Abstract
p53 is a key tumor suppressor that controls DNA damage response and genomic integrity. In response to genotoxic stress, p53 is stabilized and activated, resulting in controlled activation of genes involved in cell cycle arrest, DNA repair and/or apoptosis. ASAP is a centrosome- and spindle-associated protein, the deregulation of which induces severe mitotic defects. We show here that following double-strand break DNA formation, ASAP directly interacts with and stabilizes p53 by enhancing its p300-mediated acetylation and blocking its MDM2-mediated ubiquitination and degradation, leading to an increase of p53 transcriptional activity. Upon DNA damage, ASAP is transiently accumulated before being degraded upon persistent damage. This work links the p53 response with the cytoskeleton and confirms that the DNA-damaging signaling pathway is coordinated by centrosomal proteins. We reveal the existence of a new pathway through which ASAP signals the DNA damage response by regulating the p300-MDM2-p53 loop. These results point out ASAP as a possible target for the design of drugs to sensitize radio-resistant tumors.
Introduction
Accurate transmission of genetic information is critical for survival of living organisms. DNA damage, caused by endogenous or exogenous factors, poses a threat to the faithful transfer of the genomic information.
The tumor suppressor protein p53 is a central regulator of cell cycle arrest and apoptosis in response to genotoxic stress.Citation1 In unstressed cells, inappropriate p53 activation is prevented through a network of tight controls.Citation2 The main regulator of p53 is the E3 ubiquitin ligase MDM2, which directly binds to p53 N-terminal transcriptional activation domain, thus inhibiting its transcriptional activity and targeting p53 for proteasomal degradation.Citation2,Citation3 Under stress conditions, p53 protein is rapidly stabilized and activated by various mechanisms, including enhanced MDM2 degradation and posttranslational modifications of p53 and MDM2 (reviewed in refs. Citation4–Citation6). For instance, acetylation of p53 C-terminal lysines (K) (370, 372, 373, 381 and 382) by CREB binding protein (CBP) and p300 stabilizes p53 by preventing MDM2-mediated ubiquitination of the same residues, enhances its sequence-specific DNA binding and increases p53 recruitment of transcriptional co-activators or co-repressors.Citation2,Citation7-Citation9
We have recently characterized ASAP, a novel spindle microtubule-associated protein (MAP), which is phosphorylated by the mitotic kinase Aurora A and plays a key role in spindle assembly and mitotic progression.Citation10,Citation11 ASAP is also associated to centrosomes. Deregulation of ASAP expression leads to abnormal spindles with chromosome congression and segregation defects. In silico analyses with ASAP nucleotide or predicted protein sequences did not identify any known protein that shares global homology. The C-terminal moiety shows weak homology to various proteins involved in binding to MTs and interacts directly with MTs.Citation10,Citation12 The N-terminal part is more divergent. Different searches failed to identify any known domain in this region except a putative BRCA1 COOH terminal (BRCT) domain,Citation12 which may be indicative of a putative role of ASAP in DNA damage, since BRCT domains are usually found in proteins involved in checkpoint DNA damage response.Citation13 To date, no function was associated to this ASAP N-terminal part.
Here, we report that in response to DNA damage, ASAP interacts with p53 through its N-terminal part, contributes to its stabilization and, thus, promotes p53-dependent transcription. Following double-strand breaks (DSBs), ASAP is transiently stabilized before being degraded. Our data also suggest that ASAP stabilizes p53 both by alleviating MDM2-mediated p53 ubiquitination and by enhancing its p300-dependent acetylation. Together, our findings define ASAP as a new DDR component involved in the p300-MDM2-p53 loop regulation.
Results
ASAP interacts with p53. In order to investigate the function of the putative ASAP BRCT-like domain, the N-terminal region (aa 1–420) was used as bait for a yeast two-hybrid screen that identified the oncosuppressor Tp53 (p53) as a putative partner (). [35S]-p53 could be pulled down by recombinant GST-ASAP and the GST-ASAP-ΔCter mutant (aa 1–420), confirming the results of the two-hybrid yeast screen (). Moreover, by transient transfection in SaOS-2 cells, all the tested truncated p53 mutants () but p53 (102–393) co-immunoprecipitated with Flag-ASAP (), indicating that only the N-ter region of p53, which contains the transactivation domain and the MDM2-interacting region, interacts with ASAP. ASAP was previously shown to be localized at the centrosomes and the spindle.Citation10,Citation11 Analyzing ASAP subcellular localization by biochemical fractionation of U2OS cell extracts allowed us to show that ASAP was present in both cytosolic and nuclear fractions (with a small pool in the chromatin-enriched fraction) (Fig. S1A). In untreated cells, the ASAP nuclear pool was also detected by immunocytochemistry (Fig. S1B).
Figure 1. ASAP interacts with p53. (A) Analysis of the interaction between ASAP and p53 by yeast two-hybrid screen. The yeast strain Y187 transformed with the pGBT11-ASAP hybrid expression vector was mated with the yeast strain CG1945 transformed with the pGAD-p53 hybrid expression vector. Positive clones exhibited His+ and LacZ+ phenotypes. (B) In vitro-translated p53 was incubated with equal amounts of purified GST, GST-ASAP or GST-ASAP-Cter immobilized on Glutathione-Sepharose beads in the presence of [35S] methionine. Bound [35S]-labeled p53 was visualized by SDS-PAGE and autoradiography (upper panel). Coomassie blue staining was used to assess protein loading (lower panel). GST-ASAP-Nter did not produce any protein using different conditions. (C) Schematic drawing of full-length p53 and deletion mutants used in this study. TA, transactivation domain; DB, DNA-binding domain; OD, oligomerization domain. Numbers indicate amino acid positions. (D) SaOS-2 cells were transiently transfected with the indicated combinations of expression plasmids. Twenty-four h after transfection, whole cell lysates were prepared and immunoprecipitated with the monoclonal anti-Flag antibody followed by immunoblotting with the monoclonal anti-p53 antibody (DO-1) (left, upper panel) and with the polyclonal anti-p53 antibody (FL-393) (right, upper panel). The left and right middle and lower panels show immunoblots of whole cell lysates performed with monoclonal (left) or polyclonal (right) anti-p53 or anti-ASAP antibodies.
![Figure 1. ASAP interacts with p53. (A) Analysis of the interaction between ASAP and p53 by yeast two-hybrid screen. The yeast strain Y187 transformed with the pGBT11-ASAP hybrid expression vector was mated with the yeast strain CG1945 transformed with the pGAD-p53 hybrid expression vector. Positive clones exhibited His+ and LacZ+ phenotypes. (B) In vitro-translated p53 was incubated with equal amounts of purified GST, GST-ASAP or GST-ASAP-Cter immobilized on Glutathione-Sepharose beads in the presence of [35S] methionine. Bound [35S]-labeled p53 was visualized by SDS-PAGE and autoradiography (upper panel). Coomassie blue staining was used to assess protein loading (lower panel). GST-ASAP-Nter did not produce any protein using different conditions. (C) Schematic drawing of full-length p53 and deletion mutants used in this study. TA, transactivation domain; DB, DNA-binding domain; OD, oligomerization domain. Numbers indicate amino acid positions. (D) SaOS-2 cells were transiently transfected with the indicated combinations of expression plasmids. Twenty-four h after transfection, whole cell lysates were prepared and immunoprecipitated with the monoclonal anti-Flag antibody followed by immunoblotting with the monoclonal anti-p53 antibody (DO-1) (left, upper panel) and with the polyclonal anti-p53 antibody (FL-393) (right, upper panel). The left and right middle and lower panels show immunoblots of whole cell lysates performed with monoclonal (left) or polyclonal (right) anti-p53 or anti-ASAP antibodies.](/cms/asset/0607ef77-bf1a-4bc9-99cf-51e5c5f52244/kccy_a_10920858_f0001.gif)
ASAP level is regulated upon DNA damage.
p53 is a central effector of the DNA damage response. To investigate the potential regulation of ASAP by DNA damage signaling pathways, U2OS osteosarcoma cells were treated with sub-lethal doses (0.1 ∝M) of doxorubicin (Dox), which induces DSBs by inhibiting topoisomerase II. After 24 h of Dox treatment, cells are arrested in the G2 phase of the cell cycle (). ASAP protein started progressively to accumulate after 4 h, reaching its maximum level after 16 h of Dox treatment and then decreased progressively (, left part). No concomitant increase in ASAP mRNA level was observed, but rather a time-dependent decrease after 8 h of treatment (, right part). ASAP accumulation following Dox addition was also confirmed in primary human fibroblasts (). However, after lethal doses of Dox (1 ∝M), ASAP protein level decreased (), and no increase was observed even at earlier time points (data not shown). Treatment with etoposide, another topoisomerase II inhibitor, had a similar effect on ASAP expression (Fig. S1C). Upon Dox treatment, ASAP accumulated in both cytosolic and nuclear fractions (Fig. S1D).
Figure 2. ASAP is transiently stabilized upon DNA damage in a p53-dependent manner. (A) U2OS cells were treated with 0.1 µM Doxorubicin (Dox) for the indicated times. Cells were fixed and stained with Propidium Iodide (PI) for Flow Cytometry analysis (FACS). (B) U2OS cells were treated with 0.1 µM Dox for the indicated times. (Left) Cell lysates were analyzed by western blotting with the indicated antibodies. α-Tubulin was used as a loading control. (Right) Total RNA was extracted, reverse transcribed (RT) and ASAP expression was determined by real-time quantitative PCR (qPCR). Results were normalized to GAPDH. Experiments were performed in triplicate. (C) Cell lysates of normal human fibroblasts Hs27 treated with 0.1 µM Dox for the indicated times were analyzed by western blotting with the indicated antibodies. Actin was included as a loading control. (D) Cell lysates of U2OS cells treated with 0.1 µM or 1 µM Dox for 8 h were analyzed by western blotting using anti-ASAP and -p53 antibodies. α-Tubulin was included as a protein loading control. (E) Total cell extracts from U2OS cells treated, or not, with 0.5 µM Dox for 4 h were immunoprecipitated using pre-immune rabbit serum or rabbit ASAP anti-serum. Precipitates were analyzed by immunoblotting with antibodies against ASAP and p53. The input loaded was 10% of the IP.
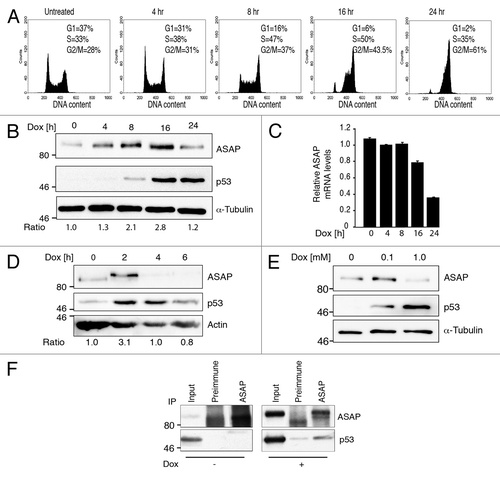
Co-immunoprecipitation experiments in vivo showed that endogenous ASAP and p53 interacted only following Dox treatment (). However, we cannot rule out that p53 level was too low in untreated cells to detect such interaction. Overexpression of Myc-p53 was sufficient to strongly increase the nuclear pool of ASAP (Fig. S1E).
These results clearly indicate that, following DNA damage, ASAP is first upregulated, independently of its mRNA level, and later on, or upon severe DNA damage, ASAP protein and mRNA levels decrease progressively.
ASAP induces p53 stabilization.
We then tested whether ASAP could directly regulate p53 activity. ASAP overexpression in p53-/- SaOS-2 cells led to a dose-dependent accumulation of ectopic p53 () that was not due to an increase of p53 mRNA level () but to p53 posttranslational stabilization. Phosphorylation at S15 and acetylation of K382 induce p53 stabilization following DNA damage.Citation2 ASAP overexpression had no effect on S15 phosphorylation, excluding a DNA damage-indirect effect, but it was sufficient to stimulate p53 acetylation at K382 (). Overexpression of p53 and Flag-HA-ASAP-ΔNter () or Flag-HA-ASAP-ΔCter () showed that ASAP-dependent stabilization of p53 requires the Nter fragment of ASAP. Finally, the half-life of endogenous p53 was increased in cycloheximide-treated ASAP-overexpressing cells compared with control cells (). Consistent with these observations, ASAP enhances p53 transcriptional activity on several p53-dependent gene promoters in a dose-dependent manner, such as p53-responsive PG13, MDM2, p21, Bax and Gadd45-luciferase reporter plasmids (; Fig. S2A and B).
Figure 3. ASAP contributes to p53 stabilization. (A) p53−/− SaOS-2 cells were co-transfected with empty pcDNA3 vector, Flag-ASAP (0.1, 0.3, 0.5 µg) or p53 (0.02 µg) as indicated. Cells lysates were collected 24 h post-transfection and analyzed by western blotting with the indicated antibodies (anti-Flag for ASAP detection). Actin was used as a loading control. (B) SaOS-2 cells were transfected with expression plasmids encoding empty vector, Flag-ASAP or p53 as indicated. Twenty-four h post-transfection, cells were lysed for RNA isolation. p53 expression was analyzed by RT-qPCR. Results were normalized to GAPDH mRNA. Experiments were performed in triplicate. (C) SaOS-2 cells were transiently transfected with constructs encoding p53, Flag-HA-ASAP or Flag-ASAP-Nter (amino acids 412–647). Lysates were analyzed by immunoblotting with anti-ASAP, -p53 and -α-Tubulin antibodies. Asterisks (*) indicate non-specific bands. (D) SaOS-2 cells were transiently transfected with expression plasmids encoding p53, Flag-ASAP or Flag-ASAP-Cter (amino acids 1–421). Cell lysates were analyzed by immunoblotting using anti-ASAP and anti-p53 antibodies. α-Tubulin was included as loading control. (E) 24 h after transfection with Flag-ASAP or empty vector, U2OS cells were treated with cycloheximide (CHX) for the indicated times, harvested and analyzed by western blotting with anti-ASAP and -p53 antibodies. α-Tubulin was used as a loading control. Band intensities were quantified with ImageJ and p53 concentration normalized to α-Tubulin (p53/α-Tubulin) is represented in the right panel. (F) SaOS-2 cells were transfected with 0.5 µg of luciferase reporter plasmids under the control of the PG13 (left panel) or MDM2 promoter (right panel) alone, or with 0.02 µg p53 and increasing amounts (0.5, 1 and 1.5 µg) of Flag-ASAP expression vector (which corresponds to the transfection conditions used in 3A). Twenty-four h after transfection, firefly luciferase activity was determined in cell lysates and normalized against Renilla luciferase activity. (G) SaOS-2 cells were transiently transfected with a luciferase reporter construct driven by the PG13 promoter and a combination of plasmids as indicated. Twenty-four h post-transfection, firefly luciferase activity was determined in cell lysates and normalized against Renilla luciferase activity. Results are presented as mean SD of three independent experiments. Asterisks (*) indicate a significant increase in pG13 luciferase activity in cells transfected with p53 and Flag-HA-ASAP-Cter compared with cells transfected with p53 alone (p < 0.00002).
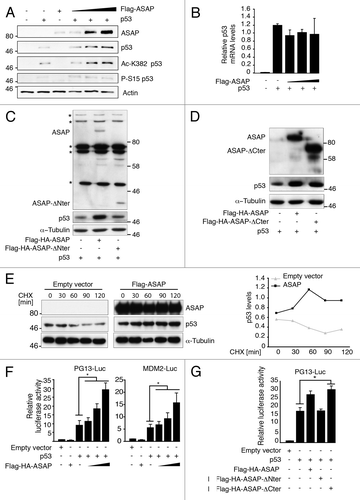
Conversely, Flag-HA-ASAP-ΔNter, which cannot interact with p53, did not boost p53 transcriptional activity on the PG13 promoter, confirming the specificity of these transactivation activities ().
ASAP-dependent effect on p53 transcriptional activity was confirmed at the protein level for MDM2 by ectopically expressing p53 and ASAP in SaOS-2 cells (). Similarly, after Dox treatment, MDM2 mRNA level () was decreased in ASAP-depleted U2OS cells in comparison to control cells. This effect was associated with a reduction in p53 acetylation at K382 but not phosphorylation at S15 (). However, surprisingly, the knockdown of endogenous ASAP also induced the mRNA levels of two targets of p53, namely p21 or Bax, in p53-independent manner (Fig. S2C).
Figure 4. MDM2 p53 target is stabilized upon ASAP induction. (A) SaOS-2 cells were transiently transfected with the indicated constructs and analyzed by western blotting using anti-ASAP, -p53 and -MDM2 antibodies. α-Tubulin was used as a loading control. (B) U2OS cells were transiently transfected with GL2 or ASAP siRNA and, 48 h later, treated or not (-) with 0.1 µM Dox for 4 h. Total RNA was extracted and MDM2 and ASAP expression was analyzed by real-time quantitative PCR (RT-qPCR). Results were normalized to GAPDH mRNA. Experiments were performed in triplicate. (C) U2OS cells were transiently transfected with GL2 or ASAP siRNA and, 48 h later, treated or not (-) with 0.1 µM Dox for 4 h. After lysis, western blot analysis was performed with the indicated antibodies. Actin was used as a loading control.
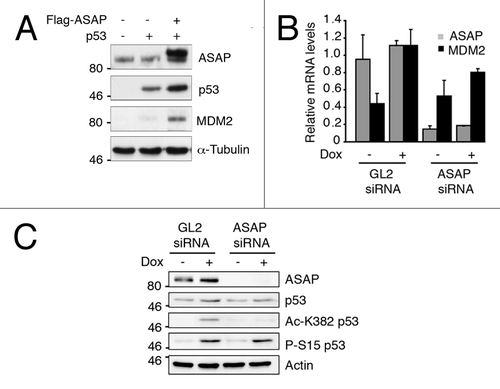
Altogether, these results demonstrate that ASAP participates in the regulation of p53 protein stability and increases its transcriptional activity during DDR.
ASAP cooperates with p300 to stabilize p53.
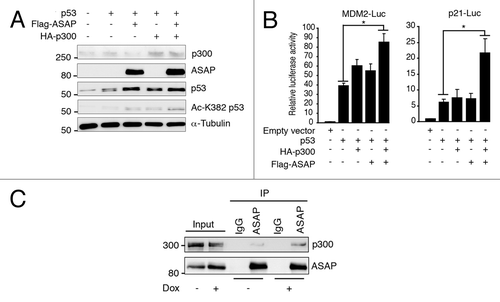
ASAP modulation was sufficient to regulate p53 acetylation at K382, which is triggered in vivo by the acetyltransferase p300.Citation14 We thus tested whether ASAP enhanced in vivo p53 acetylation by p300. K382 acetylation by ectopic p300 was significantly increased in the presence of ASAP, suggesting a cooperative effect (). Furthermore, combined ASAP and p300 overexpression resulted in significantly higher functional p53-mediated transcription (). Finally, ASAP interacts with p300, and this interaction increases upon DNA damage (). Therefore, ASAP seems to stabilize p53, at least partially, by enhancing p53 acetylation by p300. However, ASAP neither affects the in vitro p300 acetylase activity toward p53, nor modifies the endogenous p53/p300 interaction (data not shown), suggesting that ASAP-dependent p53 stabilization by p300 occurs by an indirect mechanism.
Figure 5. ASAP favors p300-dependent p53 acetylation. (A) SaOS-2 cells were transiently transfected with p53, Flag-ASAP and HA-p300 expression plasmids for 24 h and cell extracts were analyzed by western blotting with the indicated antibodies. α-Tubulin was included as loading control. (B) SaOS-2 cells were transiently transfected with 0.5 µg of luciferase reporter plasmids under the control of the MDM2 (left panel) or p21 promoter (right panel) alone, or with 0.02 µg p53, 1 µg Flag-ASAP or HA-p300 expression plasmids as indicated. Twenty-four h after transfection, firefly luciferase activity was determined in cell lysates and normalized against Renilla luciferase activity. (C) Total cell extracts from U2OS cells treated or not (-) with 0.1 µM Dox for 8 h were immunoprecipitated using mouse anti-IgG or monoclonal anti-ASAP antibodies. Precipitates were analyzed by immunoblotting with antibodies against ASAP and p300. The input loaded was 10% of the IP.
ASAP blocks MDM2-mediated p53 ubiquitination.
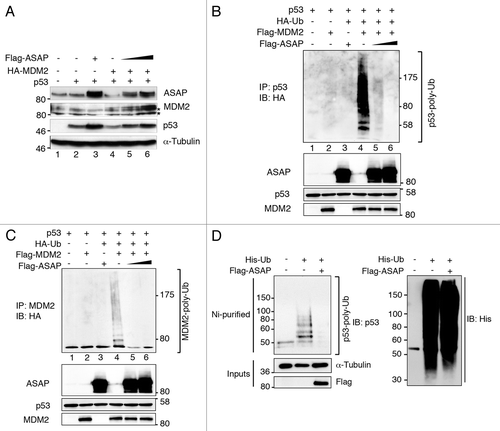
In proliferative cells, p53 is kept inactive by MDM2, which shields p53 N-terminal transactivation domain and also acts as an E3 ligase to target p53 for proteasomal degradation.Citation3,Citation15 Acetylation at K382 of p53 by p300 prevents MDM2-dependent degradation of p53. Furthermore, disruption of the MDM2-p53 complex facilitates the C terminus acetylation of p53 by p300.Citation16 Since ASAP binds to the same region of p53 as MDM2, we wondered whether ASAP might interfere with MDM2 binding to p53, thus affecting its degradation and facilitating p53 acetylation by p300. First, we checked whether ASAP affects MDM2-dependent turnover of p53. We transiently transfected p53-/- SaOS-2 cells with p53 and HA-MDM2 and/or increasing concentrations of Flag-ASAP. As reported by others, MDM2 overexpression triggered p53 degradation (, lane 4), whereas ASAP hindered MDM2-mediated p53 degradation in a dose-dependent manner (, lanes 5 and 6). To assess whether ASAP blocked MDM2-mediated ubiquitination of p53, HEK293T cells were transfected with p53, Flag-MDM2, HA-Ubiquitin and/or Flag-ASAP expression plasmids. As expected, MDM2 overexpression promoted p53 ubiquitination (, lane 4), whereas concomitant expression of ASAP inhibited it in a dose-dependent manner (, lanes 5 and 6). In parallel, ASAP overexpression also increased MDM2 stability (, lanes 5 and 6), while the presence of poly-ubiquitinated MDM2, detected using the anti-HA,Citation17 was reduced, most probably due to aborted MDM2 ubiquitination (). This ASAP-dependent regulation via MDM2 was also observed for endogenous p53 (, left), while general ubiquitination was not affected (, right). The interaction between p53 and MDM2 is necessary for MDM2 to control p53 stability. In addition, we tested whether ASAP interferes with MDM2-p53 complex formation. ASAP overexpression slightly decreased p53-MDM2 interaction (Fig. S3A). Moreover, exogenous ASAP interacted with the N-terminal part of MDM2, which is described as the region that interacts with p53 (Fig. S3B). These observations would suggest that ASAP, by its dual interaction with the N-terminal regions of MDM2 and p53, disrupts the formation of the MDM2-p53 complex, facilitating further modifications of the p53 C terminus.
Figure 6. ASAP inhibits MDM2-mediated p53 ubiquitination and degradation. (A) p53−/− SaOS-2 cells were transiently transfected with empty pcDNA3 vector, HA-MDM2, p53 and increasing amounts of Flag-ASAP (1 and 1.5 µg) expression plasmids. Immunoblotting was performed 24 h later with the indicated antibodies. Asterisks (*) indicate non-specific bands. α-Tubulin was included as loading control. (B) HEK293T cells were transiently transfected with p53 (500 ng), Flag-MDM2 (200 ng), HA-Ubiquitin (1.5 µg) and Flag-ASAP (6 and 7 µg) plasmids for 48 h, treated with 40 µM MG132 for 3 h and harvested. Cell lysates were immunoprecipitated with anti-p53 antibodies. Ubiquitinated forms of precipitated p53 were visualized using an anti-HA antibody. Western blotting of cell lysates performed with the indicated antibodies to detect the transfected proteins is shown below. (C) Cell lysates processed in were used in parallel for this experiment: HEK293T cells were transiently transfected with p53 (500 ng), Flag-MDM2 (200 ng), HA-Ubiquitin (1.5 µg) and Flag-ASAP (6 and 7 µg) plasmids for 48 h, treated with 40 µM MG132 for 3 h and harvested. Cell lysates were immunoprecipitated with anti-MDM2 antibodies. Ubiquitinated forms of precipitated MDM2 were visualized using an anti-HA antibody. Western blotting of cell lysates performed with the indicated antibodies to detect the transfected proteins is shown. (D) U2OS cells were transiently transfected with 6xHis-Ubiquitin (5 µg) and Flag-ASAP (7 µg) plasmids for 48 h, treated with 40 µM MG132 for 3h. Ni+-purified 6xHis-Ub-conjugated proteins were probed either for the presence of endogenous ubiquitinated p53 using an anti-p53 (DO-1) (left) or an anti-His antibody (right). Cell lysates were analyzed by western blotting with the indicated antibodies (Input).
Discussion
Understanding DNA damage sensing and signaling pathways has important implications for aging and cancer. The p53 tumor suppressor is a key integrator of DDR initiated by sensor kinases such as ATM and ATR.Citation18 p53 is constitutively expressed in normal cells, but its activity is regulated by a variety of posttranslational mechanisms. Of these, the autoregulatory feedback loop that intertwines p53 and MDM2 is thought to be pivotal for limiting untoward p53 activity. This feedback loop is itself inhibited by various mechanisms, among them, acetylation of p53 C-terminal domain.Citation2,Citation7
We have identified ASAP as a new component of mammalian DDR (see for a simplified model). Although ASAP was first characterized as a crucial MAP for spindle assembly and mitosis progression,Citation10,Citation11,Citation19 sequence analysis indicated that its N-terminal domain could be implicated in functions related to DDRCitation12 as we confirm in this work. Recent large-screen approaches to discover mitotic regulators have identified proteins involved in DNA/RNA pathways, such as transcription, chromatin remodeling, RNA processing or DNA repair (reviewed in ref. Citation20 and references therein). Some of these proteins localize at centrosomes and/or spindles and have mitotic-specific roles independent of their functions during interphase. For example, BRCA1 and BARD1, which are involved in DNA repair and transcription regulation, have also centrosome- and mitosis-specific functions.Citation21,Citation22 Similarly, ATM/ATR and Chk1/ Chk2 kinases are localized at centrosomesCitation23 and have recently been shown to control mitotic events and centrosome-dependent spindle assembly.Citation24
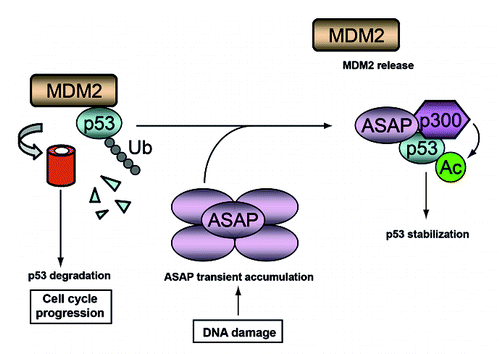
Figure 7. A schematic model describing the postulated ASAP-mediated stabilization of p53 following DNA damage. In growing cells, MDM2 degrades p53 to allow cell cycle progression. Upon DNA damage, ASAP is transiently accumulated and interacts with p53 to promote its stabilization by interfering with MDM2-p53 complex formation and suppressing its MDM2-mediated ubiquitination, thus facilitating its p300-mediated acetylation.
Here we propose a novel mechanistic pathway by which ASAP modulates p53 functions. Following DNA damage, ASAP is transiently stabilized. ASAP is degraded in case of persistent DNA damage, a fact that is reminiscent of the phenomenon involved in the so-called adaptation checkpoint. Indeed, it has been shown that Claspin degradation is needed for cell cycle re-entry and/ or apoptotic response after prolonged DNA damage.Citation25,Citation26 ASAP may have a precise function at the early step of DNA damage. We have previously shown that upon Aurora A depletion or following sustained stress, such as upon treatment with taxol or nocodazole (reviewed in ref. Citation11 and unpublished results), ASAP is degraded by the proteasome. Moreover, the mitotic centrosomal kinases Plk1 and Aurora-A that phosphorylate ASAP for functions required for correct mitosis progressionCitation11,Citation19 are also inhibited upon sustained DNA damage. Thus, ASAP degradation may be dependent on the inhibition of these kinases, which contribute to p53 stabilization and prevent the cell from progressing through mitosis.
ASAP binding to p53 was observed only in damaged cells and not in untreated cells. However, we cannot rule out that p53 level was too low to be detected in unchallenged cells.
Given the kinetics of both ASAP and p53 stabilization (), one could imagine that ASAP contributes to the early step of p53 stabilization in response to mild DNA damage.
ASAP can induce p53 acetylation in the absence of DNA damage and is required for DNA-damage-dependent acetylation and accumulation of p53. Although, double null cells revealed that CBP and p300 are dispensable for p53 targets p21 and MDM2 under defined conditions,Citation27 the combined overexpression of ASAP and p300 resulted in a higher p53-dependent transcription of these two genes. This highlights the complexity of the p53 regulation network that may vary from one condition to another. It has been demonstrated that p53 acetylation at K373 and K382 decreases ubiquitination, because these two lysines are also targeted by MDM2.Citation28 The ASAP-dependent increased p53 stabilization is probably in part achieved via an inhibition of the MDM2-dependent p53 degradation, consequently leading to an increase of p300-dependent acetylation of p53. Indeed, ASAP binds to both N-ter p53 and N-ter MDM2, thus not only disrupting p53-MDM2 interaction, but probably also facilitating C-ter p53 modifications as described.Citation16 However, since various proteins affect the p53-MDM2-p300 pathway, we cannot exclude that ASAP interacts or competes with other p53-interacting proteins that modify the outcome of the p53 response, as shown for other co-factors,Citation29 or with different drug combination treatments.Citation30 It is noteworthy that even polymorphism in the MDM2 promoter can alter its function and, consequently, disturb proper function of the p53 pathway.Citation31 In any case, the functional relationship between ASAP and the p300-MDM2-p53 complex warrants further investigation.
Although many effects seen under ASAP overexpression might be due to effects on the spindle, we suppose it is not the case in our study: ASAP-p53 interaction was first observed after a yeast two-hybrid screen that was confirmed by GST-pull down and coimmunoprecipitation, and was shown to be linked to the BRCT-like domain of ASAP. Surprinsingly, ASAP depletion also induced p53 targets such as p21 and Bax. However, this was already observed with the nucleolar protein nucleostemin (NS).Citation32 Both ectopic overexpression and depletion of NS activate p53 and its targets. In that case, NS excess or depletion either inhibits MDM2-mediated p53 ubiquitination or inhibits MDM2 function through ribosomal L5 or L11 association, respectively. Furthermore, in our case, ASAP depletion induced p21 and Bax in a p53-independent manner, since it was also observed in SaOS cells. This suggests that ASAP is involved in other pathways that warrant further investigation. If a physiological optimal level of ASAP is required for normal cell homeostasis, endogenous ASAP is also involved in the DNA damage response as shown by its transient stabilization and its p53 interaction. However, the detailed mechanistic pathway that is involved needs further investigation.
Although the last three decades have led to an incredibly detailed understanding of p53 biology, it appears that the “p53 network” defined by p53 and its interactors is getting more and more complex. Numerous coordinated mechanisms involved in the control of p53 activity need to be constantly redefined in view of new co-factors and posttranslational modifiers identified. For example, ribosomal stress can activate p53 through ribosomal protein (RP)-mediated suppression of MDM2 E3 ligase activity.Citation33 But DNA damage can initiate ribosomal stress and activate p53 through the RPL11/MDM2 pathway.Citation34 Intriguingly, in the last few years, other MAPs such as pVHLCitation35 or RASSF1ACitation36 have been implicated in the regulation of p53. More recently, Axin has been involved in both microtubule nucleationCitation37 and control of p53 activation,Citation38 and the co-transcription factor JMY regulates both cytoplasmic cytoskeletal events and, during stress response, p53 activity in the nucleus.Citation39,Citation40 This observation opens new perspective about the crosstalk of different stress-induced pathways that will need to be addressed in the future. For example, stress directed against MTs, such as nocodazole treatment, should also be viewed as part of the DNA damage response. Indeed, the centrosomal kinase LATS2 involved in the regulation of the formation of the mitotic spindle apparatusCitation41 is also crucial for the tetraploidization checkpoint in response to mitotic spindle damage in the LATS2-MDM2-p53 pathway.Citation42 It should be important to investigate ASAP function in the different loops that regulate p53 response to various types of stress through MAPs. Interestingly, the mitotic kinase Aurora A, which phosphorylates ASAP for spindle assembly,Citation11 RASSF1ACitation43 and LATS2,Citation44 also regulate the p53 pathway by direct phosphorylation, leading to MDM2-mediated p53 destabilization in normal cells.Citation45 Altogether, these observations reflect the importance of ASAP function during the cell cycle and support a link between the DNA damage checkpoint and centrosome integrity in maintaining genome stability. A more detailed analysis is necessary to fully understand ASAP regulation upon DNA damage.
Deciphering cellular responses to DNA damage constitutes one of the most important challenges in cancer biology. Indeed, genotoxic agents, which are used to cure cancer, can also induce DNA damage that is responsible for most of the side effects of anticancer therapy. What is emerging is the increasing number of centrosomal proteins involved in DNA damage pathway, highlighting the centrosome as a DNA damage regulator. With this work, we confirm that centrosomal proteins are key regulators, directly or indirectly of the DNA damage signaling pathway.
Materials and Methods
Yeast two-hybrid screen.
A cDNA library was constructed in the pADGAL4 vector with human mRNAs isolated from three complete 6- to 8-week-old human embryos.Citation46 The cDNA library was then transformed into the MATa Y187 yeast strain. The human N-ter (AA) ASAP bait plasmid was constructed in frame, with a downstream GAL4 DNA binding domain in the pGBT11 plasmid, and then transformed into MATa CG1945 yeast cells. Yeast two-hybrid screening was conducted according to the mating procedure described by Fromont-Racine et al. with minor modifications.Citation47 We obtained 52 positive clones on histidine-depleted plates, which become blue in the β-Gal test. Putative positive yeast clones were directly amplified by PCR reaction and then sequenced. To verify specific interactions between ASAP and the proteins expressed by the cDNA clones, the Y187 yeast strain transformed with full-length pGBT11-ASAP was mated with the CG1945 yeast strain transformed with a plasmid expressing p53 subcloned in frame with the GAL4 DNA activating domain (pGAD) and assayed for the His and LacZ phenotypes.
Cell culture, transfection and flow cytometry.
Cell lines (U2OS, SaOS-2, Hs27 fibroblasts and HEK293T) were obtained from the ATCC. p53-/- U2OS was kindly given by Dr. L. Linares. Cells were grown at 37°C in a 5% CO2 atmosphere in DMEM supplemented with 10% fetal bovine serum. Plasmid transient transfections were done using JetPEI (Polyplus) and siRNA duplexes transfections were performed using HiPerfect (QIAGEN), following the manufacturer’s instructions. For the Hs27 cells, transfection was performed using the Xtreme-9 reagent (Roche).
For flow cytometry analysis, U2OS cells were harvested and suspended in 0.2 ml of PBS and 0.8 ml ice-cold 100% ethanol. Cells were then incubated with RNase A (10 mg/ml) and propidium iodide (50 mg/ml), and DNA content was assessed with a FACSCalibur flow cytometer (Becton Dickinson).
Antibodies.
The following rabbit polyclonal antibodies were used: anti-ASAP,Citation10 -p300 (sc-584, Santa-Cruz and 3G230, Abcam), -(Lys-382)-Acetyl-p53 (Cell Signaling), -(Ser-15)-phospho-p53 (Cell Signaling), -Actin (20–33, Sigma), -HA (Bethyl), -GST (Abcam, ab9085). Mouse monoclonal antibodies used were: anti-ASAP 6G5 (Venoux et al. 2008a), -p53 (DO-7, DakoCytomation and DO-1, Santa-Cruz), -MDM2 (2A10, Calbiochem), -p21 (SX1118, DakoCytomation), -α-Tubulin (DM1A Sigma), -Flag (M2, Sigma), -Myc (9E10, Abcam), -HIS (BMG-His-1, Roche), -HA (12CA5, Roche).
Immunoprecipitation and western blot analysis.
Immunoprecipitations and western blotting were performed as described in reference Citation11. Where indicated, after autoradiograph scanning, band intensities were measured using the ImageJ software (http://rsb.info.nih.gov/ij). Individual values were normalized relative to control values (α-tubulin or actin).
Luciferase reporter assay.
SaOS-2 cells grown in 12-well plates were co-transfected with 500 ng of reporter construct and 50 ng of internal control Renilla construct. Twenty-four h after transfection, luciferase activity was monitored using the Dual-Luciferase Reporter Assay System (Promega) and a luminometer (Berthold). Firefly luciferase activity was normalized to Renilla luciferase activity. Experiments are presented as the mean ± SD of triplicate data representative of three independent experiments.
In vivo ubiquitination assays.
Subconfluent HEK293T cells in 10 cm plates were transfected with 1.5 ∝g HA-Ubiquitin, 0.2 ∝g Flag-MDM2, 6 or 7 ∝g Flag-ASAP and 0.5 ∝g p53 expression vectors. For each transfection, the total plasmid DNA was adjusted to 8.5 ∝g/100 mm dish by adding the parental pcDNA3 vector. 48 h after transfection, cells were treated with 40 ∝M MG132 for 3 h and then lysed in 50 mM TRIS-HCl pH 7.4, 120 mM NaCl, 5 mM EDTA, 0.5% NP40, 0.5% Triton-X100, 1% SDS, 1 mM DTT and protease inhibitors. Immunoprecipitations were performed using anti-p53 (DO-1) or -MDM2 antibodies, and the precipitated ubiquitinated forms were visualized using an anti-HA antibody and western blot analysis.
To detect endogenous p53 ubiquitination, 1.8 x 106 U2OS cells were transfected with 5 ∝g of 6xHis-tagged ubiquitin and 7 ∝g of Flag-ASAP or pcDNA3 vector (negative control). 48 h after transfection, cells were treated with 40 ∝M MG132 for 3 h and then lysed under denaturing conditions (6 M guanidinium-HCl, 10 mM TRIS-HCl pH 8.0, 100 mM sodium phosphate buffer pH 8.0). His-Ub-conjugated proteins were purified by nickel chromatography (Ni-NTA agarose, Invitrogen) as described in reference Citation48. Nickel-bound ubiquitinated proteins were subjected to western blot analysis using anti-p53 (DO-1) antibodies. Note that the levels of ubiquitination were assessed on equal amounts of proteins, as shown by the western blots of (1) the nickel-bound ubiquitinated proteins using anti-HIS antibodies and (2) an unpurified fraction of the same extracts using anti-α-tubulin antibodies (inputs).
Primers and siRNAs.
The following primers were employed for qPCR amplifications: ASAP: Forward 5'-GCC CTC CAA GCA GAA CTG TG-3', Reverse 5'-TCA GCA GGA GTG TCT GGC ATT-3'; MDM2: Forward 5'-CAG TGG CGA TTG GAG GGT AG-3', Reverse 5'-GAC TAC TAC CAA GTT CCT GTA G-3'; p53: Forward 5'-AGG CCT TGG AAC TCA AGG AT- 3', Reverse 5'-CCC TTT TTG GAC TTC AGG TG-3'; GAPDH: Forward 5'-CTC TGC TCC TCC TGT TCG AC- 3'; Reverse 5'-ACG ACC AAA TCC GTT GAC TC-3'.
siGL2 and siASAP sequences were as described in reference Citation10 and Citation11. siATM and siATR were the validated QIAGEN siRNA Hs_ATM_8_HP and Hs_ATR_12_HP, respectively.
Statistical analysis.
Results are presented as mean SD of three independent experiments. Statistical analysis was done using unpaired Student t-tests. Differences were considered statistically significant at p < 0.02 (*).
Additional material
Download Zip (2.7 MB)Acknowledgments
We thank Dr. O. Coux for helpful discussions and Drs. M. Benkirane and P. Pasero for critical reading of the manuscript. The authors are grateful to the following researchers for providing essential reagents: Drs. M. Benkirane, D. Bohmann, A.S. Coutts, O. Coux, L. Journot, M.B. Kastan, R. Kiernan, N.B. Lathangue, L. Le Cam, L. Linares, J-M. Mesnard, M. Oren, D. Spengler. SR is particularly indebted to O. Coux for helpful discussion and his constant support. We thank Dr. E. Andermarcher for the careful reading of the manuscript and the English language corrections. This work was supported by the CNRS and grants from Association de la Recherche contre le Cancer (ARC N° 4027), Ligue Nationale contre le Cancer (Comité Languedoc-Roussillon 2007, 2009), and Fondation Jérôme Lejeune to SR. DK was a recipient of a MERT fellowship.
References
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009; 137:413 - 31; http://dx.doi.org/10.1016/j.cell.2009.04.037; PMID: 19410540
- Kruse JP, Gu W. Modes of p53 regulation. Cell 2009; 137:609 - 22; http://dx.doi.org/10.1016/j.cell.2009.04.050; PMID: 19450511
- Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell 2006; 21:307 - 15; http://dx.doi.org/10.1016/j.molcel.2006.01.020; PMID: 16455486
- Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ 2006; 13:927 - 34; http://dx.doi.org/10.1038/sj.cdd.4401912; PMID: 16543935
- Oren M. Decision making by p53: life, death and cancer. Cell Death Differ 2003; 10:431 - 42; http://dx.doi.org/10.1038/sj.cdd.4401183; PMID: 12719720
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer 2006; 6:909 - 23; http://dx.doi.org/10.1038/nrc2012; PMID: 17128209
- Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell 2008; 133:612 - 26; http://dx.doi.org/10.1016/j.cell.2008.03.025; PMID: 18485870
- Teufel DP, Bycroft M, Fersht AR. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009; 28:2112 - 8; http://dx.doi.org/10.1038/onc.2009.71; PMID: 19363523
- Ferreon JC, Lee CW, Arai M, Martinez-Yamout MA, Dyson HJ, Wright PE. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc Natl Acad Sci USA 2009; 106:6591 - 6; http://dx.doi.org/10.1073/pnas.0811023106; PMID: 19357310
- Saffin JM, Venoux M, Prigent C, Espeut J, Poulat F, Giorgi D, et al. ASAP, a human microtubule-associated protein required for bipolar spindle assembly and cytokinesis. Proc Natl Acad Sci USA 2005; 102:11302 - 7; http://dx.doi.org/10.1073/pnas.0500964102; PMID: 16049101
- Venoux M, Basbous J, Berthenet C, Prigent C, Fernandez A, Lamb NJ, et al. ASAP is a novel substrate of the oncogenic mitotic kinase Aurora-A: phosphorylation on Ser625 is essential to spindle formation and mitosis. Hum Mol Genet 2008; 17:215 - 24; http://dx.doi.org/10.1093/hmg/ddm298; PMID: 17925329
- Venoux M, Delmouly K, Milhavet O, Vidal-Eychenié S, Giorgi D, Rouquier S. Gene organization, evolution and expression of the microtubule-associated protein ASAP (MAP9). BMC Genomics 2008; 9:406; http://dx.doi.org/10.1186/1471-2164-9-406; PMID: 18782428
- Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science 2003; 302:639 - 42; http://dx.doi.org/10.1126/science.1088753; PMID: 14576433
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997; 90:595 - 606; http://dx.doi.org/10.1016/S0092-8674(00)80521-8; PMID: 9288740
- Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol 2003; 13:49 - 58; http://dx.doi.org/10.1016/S1044-579X(02)00099-8; PMID: 12507556
- Poyurovsky MV, Katz C, Laptenko O, Beckerman R, Lokshin M, Ahn J, et al. The C terminus of p53 binds the N-terminal domain of MDM2. Nat Struct Mol Biol 2010; 17:982 - 9; http://dx.doi.org/10.1038/nsmb.1872; PMID: 20639885
- Cheng Q, Chen J. The phenotype of MDM2 auto-degradation after DNA damage is due to epitope masking by phosphorylation. Cell Cycle 2011; 10:1162 - 6; http://dx.doi.org/10.4161/cc.10.7.15249; PMID: 21386656
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003; 3:155 - 68; http://dx.doi.org/10.1038/nrc1011; PMID: 12612651
- Eot-Houllier G, Venoux M, Vidal-Eychenié S, Hoang MT, Giorgi D, Rouquier S. Plk1 regulates both ASAP localization and its role in spindle pole integrity. J Biol Chem 2010; 285:29556 - 68; http://dx.doi.org/10.1074/jbc.M110.144220; PMID: 20615875
- Ducat D, Kawaguchi S, Liu H, Yates JR 3rd, Zheng Y. Regulation of microtubule assembly and organization in mitosis by the AAA+ ATPase Pontin. Mol Biol Cell 2008; 19:3097 - 110; http://dx.doi.org/10.1091/mbc.E07-11-1202; PMID: 18463163
- Joukov V, Groen AC, Prokhorova T, Gerson R, White E, Rodriguez A, et al. The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell 2006; 127:539 - 52; http://dx.doi.org/10.1016/j.cell.2006.08.053; PMID: 17081976
- Parvin JD, Sankaran S. The BRCA1 E3 ubiquitin ligase controls centrosome dynamics. Cell Cycle 2006; 5:1946 - 50; http://dx.doi.org/10.4161/cc.5.17.3208; PMID: 16969086
- Löffler H, Lukas J, Bartek J, Krämer A. Structure meets function--centrosomes, genome maintenance and the DNA damage response. Exp Cell Res 2006; 312:2633 - 40; http://dx.doi.org/10.1016/j.yexcr.2006.06.008; PMID: 16854412
- Smith E, Dejsuphong D, Balestrini A, Hampel M, Lenz C, Takeda S, et al. An ATM- and ATR-dependent checkpoint inactivates spindle assembly by targeting CEP63. Nat Cell Biol 2009; 11:278 - 85; http://dx.doi.org/10.1038/ncb1835; PMID: 19182792
- Freire R, van Vugt MA, Mamely I, Medema RH. Claspin: timing the cell cycle arrest when the genome is damaged. Cell Cycle 2006; 5:2831 - 4; http://dx.doi.org/10.4161/cc.5.24.3559; PMID: 17172868
- Mamely I, van Vugt MA, Smits VA, Semple JI, Lemmens B, Perrakis A, et al. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr Biol 2006; 16:1950 - 5; http://dx.doi.org/10.1016/j.cub.2006.08.026; PMID: 16934469
- Kasper LH, Thomas MC, Zambetti GP, Brindle PK. Double null cells reveal that CBP and p300 are dispensable for p53 targets p21 and Mdm2 but variably required for target genes of other signaling pathways. Cell Cycle 2011; 10:212 - 21; http://dx.doi.org/10.4161/cc.10.2.14542; PMID: 21220944
- Carter S, Vousden KH. Modifications of p53: competing for the lysines. Curr Opin Genet Dev 2009; 19:18 - 24; http://dx.doi.org/10.1016/j.gde.2008.11.010; PMID: 19179064
- Coutts AS, Adams CJ, La Thangue NB. p53 ubiquitination by Mdm2: a never ending tail?. DNA Repair (Amst) 2009; 8:483 - 90; http://dx.doi.org/10.1016/j.dnarep.2009.01.008; PMID: 19217357
- Azmi AS, Banerjee S, Ali S, Wang Z, Bao B, Beck FW, et al. Network modeling of MDM2 inhibitor-oxaliplatin combination reveals biological synergy in wt-p53 solid tumors. Oncotarget 2011; 2:378 - 92; PMID: 21623005
- Knappskog S, Lønning PE. MDM2 promoter SNP285 and SNP309; phylogeny and impact on cancer risk. Oncotarget 2011; 2:251 - 8; PMID: 21436469
- Dai MS, Sun XX, Lu H. Aberrant expression of nucleostemin activates p53 and induces cell cycle arrest via inhibition of MDM2. Mol Cell Biol 2008; 28:4365 - 76; http://dx.doi.org/10.1128/MCB.01662-07; PMID: 18426907
- Miliani de Marval PL, Zhang Y. The RP-Mdm2-p53 pathway and tumorigenesis. Oncotarget 2011; 2:234 - 8; PMID: 21406728
- Llanos S, Serrano M. Depletion of ribosomal protein L37 occurs in response to DNA damage and activates p53 through the L11/MDM2 pathway. Cell Cycle 2010; 9:4005 - 12; http://dx.doi.org/10.4161/cc.9.19.13299; PMID: 20935493
- Roe JS, Kim H, Lee SM, Kim ST, Cho EJ, Youn HD. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol Cell 2006; 22:395 - 405; http://dx.doi.org/10.1016/j.molcel.2006.04.006; PMID: 16678111
- Song MS, Song SJ, Kim SY, Oh HJ, Lim DS. The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2-DAXX-HAUSP complex. EMBO J 2008; 27:1863 - 74; http://dx.doi.org/10.1038/emboj.2008.115; PMID: 18566590
- Fumoto K, Kadono M, Izumi N, Kikuchi A. Axin localizes to the centrosome and is involved in microtubule nucleation. EMBO Rep 2009; 10:606 - 13; http://dx.doi.org/10.1038/embor.2009.45; PMID: 19390532
- Li Q, Lin S, Wang X, Lian G, Lu Z, Guo H, et al. Axin determines cell fate by controlling the p53 activation threshold after DNA damage. Nat Cell Biol 2009; 11:1128 - 34; http://dx.doi.org/10.1038/ncb1927; PMID: 19731416
- Coutts AS, Weston L, La Thangue NB. A transcription co-factor integrates cell adhesion and motility with the p53 response. Proc Natl Acad Sci USA 2009; 106:19872 - 7; PMID: 19897726
- Coutts AS, Weston L, La Thangue NB. Actin nucleation by a transcription co-factor that links cytoskeletal events with the p53 response. Cell Cycle 2010; 9:1511 - 5; http://dx.doi.org/10.4161/cc.9.8.11258; PMID: 20372063
- Abe Y, Ohsugi M, Haraguchi K, Fujimoto J, Yamamoto T. LATS2-Ajuba complex regulates gamma-tubulin recruitment to centrosomes and spindle organization during mitosis. FEBS Lett 2006; 580:782 - 8; http://dx.doi.org/10.1016/j.febslet.2005.12.096; PMID: 16413547
- Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev 2006; 20:2687 - 700; http://dx.doi.org/10.1101/gad.1447006; PMID: 17015431
- Rong R, Jiang LY, Sheikh MS, Huang Y. Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates RASSF1A-mediated microtubule interaction and M-phase cell cycle regulation. Oncogene 2007; 26:7700 - 8; http://dx.doi.org/10.1038/sj.onc.1210575; PMID: 17563743
- Toji S, Yabuta N, Hosomi T, Nishihara S, Kobayashi T, Suzuki S, et al. The centrosomal protein Lats2 is a phosphorylation target of Aurora-A kinase. Genes Cells 2004; 9:383 - 97; http://dx.doi.org/10.1111/j.1356-9597.2004.00732.x; PMID: 15147269
- Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet 2004; 36:55 - 62; http://dx.doi.org/10.1038/ng1279; PMID: 14702041
- Zhou R, Bonneaud N, Yuan CX, de Santa Barbara P, Boizet B, Schomber T, et al. SOX9 interacts with a component of the human thyroid hormone receptor-associated protein complex. Nucleic Acids Res 2002; 30:3245 - 52; http://dx.doi.org/10.1093/nar/gkf443; PMID: 12136106
- Fromont-Racine M, Rain JC, Legrain P. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat Genet 1997; 16:277 - 82; http://dx.doi.org/10.1038/ng0797-277; PMID: 9207794
- Le Cam L, Linares LK, Paul C, Julien E, Lacroix M, Hatchi E, et al. E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell 2006; 127:775 - 88; http://dx.doi.org/10.1016/j.cell.2006.09.031; PMID: 17110336