Abstract
The ubiquitin-proteasome system (UPS) is usurped by many if not all cancers to regulate their survival, proliferation, invasion, angiogenesis and metastasis. Bioflavonoids curcumin and chalcones exhibit anti-neoplastic selectivity through inhibition of the 26S proteasome-activity within the UPS. Here, we provide evidence for a novel mechanism of action of chalcone-based derivatives AM146, RA-9 and RA-14, which exert anticancer activity by targeting deubiquitinating enzymes (DUB) without affecting 20S proteasome catalytic-core activity. The presence of the α,β-unsaturated carbonyl group susceptible to nucleophilic attack from the sulfhydryl of cysteines in the active sites of DUB determines the capacity of novel small-molecules to act as cell-permeable, partly selective DUB inhibitors and induce rapid accumulation of polyubiquitinated proteins and deplete the pool of free ubiquitin. These chalcone derivatives directly suppress activity of DUB UCH-L1, UCH-L3, USP2, USP5 and USP8, which are known to regulate the turnover and stability of key regulators of cell survival and proliferation. Inhibition of DUB-activity mediated by these compounds downregulates cell cycle promoters, e.g., cyclin D1 and upregulates tumor suppressors p53, p27Kip1 and p16Ink4A. These changes are associated with arrest in S-G2/M, abrogated anchorage-dependent growth and onset of apoptosis in breast, ovarian and cervical cancer cells without noticeable alterations in primary human cells. Altogether, this work provides evidence of antitumor activity of novel chalcone-based derivatives mediated by their DUB-targeting capacity; supports the development of pharmaceuticals to directly target DUB as a most efficient strategy compared with proteasome inhibition and also provides a clear rationale for the clinical evaluation of these novel small-molecule DUB inhibitors.
Note: Please see the Erratum, Expression of Concern, and subsequent Comment published regarding this Report.
Introduction
The usurping of the ubiquitin-proteasome pathway is a central feature of malignancy. Deubiquitinating enzymes (DUB) are crucial in regulating a variety of cellular pathways, including cell growth and proliferation, apoptosis, protein quality control, DNA repair and transcription and thus are the key molecular determinants of the aberrant cancer proteome.Citation1-Citation3 The human genome encodes over 100 putative DUB divided into five subclasses, of which the USP (ubiquitin-specific proteases) and UCH (ubiquitin C-terminal hydrolases) are the best characterized.Citation2 Evolving from our early understanding as enzymes that merely process ubiquitin precursors and scavenge ubiquitin from proteasome targeted substrates, recent studies have revealed that DUB are dynamic enzymes that partner with various interacting proteins to facilitate substrate selection and activity, ubiquitin chain editing and DUB activity.Citation1,Citation3 Additionally, published data suggest that besides participation in ubiquitination/de-ubiquitination, some DUB can regulate gene expression by acting on the regulators of transcription or on chromatin structure.Citation4 Defects associated with DUB have been implicated in a number of human pathologies, including infectious diseases, neuropathological disorders and, most notably, in cancer.Citation5-Citation7 Accordingly, DUB, being key molecular determinants of the aberrant cancer proteome, were proposed as a bona fide molecular target for therapeutic interventions offering low predicted cytotoxicity as compared with proteasome inhibitors. Currently, there are no DUB inhibitors that have been used clinically.Citation8,Citation9
The most recent efforts employing high-throughput screening and fluorescence polarization assays have led to identification of HBX 41108, a USP7-specific inhibitor,Citation10,Citation11 as well as HBX 90397 and HBX 90659,Citation10 small-molecule inhibitors of USP8, and also USP2 and UCH-L3 inhibitors.Citation12 However, specific biological data are either not available or elusive, and data on neoplastic selectivity of most of these compounds are also unavailable. Peptide-based potent, irreversible inhibitors of DUB, such as ubiquitin aldehyde (Ubal) and UbVS, have been previously described in references Citation13 and Citation14. However, their therapeutic potential is limited by their high-molecular weight and limited cell permeability.
First naturally derived small-molecule inhibitors of cellular DUB (cyclopentenone PNGs) identified using ubiquitin-PEST and z-LRGG-AMC as substrates were initially shown to inhibit ubiquitin isopeptidase activity in cells (IC50: 30 µM) and cause cellular accumulation of ubiquitinated proteins and cell death.Citation15 However, any selective inhibition on the various isopeptidases remains un-described. Based on a key molecular determinant conferring DUB inhibitory activity, an α,β-unsaturated ketone with a sterically accessible β-carbon, additional inhibitors have been described, e.g., dibenzylideneacetone (DBA, IC50: 20–40 µM), curcumin (IC50: 80–100 µM) and shikoccin (IC50: 15 µM).Citation16 Molecular analysis of WP1130, a partly selective DUB inhibitor, revealed some structural and chemical similarities to curcumin and DBA,Citation17 and the presence of the α,β-unsaturated carbonyl group determined its capacity to directly inhibit DUB activity of USP9x, USP5, USP14 and UCH37, which are known to regulate survival protein stability and 26S proteasome function.
We have recently described in reference Citation18 the synthesis of the library of the chalcone-based derivatives of the proteasome inhibitor AM114 [3,5-bis(4-boronic acid benzylidene)-1-methylpiperidin-4-one].Citation19 For some of these compounds, e.g., RA-1, we reported their anti-proliferative and apoptotic effects mediated by the inhibition of the 26S proteasome activity.Citation18 For others, like RA-9, RA-14 and AM146, the mechanism of action remained unclear. Here, we hypothesized that the presence of an α,β-unsaturated carbonyl group susceptible to nucleophilic attack from sulfhydryl groups determines the capacity of these small-molecule inhibitors to interact with the active site cysteine of DUB in a manner similar to curcumin, shikoccin, DBACitation16 and cyclopentenone PNGs.Citation15 Based on our findings, we report that chalcone derivatives, AM146, RA-9 and RA-14 act as cell-permeable DUB inhibitors, inducing a rapid and marked accumulation of poly-ubiquitinated proteins, reducing the pool of free ubiquitin monomers and resulting in the abrogated expression of cell cycle regulators, cell cycle checkpoint arrest and tumor cell apoptosis. These activities were distinct when compared with other compounds that affect protein ubiquitination, such as bortezomib, and were not observed with RA-4 incubation, used as a negative control, suggesting the divergent mechanisms and targets for AM146, RA-9 and RA-14.
Results
Growth-inhibitory properties of AM146, RA-9 and RA-14 are coupled to rapid protein ubiquitination but do not require 20S proteasome activity.
In breast, ovarian and cervical cancer cells, malignant transformation is associated with the enhanced cellular stress and pathologic redirection of the ubiquitin-mediated protein degradation. Our initial studies showed that three chalcone derivatives, AM146, RA-9 and RA-14, alter viability of breast, ovarian and cervical cancer cells in a dose-dependent manner (). Based on the analysis of cell viability, breast cancer cells compared with ovarian and cervical cancer cell lines were less susceptible to the anti-proliferative activity of AM146 and RA-14 (). Direct comparison of these compounds with proteasome inhibitor bortezomib (PS341) or structurally similar RA-4Citation18 revealed distinctions in the apoptotic onset and activity of these compounds (). Analysis of colony formation of breast, ovarian and cervical cancer cells treated with AM146, RA-9 or RA-14 revealed the capacity of these small-molecule inhibitors to suppress anchorage-dependent growth in a dose-dependent manner (not shown, IC50 < 100 nM), completely abrogating colony formation at concentration 0.3 µM ().
Figure 1. Chalcone-derivatives suppress cell viability in breast, ovarian and cervical cancers. Cells were treated with indicated doses of chalcone-derivatives for 48 h and cell viability was measured using WST-1 reagent as described in Material and Methods. Sigmoidal dose-response curves were plotted using GraphPad Prism 5.04.
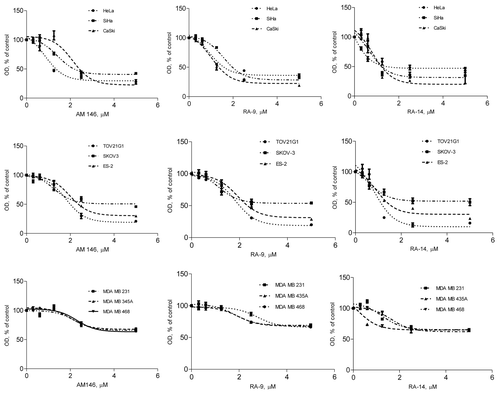
Table 1. IC50 values for chalcone-based derivatives.
Figure 2. AM146, RA-9 and RA-14 abrogate cell cycle progression and colony formation and trigger apoptosis. (A) Cell viability. HeLa cells were treated with 10 nM bortezomib (PS341), 5 µM AM146, 5 µM RA-9, 5 µM RA-14 or 5 µM RA-4. Cell viability was measured at indicated periods using WST-1 reagent as described in Material and Methods. Columns represent O.D. values as % of DMSO control ± standard error. *Indicates p ≤ 0.05. (B) Anchorage-dependent colony formation. 1x103 cells were plated in 6-well clusters and grown with or without 0.3 µM of AM146, RA-9 or RA-14. After 10 d, cells were fixed and stained. (C and D) FACS analysis. HeLa cells were plated at 2x105 in 6-well clusters. Twelve hours later, cells were treated with 10 nM PS341, 10 µM AM146, 10 µM RA-9 or 10 µM RA-14 for 24 h. Cells were then collected, stained with propidium iodide and analyzed for DNA content. (C) Progression through S or G2/M cell cycle checkpoints. Results are plotted as % of cells in S/G2/M ± standard error. *Indicates p ≤ 0.05. (D) Apoptosis frequency. Cells with hypo-diploid DNA content (sub-G1) were measured by FACS analysis. Columns reflect % of cells with hypo-diploid DNA content ± standard error. *Indicates p ≤ 0.05.
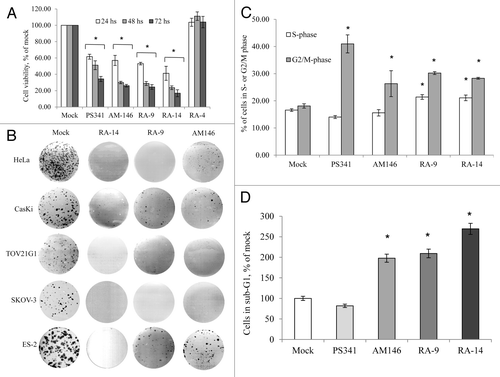
Because a prolonged and dose-dependent treatment with chalcone derivatives dramatically and selectively impeded cancer cell viability and clonogenic expansion, we sought to determine the early events underlying the capacity of these compounds to alter cancer cell growth, and thus negatively regulate the malignant phenotype, and its cellular mechanisms. Accordingly, we examined the cell cycle dynamics using FACS analysis of propidium iodide stained cells. As an early event (12 h treatment), all of the compounds impeded cell cycle progression, causing an arrest in S phase (RA9 and RA-14, p ≤ 0.05) and G2-M (AM146, RA-9 and RA-14, p ≤ 0.05) (). In addition, chalcone derivatives triggered an onset of early apoptosis as determined by the increase in sub-G1 population of cells with fragmented DNA content (p ≤ 0.05, ). These findings indicate that chalcone derivatives inhibit cancer cell growth by simultaneous blocking S-G2/M progression and induction of apoptosis.
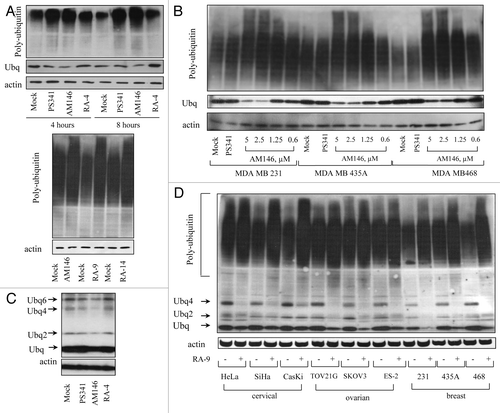
We next sought to investigate the cellular activities that may underlie the anti-proliferative/pro-apoptotic activity of these compounds. Because these compounds were originally designedCitation18 to improve the activity of previously reported chalcone-based proteasome inhibitor AM114,Citation19 this prompted a detailed analysis of the effect of AM146, RA-9 and RA-14 on protein ubiquitination as a measure of impaired proteolysis. Analysis of whole-cell extracts from inhibitor-treated HeLa (, top part) and TOV21G1 (, bottom part) cells showed a marked rapid and time-dependent accumulation of ubiqutinated proteins. We further observed a dose-dependent accumulation of ubiquitinated proteins in cells treated with AM146 (), RA-9 or RA-14 (not shown) with predominance of high molecular weight conjugates. Importantly, unlike AM146, RA-9, RA-14 and bortezomib, RA-4 treatment did not increase the level of ubiquitinated proteins in HeLa cells, which correlated with the lack of growth-inhibitory properties of RA-4 (). Remarkably, analysis of whole-cell extracts from HeLa cells treated with AM146 showed a marked decrease in expression of ubiquitin monomers, similar to RA-9 and RA-14 treatment. This, however, was not observed in PS341- or RA-4-treated samples, which served as negative controls (). The depletion in the pool of monomeric ubiquitin was dose-dependent () and was observed in cancer cell lines of different origin (). It is noteworthy that we also observed a decrease in the pool of ubiquitin dimers and unconjugated/free polyubiquitin chains (Ubq4–6) (). These findings indicate the distinct molecular targets within the ubiquitin-proteasome system mediating anti-proliferative/pro-apoptotic properties of AM-146, RA-9, RA-14 and bortezomib and their structurally relevant RA-4 compound.
Figure 3. Chalcone-derivatives induce accumulation of poly-ubiqutinated proteins in breast, ovarian and cervical cancer cell lines. Western blot analysis. Samples were probed with anti-ubiquitin antibody. Actin is shown as a loading control. (A) HeLa (top panel) or TOV21G1 (bottom panel) cells were treated with 10 nM bortezomib (PS341), 5 µM AM146, 5 µM RA-9, 5 µM RA-14 or 5 µM RA-4 for the indicated time periods. Protein samples were resolved with: top panel, 12% SDS-gel; bottom panel, 4–20% gradient SDS-gel (B). Breast cancer MDA MB 231 and MDA MB 468 cells and melanoma cancer MDA MB 435 cells (historically misidentified as breast cancer) were treated with 10 nM PS341 or indicated concentrations of AM146 for 6 h. Samples were resolved using 4–20% gradient SDS-gel. (C) HeLa cells were treated as in (A) and samples were resolved as in (B) to visualize ubiquitin monomers and unanchored ubiquitin chains (Ubq 2–6). (D) Indicated ovarian, breast and cervical cancer cells were treated with 5 µM RA-9 for 6 h and samples were resolved as in (B).
We have previously established that the accumulation of cellular poly-ubiquitinated proteins, reflecting the efficiency of proteolysis, depends on proteasome activity per se and the activity of DUB acting upstream and/or downstream (associated with the 19S subunit) of the proteasome.Citation20 To gain insight into the ability of chalcone derivatives to directly inhibit specific catalytic subunits within the 20S proteasome, we measured residual luminescent activity in AM146, RA-9 or RA-14 pre-exposed 20S purified proteasome (). The profile of proteasome inhibition shows characteristic suppression of chymotrypsin-like (chymotryptic), trypsin-like (tryptic) and peptidylglutamyl peptide hydrolyzing-like (caspase) activities by bortezomib. In contrast, chalcone derivatives did not suppress any of the 20S proteasomal activities. This result suggests that, unlike bortezomib, tested compounds do not directly inhibit the 20S proteasomal activity and indicates the distinct mechanism underlying their capacity to alter proteolysis.
Figure 4. AM146, RA-9 and RA-14 do not inhibit 20S proteasome activity but differentially modulate 26S proteasome activity in living cells. (A) 20S purified human proteasome were incubated with or without compound at indicated concentrations for one hour. Proteasome-Glo assay was performed and three activities of proteasome, chymotrypsin-like, trypsin-like and caspase-like, were measured as described in Material and Methods. Background luminescence values (DMSO + cell culture medium) were subtracted, and data plotted as relative luminescence unit (RLU). PS341 is shown as a positive control. Averages are based on the mean number for two independent experiments ± standard error. All experiments were done in triplicate. (B), (C) Stabilization of ubiquitin-luciferase bioluminescent reporter (Ub-FL). (B) Transiently transfected firefly luciferase (CMV-Luc) and Ub-FL HeLa cells were either mock-treated or treated with proteasome inhibitors bortezomib (PS341) or MG-132, non-cell-permeable DUBS inhibitor ubiquitin aldehyde (Ubal), or inhibitors AM146, or RA-9 or KVI-14 at the indicated concentrations for 8 h. Luciferase activity in cell lysates was quantified, background values were subtracted (relative luminescence units, RLU), data expressed as % of mock and the ratio of stabilized Ub-FL to baseline levels of CMV-Luc is shown on graph. Error bars are standard errors (SE) for three independent experiments. *Indicates p ≤ 0.05. (C) Transiently transfected CMV-Luc and Ub-FL HeLa cells were either mock-treated or treated with bortezomib, AM146, or a combination at the indicated concentrations for 8 h. Luciferase activity on cell lysate was quantified in relative luminescence units (RLU) and expressed as percent of control. Error bars are standard errors (SE) for three independent experiments. *Indicates p ≤ 0.05.
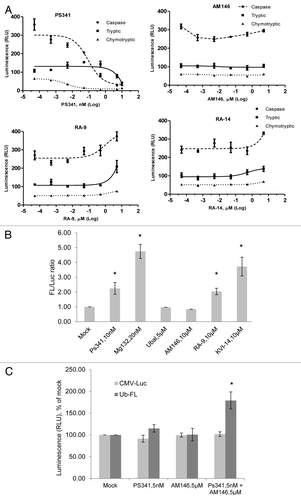
To exclude proteasome inhibitory activity in living cells, we performed a cellular assay for proteasome inhibition to verify that tested compounds have no 26S proteasome-targeting ability. To directly assay intact 26S proteasome activity in living cells, we used the ubiquitin-luciferase bioluminescence imaging reporter (Ub-FL) engineered for a robust, high-throughput screening for proteasome activity within cells.Citation21,Citation22 The Ub-FL and the control FL (CMV-Luc) expression vectors were transiently transfected into HeLa cervical cancer cells, treated as indicated, and the luminescence was recorded (). Two proteasome inhibitors, bortezomib and MG132, displayed robust stabilization of the firefly luciferase as compared with vector control (p ≤ 0.02 and 0.01, correspondingly), indicating suppression of proteasome activity. The same effect was observed for RA-9 and RA-14 (p ≤ 0.05 and 0.03, correspondingly), whereas AM146 failed to stabilize the firefly luciferase, similar to non-cell permeable DUB inhibitor ubiquitin aldehyde (Ubal) used as a negative control. Thus, these data indicate the differential effect of RA-9, RA-14 and AM146 on the 26S proteasome activity.
We and others have previously shown that proteasome activity in the cells can be modulated by deubiquitinating enzymes (DUB) functioning as positive or negative regulators of 26S proteasome activity. DUB-positive regulators of proteolysis like Doa4 (tre‑2)Citation23 and Ubp14 (IsoT)Citation20 subfamilies protect the inhibition of the 26S proteasome by free polyubiquitin chains. DUB-negative regulators of proteolysis, e.g., Uch2p (UCH37), act upstream of the proteasome by trimming ubiquitin from ubiquitinated substrates, thereby preventing their degradation.Citation24 Thus, we proposed that tested chalcone derivatives might act differentially, e.g., AM146 might predominantly inhibit DUB-negative regulators of proteasomal activity. To confirm this hypothesis, we transiently transfected HeLa cells with Ub-FL and the control CMV-Luc expression vectors and treated transfected cells with a combination of suboptimal doses of bortezomib and AM146 (). Combination treatment displayed high synergy in stabilization of the Ub-FL (80% increase compared with each compound alone, p ≤ 0.03). These findings indicate that AM146 might target DUB acting upstream of the proteasome.
AM146, RA-9 and RA-14 inhibit deubiqutinating enzymes (DUB) and alter expression of DUB-regulated oncogenes and tumor suppressors.
We and others provided ample evidence documenting the dramatic effect of cellular DUB inhibition on the level of poly-ubiquitinated proteins without impairing the proteasome activity, per se.Citation15,Citation17,Citation20,Citation25 As described earlier, chalcone derivatives contain an α,β-unsaturated carbonyl group, a feature shared with other known DUB inhibitors, like curcumin, shikoccin, DBACitation16 and WP1130.Citation17 Because the α,β-unsaturated carbonyl group is thought to be a molecular determinant conferring DUB-targeting capacity, this prompted us to assess the ability of tested compounds to inhibit DUB.
To directly evaluate the effect of AM146, RA-9 and RA-14 on DUB activity in treated HeLa cells, mock- and inhibitor-treated lysates were assayed using DUB-Glo assay (). Treatment with AM146, RA-9, RA-14 and DBA significantly reduced DUB activity. Exposure to AM146 and RA-14 treatment resulted in 30% and 36% inhibition, correspondingly, as compared with vehicle control samples, and RA-9-treatment led to 59% suppression. We further sought to explore whether specific DUB are directly targeted by chalcone derivatives. Therefore, we performed the in vitro DUB-GLO protease assay using purified human enzymes. All of the compounds significantly inhibited UCH-L1, UCH-L3 or USP8 (). Chalcone derivatives failed to suppress activity of Ataxin-3, A20CD, BAP1, Otubain 1 or USP7/HAUSP (USP7 and BAP1 are shown in ). We also determined that AM146, RA-9 and RA-14 suppress UCH-L3 activity in a dose-dependent manner and are more potent than known DUB inhibitor DBA used as a positive control (). Together, these results suggest that AM146, RA-9 and RA-14 may be partly selective DUB inhibitors.
Figure 5. Chalcone-derivatives, AM146, RA-9 and RA-15, suppress activity of deubiqutinating enzymes (DUB). (A) Cellular DUB activity. 0.6 x 105 HeLa cells were incubated with 25 nM PS341, 10 µM AM146, 10 µM RA-9, 10 µM RA-14 or 10 µM DBA for 24 h. DUB activity in whole-cell extracts was performed as described in Material and Methods using DUB-Glo™ Protease Assay reagent. Background luminescence values (DMSO + cell culture medium) were subtracted, and data plotted as relative luminescence unit (RLU) and are based on two independent experiments ± standard error. *Indicates p ≤ 0.05. All experiments were done in triplicate. (B) Activity of purified human enzymes in vitro. The activity of 500 nM UCH-L1, 50 nM UCH-L3, 200 nM BAP1, 100 nM USP7 or 50 nM USP8 was measured with/without 10 µM AM146, 10 µM RA-9 or 10 µM RA-14 using DUB-Glo™ Protease Assay reagent as described in Material and Methods. Background luminescence values (DMSO + enzyme buffer) were subtracted, and data plotted as % of untreated control (DMSO+ enzyme) ± standard error. * indicates p ≤ 0.05 Two independent experiments were done in duplicate. (C) Activity of 50 nM UCH-L3 treated with indicated concentrations of AM-146, RA-9 and RA-14 was measured as in (B). Known DUB-inhibitors, ubiquitin aldehyde (ubal) and dibenzylideneacetone (DBA), were used as a positive control. Background luminescence values (DMSO + enzyme buffer) were subtracted, and data plotted as % of untreated control (DMSO+ enzyme) ± standard error. *Indicates p ≤ 0.05.
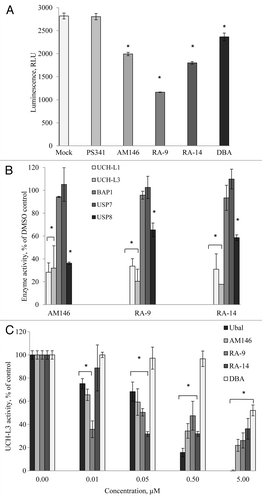
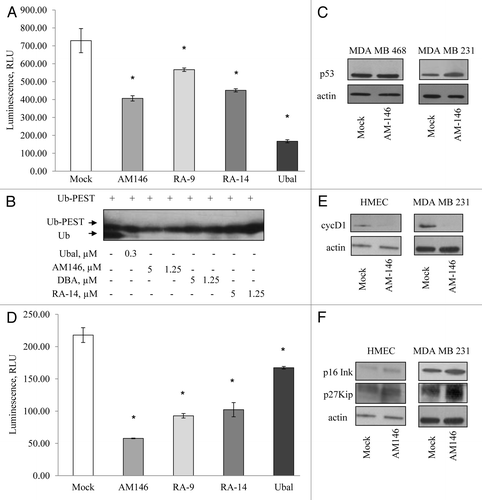
The next question we asked is whether AM146, RA-9 and RA-14 might selectively target other major cellular DUB, which are known to regulate turnover and stability of key regulators of cell survival and proliferation. Accordingly, we investigated the capacity of chalcone derivatives to inhibit the activity of two USP: Isopeptidase T (USP5) and USP2. We have previously shown that yeast Ubp14, homolog of human USP5, plays a major role in maintaining the levels of unanchored polyubiquitin chains.Citation20 Loss of USP5 stabilizes p53 due to the accumulation of free polyubiquitin chains, which compete with ubiquitinated p53.Citation26,Citation27 First, we confirmed the capacity of chalcone derivatives to suppress activity of purified USP5 (). DBA and some other small-molecule DUB inhibitors, were first identified based on their capacity to inhibit isopeptidases using ubiquitin-PEST and z-LRGG-AMC as substrates.Citation15,Citation16 Accordingly, we performed PEST-cleavage experiment with purified USP5 to evaluate the capacity of novel inhibitors to alleviate the cleavage of Ub-PEST and to confirm cell-based DUB-Glo assay findings. All of the tested compounds inhibited Ub-PEST cleavage in a dose-dependent manner. Similar suppression of Ub-PEST cleavage was achieved with 1.25 µM of chalcone derivatives compared with 5 µM DBA (). Further, in accord with published evidences, we observed accumulation of p53 in treated cells. Notably, while AM146 stabilized wild-type p53, it failed to stabilize mutant form p53 expressed in MDA MB468 cells (). Published evidence indicates that USP2 (cyclin D1-specific DUB) knockdown destabilizes cyclin D1 and induces G1/S arrest in the human cancer cell lines, where cell growth is dependent on cyclin D1 expression.Citation28 We confirmed the capacity of chalcone derivatives to suppress activity of purified USP2 (). We further observed that treatment with AM 146 downregulated cyclin D1 level (). We also documented increased levels of tumor suppressors p16Ink4A, p27Kip1 and p21 in cells treated with AM146 (). Similar results on expression of cell cycle regulators were obtained in ovarian and cervical cancers for both RA-9 and RA-14 (not shown). Although the exact trigger for apoptosis due to DUB inhibition is not fully understood, all of tested compounds disrupt DUB functions, downregulate the positive cell cycle regulator cyclin D1 and upregulate the level of pro-apoptotic proteins, e.g., p27Kip1, p16Ink4A and p53 in a dose-dependent manner. Because these events precede an onset of apoptosis, this might represent a potential mechanism for the pro-apoptotic effect of chalcone-based DUB inhibitors. Also, as others and we have shown, DUB disruption alters monoubiquitin/polyubiquitin dynamics, which could affect essential processes that rely on ubiquitin modification for function.Citation15,Citation17,Citation20
Figure 6. Chalcone-derivatives modulate expression of cell cycle promoters/tumor suppressors via altering USP activity. (A) Activity of purified human USP5 in vitro. The activity 100 nM USP5 was measured with/without 10 µM AM146, 10 µM RA-9 or 10 µM RA-14 using DUB-Glo™ Protease Assay reagent as described in Material and Methods. Treatment with 0.5 nM ubiquitin aldehyde (Ubal) was used as a positive control. Data were analyzed and plotted as in . (B) western blot analysis of Ubq-PEST cleavage in vitro. Experiment was performed as described in Material and Methods. Samples were treated with indicated concentrations of AM146, RA-14 and known DUB-inhibitors, ubiquitin aldehyde (Ubal) and dibenzylideneacetone (DBA), used as a positive control. (C) Breast cancer MDA MB231 and MDA MB468 cells were treated with 5 µM AM146 for 6 h. Whole cell extracts were resolved by western blot and probed with anti-p53 antibodies. Actin is shown as a loading control. (D) Activity of purified human USP2 (240 nM) was measured as in (A). (E,F) HMEC and breast cancer MDA MB 231 cells were treated with 5 µM AM146 for 6 h. Whole cell extracts were resolved by western blot and probed with antibodies against cell cycle promoters (cyc D1) or tumor suppressors (p16Ink and p27Kip). Actin is shown as a loading control.
AM146, RA-9 and RA-14 act as non-reversible cellular DUB inhibitors.
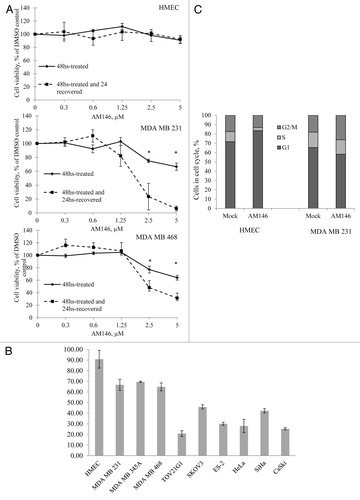
Ubiquitin aldehyde (Ubal) and UbVS, peptide-based inhibitors, have been previously described to irreversibly inhibit DUB activity.Citation13,Citation14 To test whether AM146, RA-9 and RA-14 cellular DUB inhibitory activity is reversible, we performed “recovery” experiments in primary human mammary epithelial cells HMEC and in the MDA MB 231 and MDA MB 468 breast cancer cell lines. Our results indicate that overall survival of breast cancer cells treated with 2.5–5 µM AM146 further declines 24 h after removal of compounds (p ≤ 0.01, ). This result suggests that DUB inhibition leads to irreversible cellular damage. Importantly, as shown on , primary human cells did not display a decrease in viability, indicating nonmalignant cells are less susceptible to DUB inhibitory activity. We further confirmed these findings by analyzing cell cycle dynamics in non-malignant and cancer cells (). Treatment with chalcone inhibitors triggered an onset of G2/M checkpoint arrest in tumor cells, while in primary cells, perturbation in cell cycle profile was associated with slight increase in G1 and decrease in S phase. The data presented in indicate this perturbation in cell cycle profile upon treatment of primary cells with AM146 does not translate into the reduced proliferation/viability, probably due to their overall low proliferative activity compared with cancer cell lines. These findings reinforce the proposition that DUB are suitable molecular target for anticancer drug development and indicate that inhibition of cellular DUB function by chalcone derivatives offers certain neoplastic selectivity.
Figure 7. Targeting cellular DUB by chalcone-derivative small molecule inhibitors irreversibly suppresses cancer but not normal cell growth. (A) Cell viability. Primary human mammary epithelial HMEC and breast cancer MDA MB231 or MDA MB468 cells were treated with indicated concentrations of AM146 for 48 h and cell viability was measured using WST-1 reagent as described in Material and Methods (“48-hrs-treated” curve). After the measurement, cells were washed, and fresh cell medium was added to cultures for 24 h. Cell viability of ‘recovered’ cells was recorded (“48-hrs-treated and 24-hrs-recovered” curve). Data points represent O.D. values as % of DMSO control ± standard error. *Indicates p ≤ 0.05. (B) Primary HMEC or cancer cells were treated with 5 μM AM146 for 48 h and cell viability was measured using WST-1 reagent as described in Material and Methods. (C) primary HMEC and breast cancer MDA MB 231 cells were treated with 5 µM AM146 for 12 h. Cells were collected, fixed, stained with propidium iodide and analyzed for DNA content by FACS analysis. Proliferating cells in different cell cycle phases were gated. Results are plotted as % of cells in G1, S or G2/M.
Altogether, our findings demonstrate that suppression of DUB activity by novel chalcone-based small-molecule inhibitors results in specific activation of negative cell cycle signaling in cancer cells and decreases their viability, thereby disrupting the malignant phenotype.
Discussion
The usurping of the ubiquitin-proteasome pathway governing cell survival, proliferation and metabolism at the posttranslational level is a central feature of malignancy. Various naturally derived compounds with anti-oxidative properties, including flavonoid family members curcuminCitation29-Citation35 and chalcones,Citation36 exert anticancer activity by regulating survival, proliferation, invasion, angiogenesis and metastasis of tumor cells through selective targeting of the ubiquitin-proteasome system (UPS).Citation29,Citation30 Published evidence suggests that the presence of an α,β-unsaturated carbonyl group, the key molecular determinant conferring UPS- and DUB inhibitory activity, within the curcumin molecule is responsible for its selective anti-neoplastic activity. This is associated with its capacity to reduce both the 20S proteasome proteolytic activities and the cellular deubiquitinating enzymes, leading to increased accumulation of ubiquitinated proteins and decreased levels of free ubiquitin (IC50: 80–100 µM).Citation16,Citation31-Citation34
In this study, we describe partly selective DUB inhibitory activity of chalcone-based derivatives, AM146, RA-9 and RA-14, small molecules of “AM” and “RA” series of compounds featuring the α,β-unsaturated carbonyl group that can presumably interact with the sulfhydryl of cysteines found in the active sites of DUB through a Michael addition reaction.Citation18,Citation19,Citation29 All of these three compounds induce rapid and marked accumulation of polyubiquitinated proteins, which is associated with anti-proliferative and proapoptotic effect in a variety of cancer cell lines, including breast, ovarian and cervical cancers (IC50: 1.5–12.5 µM).
We provide the evidence that AM146, RA-9 and RA-14 directly suppress activities of major cellular DUB, such as UCH-L1, UCH-L3, USP2, USP5 and USP8 ( and ), but do not inhibit Ataxin-3, A20CD, BAP1, Otubain 1, USP7/HAUSP or USP14 (). Our findings demonstrate that, among inhibitors tested, AM146 inhibits broader DUB spectrum and offers higher selectivity for neoplastic cells without significant damage to cell cycle transit or viability of primary cells when used in a range of 0.1–12 µM (, and and ). These events are associated with cellular effects, which are widely accepted as attributable to inhibition of multiple DUB activity: (1) increased accumulation of polyubiquitinated proteins (); (2) distinct pattern of polyubiquitinated proteins distribution with accumulation of higher molecular weight conjugates as compared with proteasome inhibition (); (3) depleted pool of ubiquitin monomers (); (4) an overall decrease in individual DUB activities ( and ); (5) altered expression/activity of DUB-regulated short-lived regulatory proteins, including oncoproteins and tumor suppressors (). Of note, several of the DUB targeted by AM146, RA-9 and RA-14 have been previously shown to regulate the stability and turnover of key cell cycle regulators/pro-oncogenes and proapoptotic proteins. For example, downregulation of USP2 was shown to inhibit tumor cell growth by promoting cyclin D1 degradation,Citation28 suggesting that silencing of specific DUB in tumor cells may be a safe and effective therapy in oncogene-addicted or drug-resistant cells. In accord with these studies, we found that all tested DUB inhibitors are effective in downregulating USP2 () and decreasing the expression of cyclin D1 (). These alterations were closely associated with blockage of cell cycle transit in cancer cells ( and ).
Inhibiting DUB, which are known to stabilize p53, has been recently proposed as a rational therapeutic strategy to activate p53 and promote p53-dependent apoptosis in tumors expressing wild-type p53.Citation25 In vitro, we demonstrated that AM146 and RA-14 effectively suppress activity of purified USP5 (). Following suppression of cellular USP5, known to regulate p53 stability,Citation26,Citation27 we observed stabilization of p53 (), which correlated with the induction of apoptosis. While all of tested compounds failed to stabilize mutant p53 harbored by the MDA MB468 breast cancer cells (), they were able to trigger cell cycle arrest and apoptosis independent of p53 stabilization (). This reinforces the proposition that these agents are of considerable interest, because the majority of anticancer drugs have poor efficacy in cells bearing mutant p53.Citation15 Recently, USP7/HAUSP, was reported to serve as a critical component of the p53-Mdm2 pathway, acting as a specific deubiquitinase for both p53 and Mdm2 and inducing profound stabilization of p53 due to enhanced degradation of Mdm2.Citation11,Citation37 However, chalcone-based derivatives used in this study did not suppress the USP7/HAUSP activity (). This confirms that tested inhibitors selectively target p53 stability via USP5-mediated mechanisms.
Most recently, USP17 and USP19 were reported to stabilize Skp2 and KPC1, correspondingly two known ubiquitin ligases for p27Kip1.Citation38,Citation39 Knockdown of these USP leads to perturbation in cell cycle progression and accumulation of p27Kip1. Accordingly, inhibition of USP17 and USP19 activity might be a valid mechanism explaining the effect of AM146, RA-9 and RA-14 on stabilization of p27Kip1 and their anti-proliferative activity. Many other targeted DUB and their cognate substrates in the anti-proliferative and apoptotic activity of AM146, RA-9 and RA-14 remain unknown. We were not able to confirm the complete inhibition DUB activity by AM146, RA-9 and RA-14 within the range of doses tested. However, this partial DUB inhibitory effect was sufficient to irreversibly cause cell cycle arrest in cancer cells, suppress their anchorage-dependent growth and trigger apoptosis ( and ). These findings indicate that even relatively modest downregulation of DUB activity might be efficient in curbing the neoplastic growth.
Altogether, this current work provides evidence of antitumor activity of chalcone-based derivatives, AM146, RA-9 and RA-14, based on their ability to inhibit cellular DUB and supports the development of pharmaceuticals to directly target DUB as most efficient strategy compared with proteasome inhibition; our findings also provide a clear rationale for the clinical evaluation of these small-molecule DUB inhibitors.
Materials and Methods
Cell lines, cell culture, compounds and treatment.
Ovarian (ES-2, TOV21G1 and SKOV-3) and cervical (HeLa, SiHa and CaSki) cell lines were from ATCC. Breast cancer cell lines, MDA MB 231 and MDA MB468, were generous gift from Dr. Polunovsky, University of Minnesota. The MDA MB435 cell line was provided by Dr. Sachdev, University of Minnesota. Human primary mammary epithelial cells, HMEC, were purchased from Lonza, Inc. Cancer cell lines were maintained in DMEM growth medium [Dulbecco’s modified Eagle’s high glucose (4.5 g/l) medium, Cambrex] supplemented with 10% FBS and antibiotic-antimycotic (Gibco). HMEC and HMEC/hTERT cells were grown in Clonetics® MEGM® mammary epithelial Cell Growth Medium, supplemented by growth factors (MEGM bullet kit, Lonza, Inc.).
AM146, RA-9 and RA-14 were synthesized by Dr. Ravi Anchoori, John Hopkins University.Citation18,Citation19 Ubiquitin aldehyde (Ubal) was obtained from Boston Biochem, bortezomib (PS341) was obtained from Selleck Chemicals.
WST-1 assay.
For the analysis of cell growth, 2 x 103 cells were plated in 96-well plates in 50 µl cell culture volume. Twenty-four hours later 50 µl of medium with/without compounds was added to cells to the final concentration indicated. Cells were allowed to grow for the additional indicated period of time after which 10 µl of WST-1 reagent was added to each well. Four hours later, optical density (OD) was measured at 450 nm wavelength. For the analysis, background values (DMSO + medium) were subtracted.
Inhibition of the proteasome proteolytic activitie.
To measure the three proteolytic activities associated with the proteasome, 50 µl of 1 nM (0.7 µg/ml) 20S Human proteasome (cat. #E-360, Boston Biochem) were diluted in 10 mM HEPES (pH 7.6) and incubated with or without inhibitors at indicated concentrations at room temperature for one hour. Samples were mixed with 50 µl of Proteasome-Glo 3-substrate system (Promega, cat. #G8531) and incubated at room temperature for 30 min prior to recording the luminescence with a plate-reading luminometer GloRunner 6. For the analysis, background luminescence values (DMSO + buffer) were subtracted and data points are indicated as relative luminescence units (RLU).
Inhibition of DUB enzymatic activity in living cells.
To measure the inhibition of DUB activity in living cells, 0.6 x 105 HeLa cells were incubated with 10 µM of tested compounds for 24 h. Cells were lysed in 50 µl of 25 mM HEPES, 5 mM EDTA, 0.1% CHAPS, 5 mM ATP, pH 7.5. Protein concentrations were determined with the BCA kit (Pierce) and adjusted to 1 µg/µl with 50 mM HEPES (pH 7.2), 10 mM DTT, 0.5 mM EDTA and 0.1% Prionex® buffer. 50 µl samples were then mixed with an equal volume of the DUB-GloTM Protease Assay reagent (Promega, cat. #G6261), incubated at room temperature for 30 min and luminescence was recorded with a plate-reading luminometer GloRunner 6. For the analysis, background luminescence values (DMSO + buffer) were subtracted, and data points are indicated as relative luminescence units (RLU).
Inhibition of purified DUB enzymatic activity.
To measure the ability of tested compounds to directly suppress the activity of purified human DUB (Boston Biochem), 500 nM UCH-L1, 50 nM UCH-L3, 240 nM USP2, 100 nM USP5 or 50 nM USP8 were pre-incubated in 50 mM HEPES (pH 7.2), 10 mM DTT, 0.5 mM EDTA and 0.1% Prionex® buffer at room temperature for 15 min. Enzyme samples in the presence or absence of 10 µM compound were incubated at room temperature for one hour. 50 µl of sample was mixed with 50 µl of the DUB-GloTM Protease Assay reagent (Promega, cat. #G6261), incubated at room temperature for 30 min and luminescence was recorded with a plate-reading luminometer GloRunner 6. For the analysis, background luminescence values (DMSO + buffer) were subtracted and data points are indicated as relative luminescence units (RLU).
Inhibition of Ub-PEST cleavage.
We measured the capacity of tested compounds to suppress isopeptidase enzymatic activity with Ub-PEST, a full-length ubiquitin molecule with an 18-amino acid C-terminal peptide extension (total mass, 10.5 kDa).Citation16 Ubiquitin isopeptidases specifically cleave the 18-amino acid peptide extension, releasing full-length ubiquitin (8.5 kDa). Briefly, 50 µg/ml Ub-PEST was pre-incubated with indicated concentrations of compounds in a buffer containing 25 mM HEPES, 5 mM EDTA, 0.1% CHAPS, 5 mM ATP, pH 7.5. After 1 h incubation on ice, purified 100 nM USP5 (Boston Biochem) was added to each sample. Samples were then incubated for 45 min at 25°C. Under these conditions Ub-PEST hydrolysis occurs at a linear rate. Twenty-five µl samples were then mixed with 20 µl of 2x Laemmli buffer, boiled briefly and resolved by western blot. The isopeptidase activity was monitored by determining the amount of product (8.5-kDa ubiquitin) formation.
Western assay and antibodies.
For all experiments, 2 x 105 cells were plated in 6-well plates in complete media. Twenty-four hours later, cells were treated with/without indicated concentrations of compounds for the indicated period of time. Cells were collected by trypsinization and resuspended in lysis buffer (50 mM Tris-Cl, 250 mM NaCl, 5 mM EDTA, 0.2% NP-40, 50 mM NaF) supplemented with protease inhibitor cocktail “Complete Mini” (Roche). Cells were lysed on ice for 20 min, disrupted by motor pestle (Fisher), centrifuged at 12,000 g for 10 min, and the concentration of proteins in supernatants was measured using the BCA Protein assay kit (Pierce). 5 µg of total soluble proteins was resolved by 4–20% SDS-electrophoresis and transferred to the PVDF membrane (Millipore). Blots were blocked in 0.1% Tween-20 in PBS with 10% non-fat dry milk and incubated with primary antibody according to manufacturer’s instructions. Ubiquitin antibody were from Millipore, actin antibody were from Sigma and anti-p53, p27, p16 and cyclin D1 antibodies were from BD PharMingen. Membranes were then incubated with corresponding HRP-conjugated secondary antibody and signal was detected by ECL (Amersham). Samples from at least two independent experiments were analyzed with each primary antibody.
Anchorage-dependent colony formation.
1,000 cells were seeded into 6-well clusters. Cultures were continued for 10 d in the presence or absence of 0.3 µM compounds, fixed with 4% formaldehyde and stained with Coomassie Blue.
Flow cytometry.
For quantification of DNA content, cultured cells were detached with trypsin, washed with PBS, fixed with ice-cold 70% ethanol and resuspended in propidium iodide (PI) staining mixture as previously described in reference Citation40. The percentage of cells with sub-G1 and S + G2/M DNA content was determined on a FACSCalibur flow cytometer (Becton Dickinson) with the CellQuest program.
Statistics.
Statistical analyses were performed using a Student t-test (Microsoft Office Excel data analysis pack). A p-value ≤ 0.05 was considered significant. Results were tabulated as the mean ± standard error of 2–3 separate experiments. In each experiment, all conditions were examined in triplicate. To calculate IC50 values, data were transformed and analyzed using nonlinear fit [log (inhibitor) vs. normalized response] using GraphPad Prism 5.04.
| Abbreviations: | ||
| UPS | = | ubiquitin-proteasome system |
| DUB | = | deubiquitinating enzymes |
| USPs | = | Ubiquitin-Specific Proteases |
| UCH | = | Ubiquitin C-terminal hydrolases |
| DBA | = | dibenzylideneacetone |
| HMEC | = | human mammary epithelial cells |
| hTERT | = | human telomerase reverse transcriptase |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
We are indebted to Dr. Zofia Zukowska, Department of Integrative Biology and Physiology, University of Minnesota for the encouragement, tremendous support and providing the possibility to carry out this work. We are grateful to Dr. Vitaly Polunovsky, University of Minnesota for providing breast cancer cell lines, acquisition of reagents and for the assistance with the FACS data analysis and helpful discussions. We thank Mark Peterson, University of Minnesota for English-language editing. We appreciate the help of Dr. Martha O’Brien, Promega, Inc. with troubleshooting and validating the experimental results of enzyme assays and providing DUB-Glo reagent. We thank Ann Kelly, Roche, Inc. for providing WST-1 reagent. We would like to acknowledge the assistance of the Flow Cytometry Core Facility of the Masonic Cancer Center, University of Minnesota, supported in part by P30 CA77598. Proprietary rights and synthesis of compounds - Drs. Aneta Modzelewska, Ravi K. Anchoori and Saeed R. Khan, The John Hopkins University; inquiries to S.R.K., Email: [email protected].
References
- Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell 2009; 138:389 - 403; http://dx.doi.org/10.1016/j.cell.2009.04.042; PMID: 19615732
- Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005; 123:773 - 86; http://dx.doi.org/10.1016/j.cell.2005.11.007; PMID: 16325574
- Wilkinson KD. DUBs at a glance. J Cell Sci 2009; 122:2325 - 9; http://dx.doi.org/10.1242/jcs.041046; PMID: 19571111
- Hussain S, Zhang Y, Galardy PJ. DUBs and cancer: the role of deubiquitinating enzymes as oncogenes, non-oncogenes and tumor suppressors. Cell Cycle 2009; 8:1688 - 97; http://dx.doi.org/10.4161/cc.8.11.8739; PMID: 19448430
- Luise C, Capra M, Donzelli M, Mazzarol G, Jodice MG, Nuciforo P, et al. An atlas of altered expression of deubiquitinating enzymes in human cancer. PLoS One 2011; 6:e15891; http://dx.doi.org/10.1371/journal.pone.0015891; PMID: 21283576
- Fang Y, Fu D, Shen XZ. The potential role of ubiquitin c-terminal hydrolases in oncogenesis. Biochim Biophys Acta 2010; 1806:1 - 6; PMID: 20302916
- Colland F. The therapeutic potential of deubiquitinating enzyme inhibitors. Biochem Soc Trans 2010; 38:137 - 43; http://dx.doi.org/10.1042/BST0380137; PMID: 20074048
- Atanassov BS, Koutelou E, Dent SY. The role of deubiquitinating enzymes in chromatin regulation. FEBS Lett 2010; PMID: 20974139
- Guédat P, Colland F. Patented small molecule inhibitors in the ubiquitin proteasome system. BMC Biochem 2007; 8:Suppl 1 S14; http://dx.doi.org/10.1186/1471-2091-8-S1-S14; PMID: 18047738
- Hybrigenics SA; WO2007017758 (2007)
- Colland F, Formstecher E, Jacq X, Reverdy C, Planquette C, Conrath S, et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther 2009; 8:2286 - 95; http://dx.doi.org/10.1158/1535-7163.MCT-09-0097; PMID: 19671755
- Novartis AG; WO2007009715 (2007)
- Borodovsky A, Kessler BM, Casagrande R, Overkleeft HS, Wilkinson KD, Ploegh HL. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J 2001; 20:5187 - 96; http://dx.doi.org/10.1093/emboj/20.18.5187; PMID: 11566882
- Hershko A, Rose IA. Ubiquitin-aldehyde: a general inhibitor of ubiquitin-recycling processes. Proc Natl Acad Sci U S A 1987; 84:1829 - 33; http://dx.doi.org/10.1073/pnas.84.7.1829; PMID: 3031653
- Mullally JE, Moos PJ, Edes K, Fitzpatrick FA. Cyclopentenone prostaglandins of the J series inhibit the ubiquitin isopeptidase activity of the proteasome pathway. J Biol Chem 2001; 276:30366 - 73; http://dx.doi.org/10.1074/jbc.M102198200; PMID: 11390388
- Mullally JE, Fitzpatrick FA. Pharmacophore model for novel inhibitors of ubiquitin isopeptidases that induce p53-independent cell death. Mol Pharmacol 2002; 62:351 - 8; http://dx.doi.org/10.1124/mol.62.2.351; PMID: 12130688
- Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res 2010; 70:9265 - 76; http://dx.doi.org/10.1158/0008-5472.CAN-10-1530; PMID: 21045142
- Bazzaro M, Anchoori RK, Mudiam MK, Issaenko O, Kumar S, Karanam B, et al. α,β-Unsaturated carbonyl system of chalcone-based derivatives is responsible for broad inhibition of proteasomal activity and preferential killing of human papilloma virus (HPV) positive cervical cancer cells. J Med Chem 2011; 54:449 - 56; http://dx.doi.org/10.1021/jm100589p; PMID: 21186794
- Achanta G, Modzelewska A, Feng L, Khan SR, Huang P. A boronic-chalcone derivative exhibits potent anticancer activity through inhibition of the proteasome. Mol Pharmacol 2006; 70:426 - 33; PMID: 16636137
- Amerik AYu, Swaminathan S, Krantz BA, Wilkinson KD, Hochstrasser M. In vivo disassembly of free polyubiquitin chains by yeast Ubp14 modulates rates of protein degradation by the proteasome. EMBO J 1997; 16:4826 - 38; http://dx.doi.org/10.1093/emboj/16.16.4826; PMID: 9305625
- Luker GD, Pica CM, Song J, Luker KE, Piwnica-Worms D. Imaging 26S proteasome activity and inhibition in living mice. Nat Med 2003; 9:969 - 73; http://dx.doi.org/10.1038/nm894; PMID: 12819780
- Stack JH, Whitney M, Rodems SM, Pollok BA. A ubiquitin-based tagging system for controlled modulation of protein stability. Nat Biotechnol 2000; 18:1298 - 302; http://dx.doi.org/10.1038/82422; PMID: 11101811
- Papa FR, Hochstrasser M. The yeast DOA4 gene encodes a deubiquitinating enzyme related to a product of the human tre-2 oncogene. Nature 1993; 366:313 - 9; http://dx.doi.org/10.1038/366313a0; PMID: 8247125
- Li T, Naqvi NI, Yang H, Teo TS. Identification of a 26S proteasome-associated UCH in fission yeast. Biochem Biophys Res Commun 2000; 272:270 - 5; http://dx.doi.org/10.1006/bbrc.2000.2767; PMID: 10872838
- Li Z, Melandri F, Berdo I, Jansen M, Hunter L, Wright S, et al. Delta12-Prostaglandin J2 inhibits the ubiquitin hydrolase UCH-L1 and elicits ubiquitin-protein aggregation without proteasome inhibition. Biochem Biophys Res Commun 2004; 319:1171 - 80; http://dx.doi.org/10.1016/j.bbrc.2004.05.098; PMID: 15194490
- Dayal S, Sparks A, Jacob J, Allende-Vega N, Lane DP, Saville MK. Suppression of the deubiquitinating enzyme USP5 causes the accumulation of unanchored polyubiquitin and the activation of p53. J Biol Chem 2009; 284:5030 - 41; http://dx.doi.org/10.1074/jbc.M805871200; PMID: 19098288
- Westermarck J. Regulation of transcription factor function by targeted protein degradation: an overview focusing on p53, c-Myc, and c-Jun. Methods Mol Biol 2010; 647:31 - 6; http://dx.doi.org/10.1007/978-1-60761-738-9_2; PMID: 20694659
- Shan J, Zhao W, Gu W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Mol Cell 2009; 36:469 - 76; http://dx.doi.org/10.1016/j.molcel.2009.10.018; PMID: 19917254
- Gupta SC, Kim JH, Prasad S, Aggarwal BB. Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev 2010; 29:405 - 34; http://dx.doi.org/10.1007/s10555-010-9235-2; PMID: 20737283
- Yang H, Landis-Piwowar KR, Chen D, Milacic V, Dou QP. Natural compounds with proteasome inhibitory activity for cancer prevention and treatment. Curr Protein Pept Sci 2008; 9:227 - 39; http://dx.doi.org/10.2174/138920308784533998; PMID: 18537678
- Milacic V, Banerjee S, Landis-Piwowar KR, Sarkar FH, Majumdar AP, Dou QP. Curcumin inhibits the proteasome activity in human colon cancer cells in vitro and in vivo. Cancer Res 2008; 68:7283 - 92; http://dx.doi.org/10.1158/0008-5472.CAN-07-6246; PMID: 18794115
- Dikshit P, Goswami A, Mishra A, Chatterjee M, Jana NR. Curcumin induces stress response, neurite outgrowth and prevent NF-kappaB activation by inhibiting the proteasome function. Neurotox Res 2006; 9:29 - 37; http://dx.doi.org/10.1007/BF03033305; PMID: 16464750
- Ravindran J, Prasad S, Aggarwal BB. Curcumin and cancer cells: how many ways can curry kill tumor cells selectively?. AAPS J 2009; 11:495 - 510; http://dx.doi.org/10.1208/s12248-009-9128-x; PMID: 19590964
- Si X, Wang Y, Wong J, Zhang J, McManus BM, Luo H. Dysregulation of the ubiquitin-proteasome system by curcumin suppresses coxsackievirus B3 replication. J Virol 2007; 81:3142 - 50; http://dx.doi.org/10.1128/JVI.02028-06; PMID: 17229707
- Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev 2001; 21:185 - 210; http://dx.doi.org/10.1002/med.1006; PMID: 11301410
- Yadav VR, Prasad S, Sung B, Aggarwal BB. The role of chalcones in suppression of NF-κB-mediated inflammation and cancer. Int Immunopharmacol 2011; 11:295 - 309; http://dx.doi.org/10.1016/j.intimp.2010.12.006; PMID: 21184860
- Becker K, Marchenko ND, Palacios G, Moll UM. A role of HAUSP in tumor suppression in a human colon carcinoma xenograft model. Cell Cycle 2008; 7:1205 - 13; http://dx.doi.org/10.4161/cc.7.9.5756; PMID: 18418047
- Lu Y, Adegoke OA, Nepveu A, Nakayama KI, Bedard N, Cheng D, et al. USP19 deubiquitinating enzyme supports cell proliferation by stabilizing KPC1, a ubiquitin ligase for p27Kip1. Mol Cell Biol 2009; 29:547 - 58; http://dx.doi.org/10.1128/MCB.00329-08; PMID: 19015242
- McFarlane C, Kelvin AA, de la Vega M, Govender U, Scott CJ, Burrows JF, et al. The deubiquitinating enzyme USP17 is highly expressed in tumor biopsies, is cell cycle regulated, and is required for G1-S progression. Cancer Res 2010; 70:3329 - 39; http://dx.doi.org/10.1158/0008-5472.CAN-09-4152; PMID: 20388806
- Li S, Sonenberg N, Gingras AC, Peterson M, Avdulov S, Polunovsky VA, et al. Translational control of cell fate: availability of phosphorylation sites on translational repressor 4E-BP1 governs its proapoptotic potency. Mol Cell Biol 2002; 22:2853 - 61; http://dx.doi.org/10.1128/MCB.22.8.2853-2861.2002; PMID: 11909977