Abstract
The transcription factor c-Myc strongly stimulates cell proliferation but also regulates apoptosis, senescence, cell competition and cell differentiation, and its elevated activity is a hallmark for human tumorigenesis. c-Myc induces transcription by forming heterodimers with Max and then directly binding DNA at E-box sequences. Conversely, transcription repression depends primarily on the inhibitory interaction of c-Myc/Max with Miz-1 at DNA initiator elements. We recently described a distinct mechanism of c-Myc gene regulation, in which c-Myc interacts with the retinoic acid receptor α (RARα) and is recruited to RAR DNA binding sequences (RAREs). In leukemia cells, this c-Myc/RARα complex functions either as an activator or a repressor of RARα-dependent targets through a phosphorylation switch. Unphosphorylated c-Myc interacts with RARα to repress the expression of RAR targets required for differentiation, thereby aggravating leukemia malignancy. However, if c-Myc is phosphorylated by the kinase Pak2, the c-Myc/RARα complex activates transcription of those same genes to stimulate differentiation, thus reducing tumor burden. Here, we discuss the role of c-Myc in balancing proliferation and differentiation and how modulating this previously unidentified c-Myc activity might provide alternative therapies against leukemia and possibly other types of tumors.
Introduction
c-Myc is a global transcriptional regulator that can bind to approximately 10% to 15% of the genome.1 Traditionally, the c-Myc N-terminal domain has been considered to be involved in transactivation and transrepression, and the C-terminal domain to be critical for DNA binding. While much research has focused on the role of c-Myc as a transcriptional activator, which is important for cellular transformation, less is known about its activity as a transcriptional repressor. Genome-wide analyses have indicated that c-Myc represses at least as many targets as it activates, emphasizing the importance of transcriptional repression in the biological function of c-Myc.1
As a basic helix-loop-helix leucine zipper protein (bHLH-Zip), c-Myc activates transcription of genes involved in cell proliferation, cell growth and metabolism by binding to the DNA sequence CAC GTG (termed the E box) when dimerized with the bHLH-ZIP protein Max (Myc-associated protein X).2-6 However, c-Myc also localizes to promoters of several target genes that do not contain E boxes (reviewed in ref. 1). Thus, the c-Myc/Max heterodimer can repress transcription by interacting with, and thereby inhibiting, transcriptional activators that recognize the core promoter of its target genes; to date, Miz-1 is the best characterized transcription factor that is inhibited in this way.7,8 The genes repressed by c-Myc/Max include cell cycle inhibitors and proteins involved in cell adhesion.7-16
Increasing evidence for Max-independent functions of c-Myc has recently accumulated, given that c-Myc proteins retain considerable biological activity when not bound to Max (reviewed in ref. 17). The clearest in vivo evidence for the existence of these functions was data obtained from Drosophila experiments.18 Deletion of Myc in Drosophila resulted in a more severe developmental phenotype than did that of Max. Moreover, re-expression of a Myc mutant incapable of interacting with Max in Myc-null Drosophila was able to rescue some (but not all) of the phenotypes.17,18 Interestingly, it has been reported that some core promoters are bound by c-Myc but not Miz-1, which indicates that there must also be other proteins able to recruit c-Myc to their associated DNA binding sites.19 Some candidate proteins have been suggested as c-Myc recruiters, such as the transcription factor II-I (TFII-I), yingyang-1 (YY1), specificity protein-1 (Sp1) and nuclear factor Y (NF-Y);19-23 however, it is still largely unknown how c-Myc regulates transcription independently of Max, Miz-1 and E-box elements.
Myc Coregulates RARE-containing Genes
Phosphorylation of c-Myc by Pak2 kinase at three residues within its C terminus is a naturally occurring intrinsic pathway that abrogates c-Myc/Max dimerization and its binding to E boxes in the cell.24 We performed a genome-wide analysis to study the transcriptional consequences of this c-Myc phosphorylation on leukemia, using a granulocytic cell line arrested at the promyelocytic stage of maturation (HL60 cells). We first analyzed all of the transcription factor DNA binding motifs that were overrepresented within the cohort of differentially expressed transcripts using leukemia cells that expressed a mutant form of c-Myc that mimics the constitutively phosphorylated form (aspartic acid mutant, MycD). This analysis identified retinoic acid (RA) target genes, and we subsequently confirmed that c-Myc can directly interact with RARα.25 The nature and transcriptional consequences of this interaction appear complex and seem to be dependent on a phosphorylation switch. In undifferentiated and proliferative cells, nonphosphorylated c-Myc binds to RARα through both its N- and C-terminal domains. After phosphorylation by the Pak2 kinase, c-Myc still interacts with RARα but only at its N terminus. Interestingly, at the transcriptional level, the nonphosphorylated-c-Myc/RARα⊇ complex represses RAR targets, whereas the phosphor-c‑Myc/RARα complex induces transcription of the same cohort of genes.
We hypothesize that, upon c-Myc phosphorylation, the partial loss of the interaction between c-Myc to RARα causes a conformational change similar to the one observed in RARα upon binding to its ligand (RA), which exposes otherwise hidden surfaces to co-activators while masking those required for co-repressor binding. In the absence of RA, RARα constitutively binds to RARE elements and interacts with co-repressors to regulate transcription. In contrast, upon RA stimulation, RARα undergoes a conformational change that exchanges co-repressors for co-activators, thereby stimulating transcription of target genes.25-29 Accordingly, we demonstrated by chromatin immunoprecipitation (ChIP) experiments that the unphosphorylated form of c-Myc increases co-repressor occupancy at RAREs, while phosphorylated c-Myc showed diminished binding to co-repressors and Max and also attracted co-activators. The repressor HDAC3 and the activator CBP were among those factors exchanged within the complex upon phosphorylation of c-Myc. Our hypothesis that this switch in affinity is due to the phosphorylation-dependent conformation is supported by the fact that c-Myc interacts with HDAC3 through its “Myc-box binding III,” whereas it interacts with CBP binds through its C terminus.30,31 However, this needs to be verified by structural studies.
c-Myc Actively Blocks Cell Differentiation
The RA signaling pathway plays a key role in promoting granulocytic terminal differentiation.32 While the ability of c-Myc to modulate the expression of target genes that are involved in cell cycle progression has been extensively reported in reference 33, the observed differentiation block produced by the proto-oncogene is less well understood.34 Cell cycle progression by c-Myc activation is generally incompatible with terminal differentiation, but one interesting question is whether c-Myc “passively” inhibits differentiation by enhancing proliferation. Some interesting studies have addressed this issue previously (reviewed in ref. 34). Our data now show that in addition to the ability of c-Myc to promote cell cycle progression through E boxes and Miz-1,4-6 it can also actively maintain the promyelocytic undifferentiated state of granulocytes by directly repressing RA-target genes through RARα binding. A picture emerges in which E box-containing genes, Miz-1 target genes and RARα target genes are all regulated by c-Myc ( and ). In proliferating cells, c-Myc concurrently induces E box-containing genes and represses Miz-1 targets to promote cell cycle entry. Additionally, RARα target genes are repressed in the presence of unphosphorylated c-Myc, which ensures that differentiation is blocked while cells are proliferating. This triple transcriptional action of c-Myc may contribute not only to its role in tissue homeostasis, but also to its function as an oncogene when misregulated.
Figure 1. In proliferating cells, Myc is expressed and Pak2 is inactive. Myc/Max heterodimer induces E-box-containing genes and represses both Miz-1 and RARα targets. This triple action ensures that differentiation is blocked while cells are proliferating. During RA-induced differentiation, Myc is downregulated and, at least in part, phosphorylated by active Pak2 kinase (P-Pak2). Phospho-Myc (P-Myc) cannot bind to Max, activate E-box genes or repress Miz1 targets. However, P-Myc retains RARα binding and further activates RA-target genes by recruitment of co-activators, thus favoring cell differentiation.
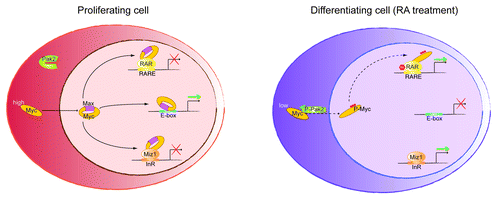
Table 1. c-Myc transcriptional regulation in proliferation and differentiation
c-Myc Phosphorylation by Pak2 in Granulocytic Differentiation
c-Myc transcript levels are downregulated during differentiation of several cell types. However, we and others35,36 have observed that c-Myc protein is still detectable in the nucleus of differentiating granulocytes upon RA administration. In addition, we showed that low levels of unphosphorylated c-Myc are enough to inhibit RA-induced differentiation.35 However, our data suggests that at least a part of the remaining nuclear c-Myc in RA-stimulated cells, in which it is phosphorylated by Pak2, enhanced RA-induced granulocytic differentiation.35 Thus, this c-Myc phosphor-switch is required for full granulocyte terminal differentiation of cells following stimulation with RA. These data fit with previous observations that showed that the Pak2 kinase is activated in late RA-induced differentiation of HL60 cells in a Rac GTPase-dependent manner.37
It is not still clear what lies upstream of Rac1-Pak2 during RA-induced differentiation. One possibility is that Pak2 is activated through Rac1 as a consequence of the cytoskeletal and morphological changes that the cell undergoes during differentiation. These changes during differentiation might activate Rac1 and Pak2, which could, in turn, phosphorylate c-Myc to complete the differentiation program. Interestingly, changes in cytoskeleton and actomyosin tension are known to regulate transcription through transcription factors essential for tissue homeostasis and tumorigenesis, including the serum response factor (SRF) and β-catenin.38-43 In fact, some of the actinomyosin-based pathways activate kinases, such as p38, which also lie downstream of Rho GTPases.42 The Rac1-Pak2 pathway has also been reported to be activated upon cellular stresses.44-46 Other examples are found in the literature, which demonstrates that kinases, such as p38, that modulate the activity of transcription factors, are activated in circumstances of cellular stress.47 Future studies will be necessary to identify the signals that activate the Pak2-phospho-c-Myc cascade that cooperates with RA to induce full differentiation of leukemia granulocytes.
Where, then, does the interaction between Pak2 and c-Myc occur within the cell, since Pak2 is cytoplasmic and c-Myc, nuclear? It has been proposed that they interact within the endoplasmatic reticulum, where active Pak2 localizes in the vicinity of nascent c-Myc protein.24 This low-abundant, phosphorylated form of c-Myc (estimated to be less than 5% of total c-Myc) could then translocate into the nucleus, where it could replace unphosphorylated c-Myc at RARα target genes. Another possibility may involve direct localization of Pak2 at the chromatin. So far, several kinases have been shown to interact directly with chromatin to catalyze posttranslational modifications within the context of the promoter under regulation, including the Jak2 kinase48 and MAPK.49-56 Intriguingly, another member of the Pak family of kinases, Pak1, directly interacts with chromatin, where it phosphorylates histone H3 at serine 10 to induce active transcription.57 Further experimental data will be needed to verify whether Pak2 locates to chromatin, where it could interact with the c-Myc/RARα complex to modulate the transcription of RAR target genes between the proliferative and differentiated states.
c-Myc Phosphorylation by Pak2 in Other Cell Types
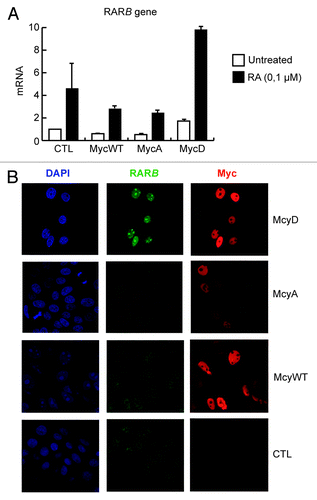
c-Myc is a ubiquitous transcription factor that exerts its function in many different tissue types. Misregulation of c-Myc is one of the most common molecular changes observed in human tumors. Conversely, RA signaling is not only important for hematopoietic differentiation, but also plays an essential role in the homeostasis of several tissue types. So far, phosphorylation of c-Myc by Pak2 has been shown to occur in fibroblasts, primary keratinocytes and leukemia cells.16,24,35,58 It is worth noting that c-Myc’s activities in transcriptional regulation and its biological consequences are largely context-dependent.16,59,60 In this regard, whereas our results indicate that phosphorylation of c-Myc by Pak2 potently stimulates granulocyte terminal differentiation of leukemia cells, previous results have shown that it can, in fact, prevent differentiation in other cellular contexts. For instance, unphosphorylated c-Myc is a potent inducer of epidermal stem cell proliferation in both murine and human skin.14‑16,61,62 However, c-Myc favors the onset of terminal differentiation by stimulating epidermal stem cell proliferation, detachment of stem cells from their niche and a permissive chromatin state required for the expression of epidermal differentiation genes.16,63 In the context of the epidermis, Pak2 phosphorylation of c-Myc prevents these biological activities of c-Myc and conversely impedes differentiation by promoting epidermal stem cell quiescence and strong adherence to the stem cell niche.58 It should be noted that this molecular cascade is stimulated by integrin α6 and Rac1 activities, thus providing epidermal stem cells with a signaling axis that connects the extracellular matrix with gene expression. Interestingly, preliminary results from our laboratory show that in primary human keratinocytes, c-Myc also regulates the bona fide RARE-containing gene RARβ at both the transcript and the protein level, dependent on its Pak2 phosphorylation (). Although the biological significance of the physical and functional interaction between c-Myc and RARα in the epidermis still needs to be studied in depth, it should be noted that RA plays important roles in maintaining epidermal growth and differentiation, which suggests that c-Myc may be important in mediating some of its effects in the skin. For instance, Watt and colleagues64 have recently characterized a novel population of predominantly quiescent epidermal stem cells that are located at the junctional zone between the hair follicles and the epidermis, and contribute to sebaceous glands and interfollicular epidermis homeostasis. Interestingly, Lrig1-positive epidermal stem cells express high levels of c-Myc and are highly sensitive to RA stimulation, which drives them to proliferate and to actively contribute to both skin compartments.64 Whether phosphorylation of c-Myc is required for the response of Lrig1 stem cells to RA remains to be determined. In addition, it will be of great interest to study whether the integrin α6/Rac1 and RA/RAR cascades, which both result in Pak2 phosphorylation of c-Myc, converge in the same population of epidermal stem cells, or whether they have specific and unique functions in different compartments of the epidermis (i.e., hair follicle stem cells, junctional stem cells or interfollicular epidermal stem cells).
Figure 2. Analysis of endogenous RARB mRNA (A) or RARB and Myc protein (B) levels in primary human keratinocytes (PHKA) stably expressing the following ER-tagged Myc constructs or empty vector (CTL): Myc wild-type (MycWT), Myc T358A/S373A/T400A (MycA) and Myc T358D/S373D/T400D (MycD). Cells were treated with 4-hydroxytamoxifen (4-OHT) for 20 h (200 nM), to induce the expression of ER-tagged Myc mutants. The quantified results in (A) are presented as means ± s.d. of triplicate samples and representative images are presented in (B).
c-Myc is also an essential factor in normal hematopoiesis, and its activity is tightly connected to its relative levels. Thus, low levels of c-Myc stimulate hematopoietic stem cells’ (HSC) self-renewal and adhesion to the niche, whereas high levels of c-Myc stimulate HSC expansion and the formation of the transit-amplifying (TA) compartment between TA cells (“Myc high”), with subsequent very low levels of c-Myc being required for cell maturation (“Myc off”). In fact, the Myc-high scenario in HSCs is similar to that in epidermal stem cells regarding detachment from their niche, given that c-Myc disrupts the adhesive interactions between hematopoietic stem cells and their niche by repressing N-cadherin and integrins.65,66 Interestingly, Rac1 activity is necessary for HSCs to be engrafted to the bone marrow niche when transplanted in lethally irradiated mice and, together with Rac2, for the homing of HSCs to their niche during normal homeostasis.67 Our results in leukemia cells showed that the expression of ICAM1 and other integrins was repressed by unphosphorylated c-Myc but activated by phosphor-c-Myc in promyelocytic-arrested (HL60 and NB4) cells. Although these data should be validated in HSCs, the results suggest that the Myc-low and Myc-high states could correspond to the phosphor-c-Myc and unphosphorylated c-Myc states, respectively. Thus, phosphor-c-Myc could play an important role in HSCs’ function by promoting their quiescence and adherence to their niche, whereas unphosphorylated c-Myc might stimulate HSC niche detachment and proliferation. Interestingly, current evidence suggests that RA is able to stimulate the activation of HSCs, although no functional link with c-Myc was investigated in these studies.68,69
In addition to the role c-Myc plays in hematopoietic and epidermal differentiation, a cleavage product of c-Myc that localizes to the cell cytoplasm, termed Myc-nick, has been recently shown to have a role in muscle differentiation.70 Myc-nick is generated by calpain-dependent proteolysis and retains the conserved c-Myc-box regions but lacks nuclear localization signals and the bHLH-Zip domain. It was proposed that Myc-nick diminishes the nuclear abundance of c-Myc and thus weakens its transcriptional block of differentiation and drives the cytoskeletal reorganization that is ultimately required for terminal muscle differentiation.70 In this context, it would be interesting to study whether the residual nuclear c-Myc remaining upon muscle cell differentiation is phosphorylated by Pak2, and whether phosphor-c-Myc is required for the expression of certain genes involved in muscle differentiation. Given that RA has been proposed to stimulate muscle differentiation,71 determining whether Myc-nick and phosphor-c-Myc cooperate in this context may help us to better understand the progressive changes in gene expression required for the process of muscle differentiation.
c-Myc Potential Crosstalk with Other Nuclear Receptors
A possibility that cannot be excluded is that c-Myc cooperates with other members of the RAR family (e.g., RARβ and RARγ) or even with other nuclear receptors. One hypothesis is that c-Myc cooperates with the predominant RAR isoform in each cell type to keep RARE-containing genes repressed. Thus, it will be of interest to study the crosstalk of c-Myc-RAR in cell types in which RARβ or RARγ play dominant roles as terminal effectors of the RA signaling pathway. To this end, mapping the binding domains of RARα to c-Myc would unveil how the interaction is achieved and, consequently, would help to anticipate whether c-Myc could potentially bind to other retinoid receptors.
Interestingly, our motif search study based on genome-wide expression analyses of c-Myc and phosphor-c-Myc in HL60 cells35 revealed that a significant subset of genes upregulated by phosphorylated c-Myc contain general retinoid responsive elements that are recognized by several members of the nuclear receptor family. Within these elements, RAR binding motifs were found together with binding motifs for RXR or PPAR. Although we focused on RARα due to its widely reported importance in granulocytic differentiation and leukemia, this finding points toward an even broader crosstalk of c-Myc with nuclear receptors. In addition, the cohort of transcripts downregulated by phosphor-c-Myc in HL60 includes DNA binding motifs specific for a significant set of transcription factors involved in the differentiation of many cell types other than granulocytes (i.e., PU.1, VDR, MyoD, MEF2 or Blimp-1).35 Further studies are necessary to elucidate whether c-Myc and its PAK2 phosphorylated form are able to regulate different sets of genes actively involved in cell differentiation by interacting with different nuclear receptors.
Unphosphorylated and Phosphorylated c-Myc in Cancer
Given the wide number of cancers in which deregulated c-Myc contributes to their neoplastic phenotype, it would be interesting to study the implications of the direct regulation of differentiation genes by c-Myc in these malignancies. We have observed inhibition of RARE-containing genes downstream of c-Myc in different transformed cell types (AML and APL leukemic cells and human embryonic kidney 293T cells),35 indicating that the interplay between c-Myc and RARα gene regulation is not limited to leukemic cells. The consequences of the repressive role of c-Myc over RARE-containing genes in conditions in which c-Myc is aberrantly active might be different depending on the role that the RAR target genes play in each cell type.
In leukemic cells, the RA signaling pathway is instrumental for triggering granulocytic differentiation, making it an especially interesting system in which to study c-Myc repression of RARE-containing genes. While in many types of cancer, eliminating (by killing) malignant cells is the only feasible antitumor strategy, stimulating the differentiation of leukemic cells into mature blood cells is an equally effective anti-leukemia therapy.72 The best proof of principle for differentiation therapy has been the treatment of APL patients with RA.73 However, RA differentiation therapy is currently only effective in a subgroup of patients with acute myeloid leukemia (AML) caused by the formation of the oncofusion protein PML-RARα (acute promyelocytic leukemia, APL).74-76 In our hands, mimicking phosphor-c-Myc also potentiated RA-induced differentiation in AML cell models. Therefore, phosphor-c-Myc inducers could be used to synergize with RA therapy on other leukemic types that do not respond to RA alone.
Recent studies from several laboratories have successfully used selective small-molecule inhibitors of BET bromodomains to treat hematological malignancies, such as MLL leukemia, multiple myeloma and Burkitt lymphoma, through c-Myc inhibition.77-80 Remarkably, and in accordance with our data, interference of c-Myc in HL60 and NB4 cells did not lead to cell differentiation, but rather to apoptosis and a proliferation decrease. In fact, our results show that even when RA was administered, APL cells could not fully express RARE-dependent terminal differentiation genes and, consequently, could not properly differentiate following interference of the expression of endogenous c-Myc. Accordingly, transient inhibition of c-Myc or its binding to Max resulted in derepression of RARE-containing promoters. On the other hand, Pak2-dependent c-Myc phosphorylation is required to activate RARE-containing promoters.35 In the future, it should be investigated whether triggering phosphor-c-Myc in other types of hematological malignancies might represent a therapeutic advantage over inhibiting Myc activity, as was shown to be the case for APL. Interestingly, frequent chromosomal rearrangements have been found at the 3q29 region that encodes for the PAK2 gene in hematological malignancies, such as chronic myeloid leukemia and B-cell lymphoma, and in cutaneous neuroendocrine tumors.81-84 These rearrangements render the kinase inactive and would, therefore, impede c-Myc phosphorylation by Pak2. Thus, loss of phosphorylation of c-Myc could be a yet-unexplored mechanism used by tumors to suppress differentiation and potentiate aggressiveness.
Stimulating c-Myc phosphorylation might be evaluated as an alternative or synergystic antitumor strategy to using c-Myc/Max dimerization mutants or small molecules that disrupt c-Myc/Max heterodimers. Inhibitors of the c-Myc/Max dimer, although very promising, may only affect one limb of c-Myc activity (that is, that required for proliferation) but without affecting the other one, required for its phosphorylation-dependent regulation of cell differentiation. Mutants of c-Myc unable to bind to Max have been developed, such as the Omomyc constructs85,86 or MycEG,87,88 which exert potent antitumor activity in mouse models of carcinogenesis. Strikingly, conditional expression of Omomyc in vivo triggered a rapid regression of incipient and established lung tumors but produced relatively mild side effects in normal tissues.86 We have also reported that phosphor-c-Myc cooperates with RA therapy to enhance differentiation and apoptosis of human xenotransplanted leukemic cells, resulting in reduced tumor dissemination and invasion. Developing phosphor-c-Myc inducers could be advantageous, mainly because they would mimic a physiological signaling pathway, thus avoiding the need for treating patients with molecules that may have additional and unpredictable side effects. On this basis, it would be now relevant to stimulate c-Myc phosphorylation by Pak2 in the available c-Myc models of cancer to evaluate its therapeutic potential.
Our results suggest that the previously reported synergistic effect of AraC with RA in stimulating leukemia differentiation is at least partially dependent on Pak2-mediated phosphorylation of c-Myc. Since AraC enhances RA-induced gene transcription, determining its effects over the transcriptome regulated by c-Myc would be interesting to further our understanding of the synergistic role AraC plays in enhancing RA-induced differentiation through c-Myc. AraC, by activating Pak2, might also decrease E-box activation and Miz-1 gene repression. In addition, the cytotoxic role of AraC in DNA synthesis inhibition and the subsequent massive induction of apoptosis should be further studied to evaluate whether this role increases its side effects or contributes to its action in the differentiation combinatory therapy.
Current evidence shows that fusion proteins found in AML induce a pre-leukemic state, which requires further genetic and epigenetic mutations to progress to leukemia.89,90 In all cases, there appears to be functional heterogeneity within the AML cell population carrying the mutations responsible for the pathogenesis. Specifically, it has been suggested that there is a subpopulation of cells with self-renewal capacity that are capable of propagating and maintaining the AML phenotype; these have, therefore, been termed leukemic stem cells (LSCs).91,92 The existence of cancer stem cells (CSCs) has also been suggested based on other types of cancer, apart from leukemia, and their origin is now being extensively investigated. Successfully developing cancer therapies that can eradicate CSCs is one of the main aims in the fight against cancer. Given the therapeutic potential of phosphor-c-Myc in cancer and c-Myc’s implications in stem cell biology, it would be interesting to measure the impact of phosphor-c-Myc expression in the population of LSCs or, even more generally, CSCs in vivo.
Acknowledgments
We are indebted to V.A. Raker for help in preparing the manuscript. This work was supported by grants from the Spanish “Ministerio de Educación y Ciencia” (BFU2010–18692), from AGAUR, and from “Fundació La Marató” to L.D.C.
References
- Patel JH, Loboda AP, Showe MK, Showe LC, McMahon SB. Analysis of genomic targets reveals complex functions of MYC. Nat Rev Cancer 2004; 4:562 - 8; http://dx.doi.org/10.1038/nrc1393; PMID: 15229481
- Blackwell TK, Kretzner L, Blackwood EM, Eisenman RN, Weintraub H. Sequence-specific DNA binding by the c-Myc protein. Science 1990; 250:1149 - 51; http://dx.doi.org/10.1126/science.2251503; PMID: 2251503
- Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991; 251:1211 - 7; http://dx.doi.org/10.1126/science.2006410; PMID: 2006410
- Amati B, Alevizopoulos K, Vlach J. Myc and the cell cycle. Front Biosci 1998; 3:d250 - 68; PMID: 9468463
- Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 1999; 19:1 - 11; PMID: 9858526
- Eilers M. Control of cell proliferation by Myc family genes. Mol Cells 1999; 9:1 - 6; PMID: 10102563
- Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massagué J. TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol 2001; 3:400 - 8; http://dx.doi.org/10.1038/35070086; PMID: 11283614
- Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol 2001; 3:392 - 9; http://dx.doi.org/10.1038/35070076; PMID: 11283613
- Inghirami G, Grignani F, Sternas L, Lombardi L, Knowles DM, Dalla-Favera R. Down-regulation of LFA-1 adhesion receptors by C-myc oncogene in human B lymphoblastoid cells. Science 1990; 250:682 - 6; http://dx.doi.org/10.1126/science.2237417; PMID: 2237417
- Seoane J, Le HV, Massagué J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 2002; 419:729 - 34; http://dx.doi.org/10.1038/nature01119; PMID: 12384701
- Herold S, Wanzel M, Beuger V, Frohme C, Beul D, Hillukkala T, et al. Negative regulation of the mammalian UV response by Myc through association with Miz-1. Mol Cell 2002; 10:509 - 21; http://dx.doi.org/10.1016/S1097-2765(02)00633-0; PMID: 12408820
- Yang W, Shen J, Wu M, Arsura M, FitzGerald M, Suldan Z, et al. Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene 2001; 20:1688 - 702; http://dx.doi.org/10.1038/sj.onc.1204245; PMID: 11313917
- Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev 2002; 16:2699 - 712; http://dx.doi.org/10.1101/gad.1021202; PMID: 12381668
- Arnold I, Watt FM. c-Myc activation in transgenic mouse epidermis results in mobilization of stem cells and differentiation of their progeny. Curr Biol 2001; 11:558 - 68; http://dx.doi.org/10.1016/S0960-9822(01)00154-3; PMID: 11369200
- Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR. Deregulated expression of c-Myc depletes epidermal stem cells. Nat Genet 2001; 28:165 - 8; http://dx.doi.org/10.1038/88889; PMID: 11381265
- Watt FM, Frye M, Benitah SA. MYC in mammalian epidermis: how can an oncogene stimulate differentiation?. Nat Rev Cancer 2008; 8:234 - 42; http://dx.doi.org/10.1038/nrc2328; PMID: 18292777
- Gallant P, Steiger D. Myc’s secret life without Max. Cell Cycle 2009; 8:3848 - 53; http://dx.doi.org/10.4161/cc.8.23.10088; PMID: 19887915
- Steiger D, Furrer M, Schwinkendorf D, Gallant P. Max-independent functions of Myc in Drosophila melanogaster. Nat Genet 2008; 40:1084 - 91; http://dx.doi.org/10.1038/ng.178; PMID: 19165923
- Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol 2005; 6:635 - 45; http://dx.doi.org/10.1038/nrm1703; PMID: 16064138
- Roy AL, Carruthers C, Gutjahr T, Roeder RG. Direct role for Myc in transcription initiation mediated by interactions with TFII-I. Nature 1993; 365:359 - 61; http://dx.doi.org/10.1038/365359a0; PMID: 8377829
- Shrivastava A, Saleque S, Kalpana GV, Artandi S, Goff SP, Calame K. Inhibition of transcriptional regulator Yin-Yang-1 by association with c-Myc. Science 1993; 262:1889 - 92; http://dx.doi.org/10.1126/science.8266081; PMID: 8266081
- Gartel AL, Ye X, Goufman E, Shianov P, Hay N, Najmabadi F, et al. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci U S A 2001; 98:4510 - 5; http://dx.doi.org/10.1073/pnas.081074898; PMID: 11274368
- Izumi H, Molander C, Penn LZ, Ishisaki A, Kohno K, Funa K. Mechanism for the transcriptional repression by c-Myc on PDGF beta-receptor. J Cell Sci 2001; 114:1533 - 44; PMID: 11282029
- Huang Z, Traugh JA, Bishop JM. Negative control of the Myc protein by the stress-responsive kinase Pak2. Mol Cell Biol 2004; 24:1582 - 94; http://dx.doi.org/10.1128/MCB.24.4.1582-1594.2004; PMID: 14749374
- Hörlein AJ, Näär AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 1995; 377:397 - 404; http://dx.doi.org/10.1038/377397a0; PMID: 7566114
- Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 1995; 377:454 - 7; http://dx.doi.org/10.1038/377454a0; PMID: 7566127
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, et al. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell 1996; 85:403 - 14; http://dx.doi.org/10.1016/S0092-8674(00)81118-6; PMID: 8616895
- Chakravarti D, LaMorte VJ, Nelson MC, Nakajima T, Schulman IG, Juguilon H, et al. Role of CBP/P300 in nuclear receptor signalling. Nature 1996; 383:99 - 103; http://dx.doi.org/10.1038/383099a0; PMID: 8779723
- Heinzel T, Lavinsky RM, Mullen TM, Söderstrom M, Laherty CD, Torchia J, et al. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature 1997; 387:43 - 8; http://dx.doi.org/10.1038/387043a0; PMID: 9139820
- Kurland JF, Tansey WP. Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res 2008; 68:3624 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-07-6552; PMID: 18483244
- Vervoorts J, Lüscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, et al. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep 2003; 4:484 - 90; http://dx.doi.org/10.1038/sj.embor.embor821; PMID: 12776737
- Gratas C, Menot ML, Dresch C, Chomienne C. Retinoid acid supports granulocytic but not erythroid differentiation of myeloid progenitors in normal bone marrow cells. Leukemia 1993; 7:1156 - 62; PMID: 8350615
- Larsson LG, Henriksson MA. The Yin and Yang functions of the Myc oncoprotein in cancer development and as targets for therapy. Exp Cell Res 2010; 316:1429 - 37; http://dx.doi.org/10.1016/j.yexcr.2010.03.025; PMID: 20382143
- Leon J, Ferrandiz N, Acosta JC, Delgado MD. Inhibition of cell differentiation: a critical mechanism for MYC-mediated carcinogenesis?. Cell Cycle 2009; 8:1148 - 57; http://dx.doi.org/10.4161/cc.8.8.8126; PMID: 19282668
- Uribesalgo I, Buschbeck M, Gutiérrez A, Teichmann S, Demajo S, Kuebler B, et al. E-box-independent regulation of transcription and differentiation by MYC. Nat Cell Biol 2011; 13:1443 - 9; http://dx.doi.org/10.1038/ncb2355; PMID: 22020439
- Yung BY. c-Myc-mediated expression of nucleophosmin/B23 decreases during retinoic acid-induced differentiation of human leukemia HL-60 cells. FEBS Lett 2004; 578:211 - 6; http://dx.doi.org/10.1016/j.febslet.2004.08.089; PMID: 15589822
- Nisimoto Y, Ogawa H. Interaction between p21-activated protein kinase and Rac during differentiation of HL-60 human promyelocytic leukemia cell induced by all-trans-retinoic acid. Eur J Biochem 2002; 269:2622 - 9; http://dx.doi.org/10.1046/j.1432-1033.2002.02939.x; PMID: 12027902
- Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and β-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 2011; 19:776 - 91; http://dx.doi.org/10.1016/j.ccr.2011.05.008; PMID: 21665151
- Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003; 113:329 - 42; http://dx.doi.org/10.1016/S0092-8674(03)00278-2; PMID: 12732141
- Vartiainen MK, Guettler S, Larijani B, Treisman R. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 2007; 316:1749 - 52; http://dx.doi.org/10.1126/science.1141084; PMID: 17588931
- Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol 2009; 11:257 - 68; http://dx.doi.org/10.1038/ncb1833; PMID: 19198601
- Connelly JT, Gautrot JE, Trappmann B, Tan DW, Donati G, Huck WT, et al. Actin and serum response factor transduce physical cues from the microenvironment to regulate epidermal stem cell fate decisions. Nat Cell Biol 2010; 12:711 - 8; http://dx.doi.org/10.1038/ncb2074; PMID: 20581838
- Connelly JT, Mishra A, Gautrot JE, Watt FM. Shape-induced terminal differentiation of human epidermal stem cells requires p38 and is regulated by histone acetylation. PLoS One 2011; 6:e27259; http://dx.doi.org/10.1371/journal.pone.0027259; PMID: 22073300
- Roig J, Traugh JA. p21-activated protein kinase gamma-PAK is activated by ionizing radiation and other DNA-damaging agents. Similarities and differences to alpha-PAK. J Biol Chem 1999; 274:31119 - 22; http://dx.doi.org/10.1074/jbc.274.44.31119; PMID: 10531298
- Roig J, Huang Z, Lytle C, Traugh JA. p21-activated protein kinase gamma-PAK is translocated and activated in response to hyperosmolarity. Implication of Cdc42 and phosphoinositide 3-kinase in a two-step mechanism for gamma-PAK activation. J Biol Chem 2000; 275:16933 - 40; http://dx.doi.org/10.1074/jbc.M001627200; PMID: 10748040
- Roig J, Traugh JA. Cytostatic p21 G protein-activated protein kinase gamma-PAK. Vitam Horm 2001; 62:167 - 98; http://dx.doi.org/10.1016/S0083-6729(01)62004-1; PMID: 11345898
- Nebreda AR, Porras A. p38 MAP kinases: beyond the stress response. Trends Biochem Sci 2000; 25:257 - 60; http://dx.doi.org/10.1016/S0968-0004(00)01595-4; PMID: 10838561
- Dawson MA, Bannister AJ, Göttgens B, Foster SD, Bartke T, Green AR, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 2009; 461:819 - 22; http://dx.doi.org/10.1038/nature08448; PMID: 19783980
- Li J, Gorospe M, Hutter D, Barnes J, Keyse SM, Liu Y. Transcriptional induction of MKP-1 in response to stress is associated with histone H3 phosphorylation-acetylation. Mol Cell Biol 2001; 21:8213 - 24; http://dx.doi.org/10.1128/MCB.21.23.8213-8224.2001; PMID: 11689710
- Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, et al. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J 2003; 22:2788 - 97; http://dx.doi.org/10.1093/emboj/cdg273; PMID: 12773393
- Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, Puri PL. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat Genet 2004; 36:738 - 43; http://dx.doi.org/10.1038/ng1378; PMID: 15208625
- Illi B, Scopece A, Nanni S, Farsetti A, Morgante L, Biglioli P, et al. Epigenetic histone modification and cardiovascular lineage programming in mouse embryonic stem cells exposed to laminar shear stress. Circ Res 2005; 96:501 - 8; http://dx.doi.org/10.1161/01.RES.0000159181.06379.63; PMID: 15705964
- Schmeck B, Beermann W, van Laak V, Zahlten J, Opitz B, Witzenrath M, et al. Intracellular bacteria differentially regulated endothelial cytokine release by MAPK-dependent histone modification. J Immunol 2005; 175:2843 - 50; PMID: 16116170
- Pokholok DK, Zeitlinger J, Hannett NM, Reynolds DB, Young RA. Activated signal transduction kinases frequently occupy target genes. Science 2006; 313:533 - 6; http://dx.doi.org/10.1126/science.1127677; PMID: 16873666
- Vicent GP, Ballaré C, Nacht AS, Clausell J, Subtil-Rodríguez A, Quiles I, et al. Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol Cell 2006; 24:367 - 81; http://dx.doi.org/10.1016/j.molcel.2006.10.011; PMID: 17081988
- Lee ER, McCool KW, Murdoch FE, Fritsch MK. Dynamic changes in histone H3 phosphoacetylation during early embryonic stem cell differentiation are directly mediated by mitogen- and stress-activated protein kinase 1 via activation of MAPK pathways. J Biol Chem 2006; 281:21162 - 72; http://dx.doi.org/10.1074/jbc.M602734200; PMID: 16728397
- Li F, Adam L, Vadlamudi RK, Zhou H, Sen S, Chernoff J, et al. p21-activated kinase 1 interacts with and phosphorylates histone H3 in breast cancer cells. EMBO Rep 2002; 3:767 - 73; http://dx.doi.org/10.1093/embo-reports/kvf157; PMID: 12151336
- Benitah SA, Frye M, Glogauer M, Watt FM. Stem cell depletion through epidermal deletion of Rac1. Science 2005; 309:933 - 5; http://dx.doi.org/10.1126/science.1113579; PMID: 16081735
- Smith AP, Verrecchia A, Fagà G, Doni M, Perna D, Martinato F, et al. A positive role for Myc in TGFbeta-induced Snail transcription and epithelial-to-mesenchymal transition. Oncogene 2009; 28:422 - 30; http://dx.doi.org/10.1038/onc.2008.395; PMID: 18978814
- Perna D, Fagà G, Verrecchia A, Gorski MM, Barozzi I, Narang V, et al. Genome-wide mapping of Myc binding and gene regulation in serum-stimulated fibroblasts. Oncogene 2011; http://dx.doi.org/10.1038/onc.2011.359; PMID: 21860422
- Gebhardt A, Frye M, Herold S, Benitah SA, Braun K, Samans B, et al. Myc regulates keratinocyte adhesion and differentiation via complex formation with Miz1. J Cell Biol 2006; 172:139 - 49; http://dx.doi.org/10.1083/jcb.200506057; PMID: 16391002
- Frye M, Watt FM. The RNA methyltransferase Misu (NSun2) mediates Myc-induced proliferation and is upregulated in tumors. Curr Biol 2006; 16:971 - 81; http://dx.doi.org/10.1016/j.cub.2006.04.027; PMID: 16713953
- Nascimento EM, Cox CL, MacArthur S, Hussain S, Trotter M, Blanco S, et al. The opposing transcriptional functions of Sin3a and c-Myc are required to maintain tissue homeostasis. Nat Cell Biol 2011; 13:1395 - 405; http://dx.doi.org/10.1038/ncb2385; PMID: 22101514
- Jensen KB, Collins CA, Nascimento E, Tan DW, Frye M, Itami S, et al. Lrig1 expression defines a distinct multipotent stem cell population in mammalian epidermis. Cell Stem Cell 2009; 4:427 - 39; http://dx.doi.org/10.1016/j.stem.2009.04.014; PMID: 19427292
- Murphy MJ, Wilson A, Trumpp A. More than just proliferation: Myc function in stem cells. Trends Cell Biol 2005; 15:128 - 37; http://dx.doi.org/10.1016/j.tcb.2005.01.008; PMID: 15752976
- Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev 2004; 18:2747 - 63; http://dx.doi.org/10.1101/gad.313104; PMID: 15545632
- Williams DA, Zheng Y, Cancelas JA. Rho GTPases and regulation of hematopoietic stem cell localization. Methods Enzymol 2008; 439:365 - 93; http://dx.doi.org/10.1016/S0076-6879(07)00427-2; PMID: 18374178
- Purton LE, Bernstein ID, Collins SJ. All-trans retinoic acid delays the differentiation of primitive hematopoietic precursors (lin-c-kit+Sca-1(+)) while enhancing the terminal maturation of committed granulocyte/monocyte progenitors. Blood 1999; 94:483 - 95; PMID: 10397716
- Purton LE, Bernstein ID, Collins SJ. All-trans retinoic acid enhances the long-term repopulating activity of cultured hematopoietic stem cells. Blood 2000; 95:470 - 7; PMID: 10627451
- Conacci-Sorrell M, Ngouenet C, Eisenman RN. Myc-nick: a cytoplasmic cleavage product of Myc that promotes alpha-tubulin acetylation and cell differentiation. Cell 2010; 142:480 - 93; http://dx.doi.org/10.1016/j.cell.2010.06.037; PMID: 20691906
- Zhu GH, Huang J, Bi Y, Su Y, Tang Y, He BC, et al. Activation of RXR and RAR signaling promotes myogenic differentiation of myoblastic C2C12 cells. Differentiation 2009; 78:195 - 204; http://dx.doi.org/10.1016/j.diff.2009.06.001; PMID: 19560855
- Nowak D, Stewart D, Koeffler HP. Differentiation therapy of leukemia: 3 decades of development. Blood 2009; 113:3655 - 65; http://dx.doi.org/10.1182/blood-2009-01-198911; PMID: 19221035
- Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988; 72:567 - 72; PMID: 3165295
- Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 2002; 295:1079 - 82; http://dx.doi.org/10.1126/science.1065173; PMID: 11834837
- Di Croce L. Chromatin modifying activity of leukaemia associated fusion proteins. Hum Mol Genet 2005; 14:Spec No 1 R77 - 84; http://dx.doi.org/10.1093/hmg/ddi109; PMID: 15809276
- Uribesalgo I, Di Croce L. Dynamics of epigenetic modifications in leukemia. Brief Funct Genomics 2011; 10:18 - 29; http://dx.doi.org/10.1093/bfgp/elr002; PMID: 21258047
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011; 478:524 - 8; http://dx.doi.org/10.1038/nature10334; PMID: 21814200
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011; 478:529 - 33; http://dx.doi.org/10.1038/nature10509; PMID: 21964340
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011; 146:904 - 17; http://dx.doi.org/10.1016/j.cell.2011.08.017; PMID: 21889194
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A 2011; 108:16669 - 74; http://dx.doi.org/10.1073/pnas.1108190108; PMID: 21949397
- Slavutsky I, de Vinuesa ML, Larripa I, Dupont J, de Salum SB. Translocation (2;3) in hematologic malignancies. Cancer Genet Cytogenet 1986; 21:335 - 42; http://dx.doi.org/10.1016/0165-4608(86)90214-1; PMID: 3456824
- Dierlamm J, Rosenberg C, Stul M, Pittaluga S, Wlodarska I, Michaux L, et al. Characteristic pattern of chromosomal gains and losses in marginal zone B cell lymphoma detected by comparative genomic hybridization. Leukemia 1997; 11:747 - 58; http://dx.doi.org/10.1038/sj.leu.2400635; PMID: 9180302
- Lafage-Pochitaloff M, Courcoul M, Simonetti J, Sainty D, Dastugue N, Tabilio A, et al. Expression of the ETS2 and transferrin receptor genes in Philadelphia-positive chronic myeloid leukemia patients with a reciprocal t(3;21). Genes Chromosomes Cancer 1992; 5:1 - 13; http://dx.doi.org/10.1002/gcc.2870050102; PMID: 1384656
- Perlman EJ, Lumadue JA, Hawkins AL, Cohen K, Colombani P, Griffin CA. Primary cutaneous neuroendocrine tumors. Diagnostic use of cytogenetic and MIC2 analysis. Cancer Genet Cytogenet 1995; 82:30 - 4; http://dx.doi.org/10.1016/0165-4608(94)00271-C; PMID: 7627931
- Soucek L, Helmer-Citterich M, Sacco A, Jucker R, Cesareni G, Nasi S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 1998; 17:2463 - 72; http://dx.doi.org/10.1038/sj.onc.1202199; PMID: 9824157
- Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, et al. Modelling Myc inhibition as a cancer therapy. Nature 2008; 455:679 - 83; http://dx.doi.org/10.1038/nature07260; PMID: 18716624
- Amati B, Brooks MW, Levy N, Littlewood TD, Evan GI, Land H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 1993; 72:233 - 45; http://dx.doi.org/10.1016/0092-8674(93)90663-B; PMID: 8425220
- Amati B, Littlewood TD, Evan GI, Land H. The c-Myc protein induces cell cycle progression and apoptosis through dimerization with Max. EMBO J 1993; 12:5083 - 7; PMID: 8262051
- Yuan Y, Zhou L, Miyamoto T, Iwasaki H, Harakawa N, Hetherington CJ, et al. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc Natl Acad Sci U S A 2001; 98:10398 - 403; http://dx.doi.org/10.1073/pnas.171321298; PMID: 11526243
- Villa R, Pasini D, Gutierrez A, Morey L, Occhionorelli M, Viré E, et al. Role of the polycomb repressive complex 2 in acute promyelocytic leukemia. Cancer Cell 2007; 11:513 - 25; http://dx.doi.org/10.1016/j.ccr.2007.04.009; PMID: 17560333
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994; 367:645 - 8; http://dx.doi.org/10.1038/367645a0; PMID: 7509044
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3:730 - 7; http://dx.doi.org/10.1038/nm0797-730; PMID: 9212098