Neurofibromatosis 1: closing the GAP between mice and men. Curr Opin Genet Dev 2003; 13:20 - 27; PMID: 12573431; http://dx.doi.org/10.1016/S0959-437X(02)00015-1
- Banerjee S, Crouse NR, Emnett RJ, Gianino SM, Gutmann DH. Neurofibromatosis-1 regulates mTOR-mediated astrocyte growth and glioma formation in a TSC/Rheb-independent manner. Proc Natl Acad Sci USA 2011; 108:15996 - 16001; PMID: 21896734; http://dx.doi.org/10.1073/pnas.1019012108
- Hegedus B, Dasgupta B, Shin JE, Emnett RJ, Hart-Mahon EK, Elghazi L, et al. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell 2007; 1:443 - 457; PMID: 18371380; http://dx.doi.org/10.1016/j.stem.2007.07.008
- Hegedus B, Banerjee D, Yeh TH, Rothermich S, Perry A, Rubin JB, et al. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res 2008; 68:1520 - 1528; PMID: 18316617; http://dx.doi.org/10.1158/0008-5472.CAN-07-5916
- Johannessen CM, Johnson BW, Williams SM, Chan AW, Reczek EE, Lynch RC, et al. TORC1 is essential for NF1-associated malignancies. Curr Biol 2008; 18:56 - 62; PMID: 18164202; http://dx.doi.org/10.1016/j.cub.2007.11.066
- Johansson G, Mahller YY, Collins MH, Kim MO, Nobukuni T, Perentesis J, et al. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Mol Cancer Ther 2008; 7:1237 - 1245; PMID: 18483311; http://dx.doi.org/10.1158/1535-7163.MCT-07-2335
- Crino PB, Henske EP. New developments in the neurobiology of the tuberous sclerosis complex. Neurology 1999; 53:1384 - 1390; PMID: 10534239
- Franz DN, Bissler JJ, McCormack FX. Tuberous sclerosis complex: neurological, renal and pulmonary manifestations. Neuropediatrics 2010; 41:199 - 208; PMID: 21210335; http://dx.doi.org/10.1055/s-0030-1269906
- Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci USA 2000; 97:6085 - 6090; PMID: 10823953; http://dx.doi.org/10.1073/pnas.97.11.6085
- van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997; 277:805 - 808; PMID: 9242607; http://dx.doi.org/10.1126/science.277.5327.805
- European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993; 75:1305 - 1315; PMID: 8269512; http://dx.doi.org/10.1016/0092-8674(93)90618-Z
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 2003; 11:1457 - 1466; PMID: 12820960; http://dx.doi.org/10.1016/S1097-2765(03)00220-X
- Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003; 17:1829 - 1834; PMID: 12869586; http://dx.doi.org/10.1101/gad.1110003
- Nellist M, van Slegtenhorst MA, Goedbloed M, van den Ouweland AM, Halley DJ, van der Sluijs P. Characterization of the cytosolic tuberinhamartin complex. Tuberin is a cytosolic chaperone for hamartin. J Biol Chem 1999; 274:35647 - 35652; PMID: 10585443; http://dx.doi.org/10.1074/jbc.274.50.35647
- Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 2003; 13:1259 - 1268; PMID: 12906785; http://dx.doi.org/10.1016/S0960-9822(03)00506-2
- van Slegtenhorst M, Nellist M, Nagelkerken B, Cheadle J, Snell R, van den Ouweland A, et al. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet 1998; 7:1053 - 1057; PMID: 9580671; http://dx.doi.org/10.1093/hmg/7.6.1053
- Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol 2003; 5:578 - 581; PMID: 12771962; http://dx.doi.org/10.1038/ncb999
- Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol 2006; 59:490 - 498; PMID: 16453317; http://dx.doi.org/10.1002/ana.20784
- Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 2010; 363:1801 - 1811; PMID: 21047224; http://dx.doi.org/10.1056/NEJMoa1001671
- Lam C, Bouffet E, Tabori U, Mabbott D, Taylor M, Bartels U. Rapamycin (sirolimus) in tuberous sclerosis associated pediatric central nervous system tumors. Pediatr Blood Cancer 2010; 54:476 - 479; PMID: 19856393; http://dx.doi.org/10.1002/pbc.22298
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 2005; 42:318 - 321; PMID: 15805158; http://dx.doi.org/10.1136/jmg.2004.024646
- Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 1997; 16:64 - 67; PMID: 9140396; http://dx.doi.org/10.1038/ng059764
- Nelen MR, van Staveren WC, Peeters EA, Hassel MB, Gorlin RJ, Hamm H, et al. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Hum Mol Genet 1997; 6:1383 - 1387; PMID: 9259288; http://dx.doi.org/10.1093/hmg/6.8.1383
- Zhou XP, Marsh DJ, Morrison CD, Chaudhury AR, Maxwell M, Reifenberger G, et al. Germline inactivation of PTEN and dysregulation of the phosphoinositol-3-kinase/Akt pathway cause human Lhermitte-Duclos disease in adults. Am J Hum Genet 2003; 73:1191 - 1198; PMID: 14566704; http://dx.doi.org/10.1086/379382
- Zhou XP, Waite KA, Pilarski R, Hampel H, Fernandez MJ, Bos C, et al. Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. Am J Hum Genet 2003; 73:404 - 411; PMID: 12844284; http://dx.doi.org/10.1086/377109
- Kwon CH, Zhu X, Zhang J, Baker SJ. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci USA 2003; 100:12923 - 12928; PMID: 14534328; http://dx.doi.org/10.1073/pnas.2132711100
- Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, et al. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci 2009; 29:1773 - 1783; PMID: 19211884
- Sunnen CN, Brewster AL, Lugo JN, Vanegas F, Turcios E, Mukhi S, et al. Inhibition of the mammalian target of rapamycin blocks epilepsy progression in NS-Pten conditional knockout mice. Epilepsia 2011; 52:2065 - 2075; PMID: 21973019; http://dx.doi.org/10.1111/j.1528-167.2011.03280.x
- Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998; 391:184 - 187; PMID: 9428765; http://dx.doi.org/10.1038/34432
- Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev 2004; 18:1533 - 1538; PMID: 15231735; http://dx.doi.org/10.1101/gad.1199104
- Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004; 6:91 - 99; PMID: 15261145; http://dx.doi.org/10.1016/j.ccr.2004.06.007
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 2003; 13:2004 - 2008; PMID: 14614828; http://dx.doi.org/10.1016/j.cub.2003.10.031
- Fu A, Ng AC, Depatie C, Wijesekara N, He Y, Wang GS, et al. Loss of Lkb1 in adult beta cells increases beta cell mass and enhances glucose tolerance in mice. Cell Metab 2009; 10:285 - 295; PMID: 19808021; http://dx.doi.org/10.1016/j.cmet.2009.08.008
- Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature 2010; 468:701 - 704; PMID: 21124456; http://dx.doi.org/10.1038/nature09595
- Abraham RT, Wiederrecht GJ. Immunopharmacology of rapamycin. Annu Rev Immunol 1996; 14:483 - 510; PMID: 8717522; http://dx.doi.org/10.1146/annurev.immunol.14.1.483
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991; 253:905 - 909; PMID: 1715094; http://dx.doi.org/10.1126/science.1715094
- Heitman J, Movva NR, Hiestand PC, Hall MN. FK 506-binding protein proline rotamase is a target for the immunosuppressive agent FK 506 in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 1991; 88:1948 - 1952; PMID: 1705713; http://dx.doi.org/10.1073/pnas.88.5.1948
- Koltin Y, Faucette L, Bergsma DJ, Levy MA, Cafferkey R, Koser PL, et al. Rapamycin sensitivity in Saccharomyces cerevisiae is mediated by a peptidylprolyl cis-trans isomerase related to human FK506-binding protein. Mol Cell Biol 1991; 11:1718 - 1723; PMID: 1996117
- Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva NR, Hall MN. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 1993; 73:585 - 596; PMID: 8387896; http://dx.doi.org/10.1016/0092-8674(93)90144-F
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994; 369:756 - 758; PMID: 8008069; http://dx.doi.org/10.1038/369756a0
- Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994; 78:35 - 43; PMID: 7518356; http://dx.doi.org/10.1016/0092-8674(94)90570-3
- Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem 1995; 270:815 - 822; PMID: 7822316; http://dx.doi.org/10.1074/jbc.270.2.815
- Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, Houghton PJ, et al. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 1997; 277:99 - 101; PMID: 9204908; http://dx.doi.org/10.1126/science.277.5322.99
- Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci USA 1998; 95:1432 - 1437; PMID: 9465032; http://dx.doi.org/10.1073/pnas.95.4.1432
- Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev 2000; 14:2712 - 2724; PMID: 11069888; http://dx.doi.org/10.1101/gad.835000
- Oldham S, Montagne J, Radimerski T, Thomas G, Hafen E. Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev 2000; 14:2689 - 2694; PMID: 11069885; http://dx.doi.org/10.1101/gad.845700
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002; 110:163 - 175; PMID: 12150925; http://dx.doi.org/10.1016/S0092-8674(02)00808-5
- Risson V, Mazelin L, Roceri M, Sanchez H, Moncollin V, Corneloup C, et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J Cell Biol 2009; 187:859 - 874; PMID: 20008564; http://dx.doi.org/10.1083/jcb.200903131
- Avruch J, Belham C, Weng Q, Hara K, Yonezawa K. The p70 S6 kinase integrates nutrient and growth signals to control translational capacity. Prog Mol Subcell Biol 2001; 26:115 - 154; PMID: 11575164; http://dx.doi.org/10.1007/978-3-642-56688-2_5
- Thomas G. The S6 kinase signaling pathway in the control of development and growth. Biol Res 2002; 35:305 - 313; PMID: 12415748; http://dx.doi.org/10.4067/S0716-97602002000200022
- Price DJ, Grove JR, Calvo V, Avruch J, Bierer BE. Rapamycin-induced inhibition of the 70-kilodalton S6 protein kinase. Science 1992; 257:973 - 977; PMID: 1380182; http://dx.doi.org/10.1126/science.1380182
- Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 1992; 69:1227 - 1236; PMID: 1377606; http://dx.doi.org/10.1016/0092-8674(92)90643-Q
- Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 kinase: a regulator of cell size. Science 1999; 285:2126 - 2129; PMID: 10497130; http://dx.doi.org/10.1126/science.285.5436.2126
- Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J 1998; 17:6649 - 6659; PMID: 9822608; http://dx.doi.org/10.1093/emboj/17.22.6649
- Lin TA, Kong X, Haystead TA, Pause A, Belsham G, Sonenberg N, et al. PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science 1994; 266:653 - 656; PMID: 7939721; http://dx.doi.org/10.1126/science.7939721
- Pause A, Belsham GJ, Gingras AC, Donze O, Lin TA, Lawrence J Jr, et al. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 1994; 371:762 - 767; PMID: 7935836; http://dx.doi.org/10.1038/371762a0
- Graves LM, Bornfeldt KE, Argast GM, Krebs EG, Kong X, Lin TA, et al. cAMP- and rapamycin-sensitive regulation of the association of eukaryotic initiation factor 4E and the translational regulator PHAS-I in aortic smooth muscle cells. Proc Natl Acad Sci USA 1995; 92:7222 - 7226; PMID: 7638171; http://dx.doi.org/10.1073/pnas.92.16.7222
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002; 110:177 - 189; PMID: 12150926; http://dx.doi.org/10.1016/S0092-8674(02)00833-4
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 2004; 14:1296 - 1302; PMID: 15268862; http://dx.doi.org/10.1016/j.cub.2004.06.054
- Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell 2003; 11:895 - 904; PMID: 12718876; http://dx.doi.org/10.1016/S1097-2765(03)00114-X
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098 - 1101; PMID: 15718470; http://dx.doi.org/10.1126/science.1106148
- Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol 2006; 16:1865 - 1870; PMID: 16919458; http://dx.doi.org/10.1016/j.cub.2006.08.001
- Pearce LR, Huang X, Boudeau J, Pawlowski R, Wullschleger S, Deak M, et al. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J 2007; 405:513 - 522; PMID: 17461779; http://dx.doi.org/10.1042/BJ20070540
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 2007; 25:903 - 915; PMID: 17386266; http://dx.doi.org/10.1016/j.molcel.2007.03.003
- Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 2007; 9:316 - 323; PMID: 17277771; http://dx.doi.org/10.1038/ncb1547
- Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009; 137:873 - 886; PMID: 19446321; http://dx.doi.org/10.1016/j.cell.2009.03.046
- Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR Inhibitor, Is a Physiological Substrate of SCF(betaTrCP) E3 Ubiquitin Ligase and Regulates Survival and Autophagy. Mol Cell 2011; 44:304 - 316; PMID: 22017876; http://dx.doi.org/10.1016/j.molcel.2011.08.029
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008; 320:1496 - 1501; PMID: 18497260; http://dx.doi.org/10.1126/science.1157535
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 2008; 10:935 - 945; PMID: 18604198; http://dx.doi.org/10.1038/ncb1753
- Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol 2003; 5:566 - 571; PMID: 12766776; http://dx.doi.org/10.1038/ncb996
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002; 4:648 - 657; PMID: 12172553; http://dx.doi.org/10.1038/ncb839
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide-3-kinase/akt pathway. Mol Cell 2002; 10:151 - 162; PMID: 12150915; http://dx.doi.org/10.1016/S1097-2765(02)00568-3
- Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol 2002; 4:658 - 665; PMID: 12172554; http://dx.doi.org/10.1038/ncb840
- Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem 2003; 278:32493 - 32496; PMID: 12842888; http://dx.doi.org/10.1074/jbc.C300226200
- Tee AR, Anjum R, Blenis J. Inactivation of the tuberous sclerosis complex-1 and -2 gene products occurs by phosphoinositide-3-kinase/Akt-dependent and -independent phosphorylation of tuberin. J Biol Chem 2003; 278:37288 - 37296; PMID: 12867426; http://dx.doi.org/10.1074/jbc.M303257200
- Fonseca BD, Smith EM, Lee VH, MacKintosh C, Proud CG. PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J Biol Chem 2007; 282:24514 - 24524; PMID: 17604271; http://dx.doi.org/10.1074/jbc.M704406200
- Oshiro N, Takahashi R, Yoshino K, Tanimura K, Nakashima A, Eguchi S, et al. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J Biol Chem 2007; 282:20329 - 20339; PMID: 17517883; http://dx.doi.org/10.1074/jbc.M702636200
- Wang L, Harris TE, Lawrence J Jr. Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J Biol Chem 2008; 283:15619 - 15627; PMID: 18372248; http://dx.doi.org/10.1074/jbc.M800723200
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006; 126:955 - 968; PMID: 16959574; http://dx.doi.org/10.1016/j.cell.2006.06.055
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008; 30:214 - 226; PMID: 18439900; http://dx.doi.org/10.1016/j.molcel.2008.03.003
- Kemp BE, Mitchelhill KI, Stapleton D, Michell BJ, Chen ZP, Witters LA. Dealing with energy demand: the AMP-activated protein kinase. Trends Biochem Sci 1999; 24:22 - 25; PMID: 10087918; http://dx.doi.org/10.1016/S0968-0004(98)01340-1
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005; 18:283 - 293; PMID: 15866171; http://dx.doi.org/10.1016/j.molcel.2005.03.027
- Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008; 134:451 - 460; PMID: 18692468; http://dx.doi.org/10.1016/j.cell.2008.06.028
- Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle 2011; 10:2305 - 2316; PMID: 21670596; http://dx.doi.org/10.4161/cc.10.14.16586
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011; 12:21 - 35; PMID: 21157483; http://dx.doi.org/10.1038/nrm3025
- Hornstein E, Tang H, Meyuhas O. Mitogenic and nutritional signals are transduced into translational efficiency of TOP mRNAs. Cold Spring Harb Symp Quant Biol 2001; 66:477 - 484; PMID: 12762050; http://dx.doi.org/10.1101/sqb.2001.66.477
- Tang H, Hornstein E, Stolovich M, Levy G, Livingstone M, Templeton D, et al. Amino acid-induced translation of TOP mRNAs is fully dependent on phosphatidylinositol-3-kinase-mediated signaling, is partially inhibited by rapamycin, and is independent of S6K1 and rpS6 phosphorylation. Mol Cell Biol 2001; 21:8671 - 8683; PMID: 11713299; http://dx.doi.org/10.1128/MCB.21.24.8671-83.2001
- Meyuhas O. Synthesis of the translational apparatus is regulated at the translational level. Eur J Biochem 2000; 267:6321 - 6330; PMID: 11029573; http://dx.doi.org/10.1046/j.1432-327.2000.01719.x
- Stolovich M, Tang H, Hornstein E, Levy G, Cohen R, Bae SS, et al. Transduction of growth or mitogenic signals into translational activation of TOP mRNAs is fully reliant on the phosphatidylinositol-3-kinase-mediated pathway but requires neither S6K1 nor rpS6 phosphorylation. Mol Cell Biol 2002; 22:8101 - 8113; PMID: 12417714; http://dx.doi.org/10.1128/MCB.22.23.8101-13.2002
- Beretta L, Gingras AC, Svitkin YV, Hall MN, Sonenberg N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J 1996; 15:658 - 664; PMID: 8599949
- Haghighat A, Mader S, Pause A, Sonenberg N. Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J 1995; 14:5701 - 5709; PMID: 8521827
- Mader S, Lee H, Pause A, Sonenberg N. The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4gamma and the translational repressors 4E-binding proteins. Mol Cell Biol 1995; 15:4990 - 4997; PMID: 7651417
- Raught B, Peiretti F, Gingras AC, Livingstone M, Shahbazian D, Mayeur GL, et al. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J 2004; 23:1761 - 1769; PMID: 15071500; http://dx.doi.org/10.1038/sj.emboj.7600193
- Shahbazian D, Parsyan A, Petroulakis E, Topisirovic I, Martineau Y, Gibbs BF, et al. Control of cell survival and proliferation by mammalian eukaryotic initiation factor 4B. Mol Cell Biol 2010; 30:1478 - 1485; PMID: 20086100; http://dx.doi.org/10.1128/MCB.01218-09
- Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006; 314:467 - 471; PMID: 17053147; http://dx.doi.org/10.1126/science.1130276
- Yang HS, Cho MH, Zakowicz H, Hegamyer G, Sonenberg N, Colburn NH. A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol Cell Biol 2004; 24:3894 - 3906; PMID: 15082783; http://dx.doi.org/10.1128/MCB.24.9.3894-906.2004
- Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 2006; 25:6384 - 6391; PMID: 17041624; http://dx.doi.org/10.1038/sj.onc.1209883
- Pelletier CL, Maggi L Jr, Brady SN, Scheidenhelm DK, Gutmann DH, Weber JD. TSC1 sets the rate of ribosome export and protein synthesis through nucleophosmin translation. Cancer Res 2007; 67:1609 - 1617; PMID: 17308101; http://dx.doi.org/10.1158/0008-5472.CAN-06-2875
- Olanich ME, Moss BL, Piwnica-Worms D, Townsend RR, Weber JD. Identification of FUSE-binding protein 1 as a regulatory mRNA-binding protein that represses nucleophosmin translation. Oncogene 2011; 30:77 - 86; PMID: 20802533; http://dx.doi.org/10.1038/onc.2010.404
- Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res 2005; 65:2755 - 2760; PMID: 15805275; http://dx.doi.org/10.1158/0008-5472.CAN-04-4058
- Maggi L Jr, Kuchenruether M, Dadey DY, Schwope RM, Grisendi S, Townsend RR, et al. Nucleophosmin serves as a rate-limiting nuclear export chaperone for the Mammalian ribosome. Mol Cell Biol 2008; 28:7050 - 7065; PMID: 18809582; http://dx.doi.org/10.1128/MCB.01548-07
- Yu Y, Maggi L Jr, Brady SN, Apicelli AJ, Dai MS, Lu H, et al. Nucleophosmin is essential for ribosomal protein L5 nuclear export. Mol Cell Biol 2006; 26:3798 - 3809; PMID: 16648475; http://dx.doi.org/10.1128/MCB.26.10.3798-809.2006
- Sandsmark DK, Zhang H, Hegedus B, Pelletier CL, Weber JD, Gutmann DH. Nucleophosmin mediates mammalian target of rapamycin-dependent actin cytoskeleton dynamics and proliferation in neurofibromin-deficient astrocytes. Cancer Res 2007; 67:4790 - 4799; PMID: 17510408; http://dx.doi.org/10.1158/0008-5472.CAN-06-4470
- Pearce LR, Sommer EM, Sakamoto K, Wullschleger S, Alessi DR. Protor-1 is required for efficient mTORC2-mediated activation of SGK1 in the kidney. Biochem J 2011; 436:169 - 179; PMID: 21413931; http://dx.doi.org/10.1042/BJ20102103
- Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, et al. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J 2001; 20:7052 - 7059; PMID: 11742982; http://dx.doi.org/10.1093/emboj/20.24.7052
- Pearce D. The role of SGK1 in hormone-regulated sodium transport. Trends Endocrinol Metab 2001; 12:341 - 347; PMID: 11551807; http://dx.doi.org/10.1016/S1043-2760(01)00439-8
- Masri J, Bernath A, Martin J, Jo OD, Vartanian R, Funk A, et al. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via over-expression of rictor. Cancer Res 2007; 67:11712 - 11720; PMID: 18089801; http://dx.doi.org/10.1158/0008-5472.CAN-07-2223
- Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J 2008; 27:1932 - 1943; PMID: 18566586; http://dx.doi.org/10.1038/emboj.2008.120
- Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J 2008; 27:1919 - 1931; PMID: 18566587; http://dx.doi.org/10.1038/emboj.2008.119
- Gulhati P, Bowen KA, Liu J, Stevens PD, Rychahou PG, Chen M, et al. mTORC1 and mTORC2 regulate EMT, motility and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res 2011; 71:3246 - 3256; PMID: 21430067; http://dx.doi.org/10.1158/0008-5472.CAN-10-4058
- Hernández-Negrete I, Carretero-Ortega J, Rosenfeldt H, Hernandez-Garcia R, Calderon-Salinas JV, Reyes-Cruz G, et al. P-Rex1 links mammalian target of rapamycin signaling to Rac activation and cell migration. J Biol Chem 2007; 282:23708 - 23715; PMID: 17565979; http://dx.doi.org/10.1074/jbc.M703771200
- Oh WJ, Wu CC, Kim SJ, Facchinetti V, Julien LA, Finlan M, et al. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J 2010; 29:3939 - 3951; PMID: 21045808; http://dx.doi.org/10.1038/emboj.2010.271
- Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell 2011; 144:757 - 768; PMID: 21376236; http://dx.doi.org/10.1016/j.cell.2011.02.014
- Ghosh D, Srivastava GP, Xu D, Schulz LC, Roberts RM. A link between SIN1 (MAPKAP1) and poly(rC) binding protein 2 (PCBP2) in counteracting environmental stress. Proc Natl Acad Sci USA 2008; 105:11673 - 11678; PMID: 18687895; http://dx.doi.org/10.1073/pnas.0803182105
- Banerjee S, Byrd JN, Gianino SM, Harpstrite SE, Rodriguez FJ, Tuskan RG, et al. The neurofibromatosis type 1 tumor suppressor controls cell growth by regulating signal transducer and activator of transcription-3 activity in vitro and in vivo. Cancer Res 2010; 70:1356 - 1366; PMID: 20124472; http://dx.doi.org/10.1158/0008-5472.CAN-09-2178
- Saci A, Cantley LC, Carpenter CL. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol Cell 2011; 42:50 - 61; PMID: 21474067; http://dx.doi.org/10.1016/j.molcel.2011.03.017
- Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, et al. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest 2003; 112:1223 - 1233; PMID: 14561707
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005; 436:725 - 730; PMID: 16079851; http://dx.doi.org/10.1038/nature03918
- Cichowski K, Santiago S, Jardim M, Johnson BW, Jacks T. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumor suppressor. Genes Dev 2003; 17:449 - 454; PMID: 12600938; http://dx.doi.org/10.1101/gad.1054703
- Lee da Y, Yeh TH, Emnett RJ, White CR, Gutmann DH. Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev 2010; 24:2317 - 2329; PMID: 20876733; http://dx.doi.org/10.1101/gad.1957110