Abstract
Here, we set out to test the novel hypothesis that increased mitochondrial biogenesis in epithelial cancer cells would “fuel” enhanced tumor growth. For this purpose, we generated MDA-MB-231 cells (a triple-negative human breast cancer cell line) overexpressing PGC-1α and MitoNEET, which are established molecules that drive mitochondrial biogenesis and increased mitochondrial oxidative phosphorylation (OXPHOS). Interestingly, both PGC-1α and MitoNEET increased the abundance of OXPHOS protein complexes, conferred autophagy resistance under conditions of starvation and increased tumor growth by up to ~3-fold. However, this increase in tumor growth was independent of neo-angiogenesis, as assessed by immunostaining and quantitation of vessel density using CD31 antibodies. Quantitatively similar increases in tumor growth were also observed by overexpression of PGC-1β and POLRMT in MDA-MB-231 cells, which are also responsible for mediating increased mitochondrial biogenesis. Thus, we propose that increased mitochondrial “power” in epithelial cancer cells oncogenically promotes tumor growth by conferring autophagy resistance. As such, PGC-1α, PGC-1β, mitoNEET and POLRMT should all be considered as tumor promoters or “metabolic oncogenes.” Our results are consistent with numerous previous clinical studies showing that metformin (a weak mitochondrial “poison”) prevents the onset of nearly all types of human cancers in diabetic patients. Therefore, metformin (a complex I inhibitor) and other mitochondrial inhibitors should be developed as novel anticancer therapies, targeting mitochondrial metabolism in cancer cells.
Introduction
Mitochondria are not only the powerhouse of the cell, but they also play a critical role in the metabolism of proliferating cells.Citation1 Despite these known function(s) in cell growth and division, the role of mitochondria in carcinogenesis has been largely ignored for many years because of the “Warburg hypothesis.”
Warburg’s hypothesis, also known as “aerobic glycolysis,” postulates that tumor cells shut down their oxidative mitochondrial metabolism and depend only on glycolysis, even under oxygen-rich conditions.Citation2 This hypothesis discounts the fact that glycolysis produces a very low-yield of ATP, while cancer cells have high-energy demands for ATP. This implies that cancer cells cannot really depend solely on the glycolytic pathway to produce their energy. Utilizing the end products of glycolysis via oxidative mitochondrial metabolism [the TCA/citric acid cycle and oxidative phosphorylation (OXPHOS)] would be a much more efficient method of providing energy to cancer cells, which are in desperate need of ATP molecules.
Recently, we have directly shown that that oxidative mitochondrial activity is fact induced in cancer cells when they are co-cultured with fibroblasts.Citation3-Citation5 As a consequence, we have proposed a new hypothesis, termed “two-compartment tumor metabolism,” to resolve the Warburg paradox.Citation3-Citation5 Our previous studies also indicated that cancer cells are able to recruit stromal cells by releasing reactive oxygen species (ROS) into their local microenvironment. As a result, cancer-associated fibroblasts (CAFs) activate autophagic signaling pathways in order to recycle their proteins and organelles, such as mitochondria (mitophagy). Without functional mitochondria, CAFs become glycolytic and secrete L-lactate and ketone bodies, which are then taken-up by nearby cancer cells.Citation6 Subsequently, these energy-rich nutrients are “burned” via oxidative mitochondrial metabolism in the cancer cells to produce energy and anabolic products.
MitoNEET is an integral membrane protein component of the outer mitochondrial membrane that was found to be co-localized with pioglitazone.Citation7 It was named based on its mitochondrial localization and the presence of a unique amino acid sequence Asn-Glu-Glu-Thr (NEET) at its C terminus.Citation7 Recently, mitoNEET has been demonstrated to be a regulator of mitochondrial respiration.Citation8 Wiley et al. showed that mitochondria isolated from MitoNEET-null murine hearts displayed lower oxidative capacity.Citation8
Furthermore, members of the peroxisome proliferator-activated receptor-γ coactivators-1 (PGC-1) family play a central role in cellular and systemic metabolism.Citation9,Citation10 The family consists of PGC-1α, PGC-1β and the PGC-1-related co-activator (PRC). All three proteins share a unique co-activator LXXLL motif.Citation10,Citation11 Their role in mitochondrial biogenesis is via the regulation of nuclear respiratory factor-1 (NRF-1), NRF-2 and ERRα (estrogen-related receptor α). PGC-1α and PGC-1β proteins share high similarity in structure and function,Citation11 although PGC-1β demonstrates a higher coupling respiration due to differences in proton leakage.Citation11
Another important molecule for mitochondrial function and biogenesis is mitochondrial RNA polymerase, POLMRT.Citation12 POLRMT is responsible for the transcription of the 13 subunits of the OXPHOS complexes and acts as a RNA primase for mitochondrial DNA replication.Citation13,Citation14 POLRMT requires transcription factor B2M (TFB2M) and transcription factor AM (TFAM) for mitochondrial RNA transcription.Citation14
Here, we examined the role of mitochondrial biogenesis and function in breast cancer tumor growth. Using a preclinical animal model, we determined how the overexpression of mitoNEET, PGC-1α, PGC-1β and POLMRT in epithelial breast cancer cells (MDA-MB-231) affects tumor growth. Remarkably, overexpression of these key molecules that drive mitochondrial biogenesis promoted tumor growth in nude mice. However, this increased growth was clearly independent of tumor vascularization. Our results demonstrate that mitochondrial biogenesis in epithelial cancer cells promotes breast cancer tumor growth, likely via increased energy production and nutrient reserves, which confers autophagy resistance.
Thus, we have identified oxidative mitochondrial metabolism as a new “druggable” therapeutic target for the development of novel anticancer agents.
In addition, our findings directly support clinical studies demonstrating that metformin (a mitochondrial “poison” and complex I inhibitor) prevents the onset of nearly all types of human cancers in diabetic patients (reviewed within ref. Citation15). As a consequence, metformin, and other mitochondrial inhibitors, should be used to target mitochondrial metabolism in epithelial cancer cells.
Results
Mitochondrial OXPHOS is increased in breast cancer cells during overexpression of PGC-1α and mitoNEET
Recently, we have suggested that increased mitochondrial function in cancer cells may directly promote tumor growth.Citation3 Here, to test this hypothesis more directly, we stably overexpressed PGC-1α and MitoNEET in MDA-MB-231 cells, a human triple-negative breast cancer cell line. Then, immunoblot analysis was used to confirm the successful overexpression of both proteins ().
Figure 1. Overexpression of PGC-1α and MitoNEET induces the expression of mitochondrial OXPHOS complexes. (A) Using a lentiviral vector, we established MDA-MB-231 cell lines that stably overexpress MitoNEET, as seen by immunoblotting. β-actin is shown as a control for equal protein loading. (B) PGC-1α and MitoNEET induce expression of mitochondrial OXPHOS complexes. Both PGC-1α and mitoNEET-overexpressing MDA-MD-231 cells displayed elevated PGC-1α expression. Most importantly, they also showed an elevation of mitochondrial complexes (OXPHOS). β-actin is shown as a control for equal protein loading. EV, empty vector (Lv105).
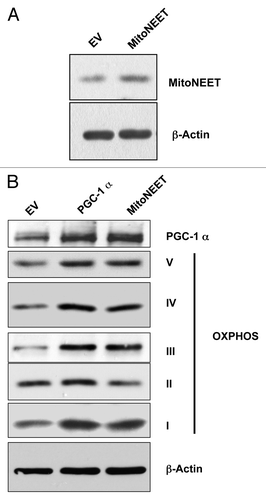
PGC-1α is a master transcription factor that stimulates mitochondrial biogenesis and respiratory function.Citation8 Moreover, mitoNEET, a mitochondrial outer membrane protein, regulates mitochondrial oxidative phosphorylation.Citation8 Interestingly, mitoNEET overexpressing MDA-MB-231 cells displayed elevated PGC-1α expression. As expected, immunoblotting against various OXPHOS components showed that these mitochondrial complexes (I, III, IV and V) were increased in both PGC-1α and MitoNEET-overexpressing MDA-MB-231 cells ().
PGC-1α and mitoNEET confer autophagy resistance in breast cancer cells
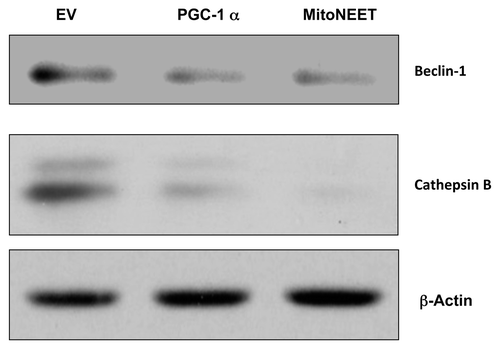
Next, we wanted to test if PGC-1α and mitoNEET will reduce autophagy in cancer cells, as they do in skeletal muscles.Citation16 Interestingly, PGC-1α and mitoNEET both protected cancer cells from autophagy, as Beclin-1 and Cathepsin B (markers of autophagy) were not upregulated under conditions of starvation ().
Figure 2. PGC-1α and MitoNEET protect cancer cells against autophagy. Under conditions of starvation, PGC-1α and MitoNEET-overexpressing MDA-MB-231 cells fail to upregulate autophagy markers, such as Beclin-1 and Cathepsin B. EV, empty vector (Lv105).
PGC-1 α and mitoNEET increase tumor growth in vivo, independently of angiogenesis
Traditionally, altered mitochondrial metabolism was considered as a secondary effect of proliferation. Here, we wanted to test if increased mitochondrial biogenesis and OXPHOS could directly promote tumor growth. PGC-1α and mitoNEET-overexpressing MDA cells were injected in nude mice. Tumor volumes were then measured at various time points post-injection.
Tumors in which PGC-1α and mitoNEET were overexpressed were significantly larger, as compared with control tumors (). At 3 wk post-injection, PGC-1α tumor volumes and weights were on average approximately 3-fold larger in than control tumors, which harbored the expression vector alone. Moreover, MitoNEET tumor volumes and weights were 2.9- and 2.25-fold larger than control tumors, respectively ().
Figure 3. PGC-1α and MitoNEET overexpression increases tumor growth. PGC-1α and MitoNEET overexpressing MDA-MB-231 cells were subcutaneously injected into the flanks of nude mice. (A) Tumor volumes were measured on days 14, 19 and 21. Note that PGC-1α and MitoNEET tumors demonstrate a trend of significantly larger volumes, as compared with control tumors. (B) Tumors were excised, weighed, and their volumes were measured. PGC-1α tumor volumes and weights were 3- and 2.7-fold larger, as compared with control tumors, respectively. Also, MitoNEET tumor volumes and weights were 2.9- and 2.25-fold larger, as compared with control tumors, respectively. (C) Tumor angiogenesis and vessel quantification (number of vessels per field). Frozen sections were cut and immunostained with anti-CD31 antibodies. Quantification of CD31-positive vessels shows a non-significant reduction in angiogenesis in PGC-1α tumors, while MitoNEET tumors displayed a significant reduction in CD31-positive vessels. These results indicate that PGC-1α and MitoNEET stimulate tumor growth, but independently of neo-angiogenesis. EV, empty vector (Lv105).
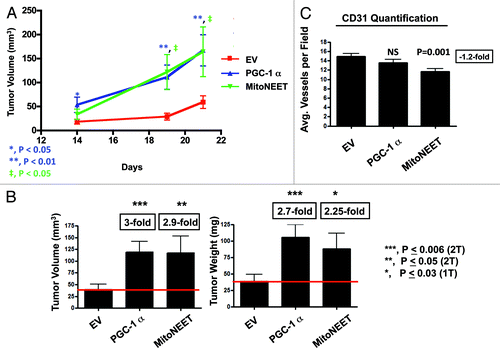
To examine the possibility that the observed increase in tumor growth was due to increased angiogenesis, we immunostained tumor sections with CD31, a well-established marker used to quantitate vessel density. Interestingly, PGC-1α tumors did not show any significant differences in vessel number, while mitoNEET tumors displayed decreased tumor vascularization (). These results imply that the increased tumor growth caused by PGC-1α and/or mitoNEET overexpression is independent of tumor vascularization.
PGC-1β and POLRMT also promote breast cancer tumor growth
Given the growth-promoting effects of PGC-1α and mitoNEET, we decided to also study the functional effects of PGC-1β and POLRMT, a mitochondrial RNA polymerase,Citation17 on breast cancer tumor growth.
Using lentiviral vectors, MDA-MB-231 cells were stably transduced to overexpress either PGC-1β or POLRMT. Successful overexpression was evident by immunoblotting for total PGC-1β or POLRMT (). To test if overexpression of PGC-1β or POLRMT increases tumor growth, we subcutaneously injected PGC-1β or POLRMT-overexpressing MDA-MB-231 cells into the flanks of nude mice. The tumor growth curves for PGC-1β and POLRMT show a significant increase in tumor size (). At 3 wk post-injection, PGC-1β and POLRMT tumor volumes were 2-fold larger than control tumors, which harbored the vector alone ().
Figure 4. Overexpression of PGC-1β and POLRMT. Immunoblot analysis shows successful overexpression of PGC-1β and POLRMT in transduced MDA-MB-231 cells. β-actin is shown as a control for equal protein loading. EV, empty vector (Lv105).
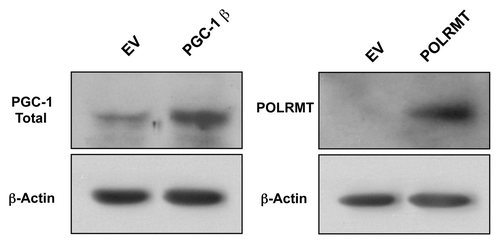
Figure 5. PGC-1β and POLRMT increase tumor growth in a pre-clinical animal model. PGC-1β- and POLRMT-overexpressing MDA-MB-231 cells were injected into the flanks of nude mice. (A) Tumor growth curve. Tumor volumes were measured on days 11, 14, 18 and 20. Note that PGC-1β and POLRMT tumors are significantly larger in volume vs. control tumors. (B) Excised tumor volumes. PGC-1β and POLRMT tumors were 2.2- and 2-fold larger than control tumors. (C) Tumor angiogenesis and vessel quantification (number of vessels per field). PGC-1 β and POLRMT tumors displayed a significant reduction in tumor vascularization, showing that PGC-1β and POLRMT increase tumor growth, independently of angiogenesis. EV, empty vector (Lv105).
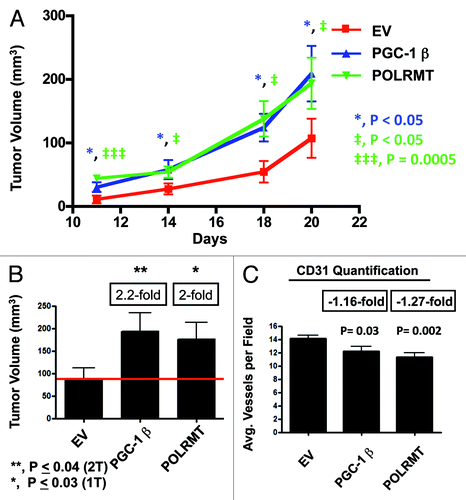
Finally, we examined if this observed increase in tumor growth is related to increased tumor angiogenesis. In fact, these tissue sections stained for CD31 displayed a significant decrease in vascularization in PGC-1β and POLRMT tumors (). Thus, these data show that enhanced tumor growth induced by PGC-1β and POLRMT is not due to increases in neo-vascularization.
Discussion
Traditionally, the “Warburg effect” has been used to describe cancer metabolism, whereby cancer cells become more dependent on glycolysis and less dependent on OXPHOS to produce their ATP.Citation2 However, this theory does not resolve the issue that cancer cells could not possibly depend solely on glycolysis, which has an extremely low yield of ATP.
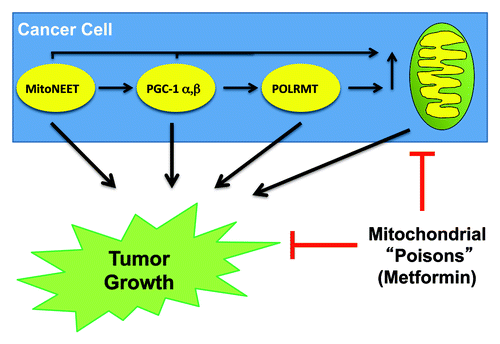
Recently, we and other researchers have highlighted the crucial role of mitochondrial metabolism in tumorgenesis.Citation18-Citation21 In this study, we demonstrate the effect of elevated mitochondrial biogenesis and metabolism on tumor growth, using pre-clinical animal models. This was achieved by stably transducing MDA-MB-231 breast cancer cells to overexpress several positive regulators of mitochondrial biogenesis, including PGC-1α, PGC-1β, mitoNEET and POLRMT. Interestingly, PGC-1α and mitoNEET induced mitochondrial OXPHOS complexes and protected cancer cells from autophagy. Moreover, PGC-1α, PGC-1β, mitoNEET and POLRMT overexpression all significantly increased tumor growth in nude mouse xenografts ().
Figure 6. Mitochondrial biogenesis increases tumor growth. Schematic diagram highlighting that MitoNEET, PGC-1 (α- and β-isoforms) and POLRMT all drive mitochondrial biogenesis, and effectively promote tumor growth, when expressed in cancer cells. Thus, the therapeutic use of mitochondrial inhibitors or “poisons” (such as metformin) will inhibit tumor growth. Importantly, numerous clinical studies have now shown that metformin (a mitochondrial compex I inhibitor) prevents the onset of nearly all types of human cancers in diabetic patients.
PGC-1α and PGC-1β are transcriptional co-activators that have a central role in maintaining metabolic pathways, such as oxidative metabolism, energy homeostasis and glucose and lipid metabolism in normal cells.Citation22 Recently, PGC-1β knockdown in tamoxifen-resistant MCF-7 cells resulted in a cell line that was sensitive to tamoxifen-treatment.Citation23 Similarly, Shiota et al. showed that silencing of PGC-1α inhibited LNCaP prostate cancer cell proliferation.Citation24
It is worth pointing out that PGC-1 family members induce superoxide dismutase (SOD), catalase (CAT) and glutathione-peroxidase (GPX), all critical protein antioxidants.Citation25,Citation26 This action protects cancer cells against elevated mitochondrial ROS. Alternatively, PGC-1α has been shown to inhibit ROS by inducing uncoupling proteins, which leak protons across the inner membrane of mitochondria to lower their membrane potential, eventually decreasing ROS produced by the mitochondria. Interestingly, this antioxidant role can protect cancer cells from self-released ROS, allowing them to activate neighboring CAFs via oxidative stress.
Interestingly, type 2 diabetic patients suffer from oxidative stress due to reduced mitochondrial mass and oxidative capacity in skeletal muscle.Citation27-Citation29 Pioglitazone, a thiazolidinedione member that is used as an insulin-sensitizing drug for type 2 diabetes, improves mitochondrial respiration in type 2 diabetic patients.Citation29 Classically, thiazolidinedione family members are thought to be agonists for peroxisomal proliferator-activated receptor (PPAR)-γ expression, which, in turn, induces the expression of PGC-1α.Citation30
Recently, pioglitazone was found to co-localize with an iron-containing outer mitochondrial membrane protein, mitoNEET.Citation7 It is possible that pioglitazone induces mitochondrial respiration through mitoNEET binding, as mitoNEET-null mice demonstrated lower mitochondrial capacity.Citation8,Citation29 Accordingly, our results demonstrate that mitoNEET-overexpressing cancer cells exhibited an increase in mitochondrial complexes I, III, IV, V and a decrease in autophagy. Strikingly, mitoNEET tumors were ~3-fold larger than control tumors. Thus, we established a novel role for mitoNEET in promoting tumor growth. These results provide evidence that mitochondrial metabolism and OXPHOS are driving factors for cancer growth and are not a side effect of increased cellular proliferation.
Moreover, POLMRT is a mitochondrial bacteriophage-related RNA polymerase that is needed for the transcription and replication of mtDNA.Citation31,Citation32 POLRMT is responsible for transcription of several OXPHOS subunits. Interestingly, nuclear factor NRF-1, which is regulated by PGC-1 family members, stimulates POLRMT to increase mitochondrial replication.Citation14 Moreover, POLRMT depletion affects mtDNA replication.Citation32 Our results demonstrate that POLRMT-overexpressing cancer cells drive a 2-fold increase in tumor growth in nude mice. This growth was independent of tumor angiogenesis. Thus, our current studies are consistent with a new role for POLRMT as a tumor promoter.
Finally, we observed that overexpression of PGC-1α and mitoNEET, which induce mitochondrial biogenesis, conferred autophagy resistance under conditions of starvation. In addition, overexpression of PGC-1α and mitoNEET increased tumor growth by ~3-fold. Similarly, loss of Beclin-1, a required autophagy gene and tumor suppressor, is known to confer autophagy resistance in cancer cells and increase tumor growth.Citation33-Citation35 Thus, autophagy resistance in cancer cells—mediated by either (1) increased mitochondrial power or (2) genetic loss of required autophagy genes—has the same net effect, to increase tumor growth rates.
In conclusion, we studied several molecules that induce mitochondrial biogenesis in cancer cells. We have shown that all four of these molecules promote tumor growth. Thus, we propose that increased mitochondrial biogenesis functions as an “oncogene” or tumor promoter in cancer cells. Induced oxidative mitochondrial metabolism would allow cancer cells to more efficiently utilize imported L-lactate, ketone bodies, glutamine and fatty acids, which are all produced by neighboring activated CAFs or related stromal cells. Thus, targeting cancer cell mitochondria with mitochondrial “poisons” could halt this metabolic coupling, effectively shutting-down the “engine” or “powerhouse” driving tumor growth.
Materials and Methods
Materials
Antibodies were all purchased from commercial sources: anti-PGC-1α (NBP-1–04676, Novus), anti-mitoNEET (ab118027, Abcam), anti-PGC-1 total (ab72230, Abcam), anti-POLRMT (HPA006366, Sigma), anti-OXPHOS (MS601, Mitoscience), anti-β actin (A5441, Sigma), anti-Beclin-1 (NBP1–00085, Novus) and anti-Cathepsin B (FL-339, Santa Cruz Biotechnology).
Cell culture and transfection
MDA-MB-231 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum in a 37°C humidified atmosphere containing 5% CO2, unless otherwise noted. Lentiviral particles were produced after 48 h of transfecting the GeneCopoeia 293Ta lentiviral packaging cell line with lentiviral plasmids (EX-NEG-Lv105) (empty vector), PGC-1α (EX-U0564-Lv105), MitoNEET (EX-V0831-Lv105), PGC-1β (EX-Mm13777-Lv105) and POLRMT (EX-Z7565-Lv105) (all obtained from GeneCopoeia, Inc.) using the Lenti-Pac HIV Expression Packing Kit (GeneCopoeia, Inc.) according to manufacturer’s recommendation. MDA-MB-231 breast cancer cells were transduced with lentivirual particles in the presence of 5 μg/ml of Polybrene (Santa Cruz Biotech). Successfully transduced cells were selected with 2 μg/ml of puromycin.
Immunoblotting
Cells were lysed by scraping into cell lysis buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100 and 60 mM n-octyl-glucoside), containing protease inhibitors (Boehringer Mannheim). To induce autophagy, cells were starved for 5 h using Hank's Balanced Salt Solution (HBSS) supplemented with 40 mM HEPES. Samples were incubated on a rotating platform at 4°C and were then centrifuged at 12,000 × g for 10 min (at 4°C) to remove insoluble debris. Protein concentrations were analyzed using the BCA reagent (Pierce). Samples were then separated by SDS-PAGE (12% acrylamide) and transferred to nitrocellulose. All subsequent wash buffers contained 10 mM Tris, pH 8.0, 150 mM NaCl, 0.1% Tween 20, which was supplemented with 5% nonfat dry milk (Carnation) for the blocking solution and 1% bovine serum albumin (Sigma) for the antibody diluent. Horseradish peroxidase-conjugated secondary antibodies were used to visualize bound primary antibodies with an ECL detection kit (Pierce).
Animal studies
All animals studied were maintained in a pathogen-free environment/barrier facility at the Kimmel Cancer Center at Thomas Jefferson University under National Institutes of Health (NIH) guidelines. All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC). Tumor cells (MDA-MB-231; 1 × 106 cells) in 100 μl of sterile PBS were injected into the flanks of athymic NCr nude mice (NCRNU; Taconic Farms; 6–8 wk of age). Mice were then sacrificed at 21 d post-injection; tumors were excised to determine their weights and volumes.
Angiogenesis quantification
Frozen sections were processed, as we described previously.Citation36 Then the anti-CD31 antibody was incubated overnight at 4°C. Finally, the sections were counter-stained with hematoxylin for 5–10 sec, air-dried and mounted with cover-slips. To quantify tumor angiogenesis, CD31-positive vessels were enumerated in 8–10 fields within the central area of each tumor using a 20X objective lens and an ocular grid (0.25 mm2 per field). The total numbers of vessel per unit area was calculated using ImageJ, and the data was represented graphically.
Statistical analysis
Statistical significance was examined by the Student’s t-test. Values of p ≤ 0.05 were considered significant. All p values were two-tailed (2T) unless otherwise noted.
Acknowledgments
F.S. was the recipient of a Young Investigator Award from the Breast Cancer Alliance. Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the U.K. and an Advanced ERC Grant from the European Research Council.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21:297 - 308; http://dx.doi.org/10.1016/j.ccr.2012.02.014; PMID: 22439925
- Warburg O. On the origin of cancer cells. Science 1956; 123:309 - 14; http://dx.doi.org/10.1126/science.123.3191.309; PMID: 13298683
- Salem AF, Whitaker-Menezes D, Lin Z, Martinez-Outschoorn UE, Tanowitz HB, Al-Zoubi MS, et al. Two-compartment tumor metabolism: autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle 2012; 11:2545 - 56; http://dx.doi.org/10.4161/cc.20920; PMID: 22722266
- Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab 2012; 15:4 - 5; http://dx.doi.org/10.1016/j.cmet.2011.12.011; PMID: 22225869
- Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011; 10:1772 - 83; http://dx.doi.org/10.4161/cc.10.11.15659; PMID: 21558814
- Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011; 10:1772 - 83; http://dx.doi.org/10.4161/cc.10.11.15659; PMID: 21558814
- Colca JR, McDonald WG, Waldon DJ, Leone JW, Lull JM, Bannow CA, et al. Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am J Physiol Endocrinol Metab 2004; 286:E252 - 60; http://dx.doi.org/10.1152/ajpendo.00424.2003; PMID: 14570702
- Wiley SE, Murphy AN, Ross SA, van der Geer P, Dixon JE. MitoNEET is an iron-containing outer mitochondrial membrane protein that regulates oxidative capacity. Proc Natl Acad Sci USA 2007; 104:5318 - 23; http://dx.doi.org/10.1073/pnas.0701078104; PMID: 17376863
- Liu C, Lin JD. PGC-1 coactivators in the control of energy metabolism. Acta Biochim Biophys Sin (Shanghai) 2011; 43:248 - 57; http://dx.doi.org/10.1093/abbs/gmr007; PMID: 21325336
- Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 2011; 1813:1269 - 78; http://dx.doi.org/10.1016/j.bbamcr.2010.09.019; PMID: 20933024
- Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta ), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem 2002; 277:1645 - 8; http://dx.doi.org/10.1074/jbc.C100631200; PMID: 11733490
- Arnold JJ, Smidansky ED, Moustafa IM, Cameron CE. Human mitochondrial RNA polymerase: structure-function, mechanism and inhibition. Biochim Biophys Acta 2012; 1819:948 - 60; http://dx.doi.org/10.1016/j.bbagrm.2012.04.002; PMID: 22551784
- Wanrooij S, Falkenberg M. The human mitochondrial replication fork in health and disease. Biochim Biophys Acta 2010; 1797:1378 - 88; http://dx.doi.org/10.1016/j.bbabio.2010.04.015; PMID: 20417176
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 2008; 88:611 - 38; http://dx.doi.org/10.1152/physrev.00025.2007; PMID: 18391175
- Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011; 10:4047 - 64; http://dx.doi.org/10.4161/cc.10.23.18151; PMID: 22134189
- Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci USA 2009; 106:20405 - 10; http://dx.doi.org/10.1073/pnas.0911570106; PMID: 19918075
- Asin-Cayuela J, Gustafsson CM. Mitochondrial transcription and its regulation in mammalian cells. Trends Biochem Sci 2007; 32:111 - 7; http://dx.doi.org/10.1016/j.tibs.2007.01.003; PMID: 17291767
- Salem AF, Whitaker-Menezes D, Lin Z, Martinez-Outschoorn UE, Tanowitz HB, Al-Zoubi MS, et al. Two-compartment tumor metabolism: autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle 2012; 11:2545 - 56; http://dx.doi.org/10.4161/cc.20920; PMID: 22722266
- Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab 2012; 15:4 - 5; http://dx.doi.org/10.1016/j.cmet.2011.12.011; PMID: 22225869
- Fogal V, Richardson AD, Karmali PP, Scheffler IE, Smith JW, Ruoslahti E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol Cell Biol 2010; 30:1303 - 18; http://dx.doi.org/10.1128/MCB.01101-09; PMID: 20100866
- Funes JM, Quintero M, Henderson S, Martinez D, Qureshi U, Westwood C, et al. Transformation of human mesenchymal stem cells increases their dependency on oxidative phosphorylation for energy production. Proc Natl Acad Sci USA 2007; 104:6223 - 8; http://dx.doi.org/10.1073/pnas.0700690104; PMID: 17384149
- Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev 2006; 27:728 - 35; PMID: 17018837
- Deblois G, Chahrour G, Perry MC, Sylvain-Drolet G, Muller WJ, Giguère V. Transcriptional control of the ERBB2 amplicon by ERRalpha and PGC-1beta promotes mammary gland tumorigenesis. Cancer Res 2010; 70:10277 - 87; http://dx.doi.org/10.1158/0008-5472.CAN-10-2840; PMID: 20961995
- Shiota M, Yokomizo A, Tada Y, Inokuchi J, Tatsugami K, Kuroiwa K, et al. Peroxisome proliferator-activated receptor gamma coactivator-1alpha interacts with the androgen receptor (AR) and promotes prostate cancer cell growth by activating the AR. Mol Endocrinol 2010; 24:114 - 27; http://dx.doi.org/10.1210/me.2009-0302; PMID: 19884383
- Chen SD, Yang DI, Lin TK, Shaw FZ, Liou CW, Chuang YC. Roles of Oxidative Stress, Apoptosis, PGC-1α and Mitochondrial Biogenesis in Cerebral Ischemia. Int J Mol Sci 2011; 12:7199 - 215; http://dx.doi.org/10.3390/ijms12107199; PMID: 22072942
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006; 127:397 - 408; http://dx.doi.org/10.1016/j.cell.2006.09.024; PMID: 17055439
- Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005; 54:8 - 14; http://dx.doi.org/10.2337/diabetes.54.1.8; PMID: 15616005
- Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002; 51:2944 - 50; http://dx.doi.org/10.2337/diabetes.51.10.2944; PMID: 12351431
- Rabøl R, Boushel R, Almdal T, Hansen CN, Ploug T, Haugaard SB, et al. Opposite effects of pioglitazone and rosiglitazone on mitochondrial respiration in skeletal muscle of patients with type 2 diabetes. Diabetes Obes Metab 2010; 12:806 - 14; http://dx.doi.org/10.1111/j.1463-1326.2010.01237.x; PMID: 20649633
- Mensink M, Hesselink MK, Russell AP, Schaart G, Sels JP, Schrauwen P. Improved skeletal muscle oxidative enzyme activity and restoration of PGC-1 alpha and PPAR beta/delta gene expression upon rosiglitazone treatment in obese patients with type 2 diabetes mellitus. Int J Obes (Lond) 2007; 31:1302 - 10; http://dx.doi.org/10.1038/sj.ijo.0803567; PMID: 17310221
- Arnold JJ, Smidansky ED, Moustafa IM, Cameron CE. Human mitochondrial RNA polymerase: structure-function, mechanism and inhibition. Biochim Biophys Acta 2012; 1819:948 - 60; http://dx.doi.org/10.1016/j.bbagrm.2012.04.002; PMID: 22551784
- Fusté JM, Wanrooij S, Jemt E, Granycome CE, Cluett TJ, Shi Y, et al. Mitochondrial RNA polymerase is needed for activation of the origin of light-strand DNA replication. Mol Cell 2010; 37:67 - 78; http://dx.doi.org/10.1016/j.molcel.2009.12.021; PMID: 20129056
- Li Z, Chen B, Wu Y, Jin F, Xia Y, Liu X. Genetic and epigenetic silencing of the beclin 1 gene in sporadic breast tumors. BMC Cancer 2010; 10:98; http://dx.doi.org/10.1186/1471-2407-10-98; PMID: 20230646
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672 - 6; http://dx.doi.org/10.1038/45257; PMID: 10604474
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 2003; 100:15077 - 82; http://dx.doi.org/10.1073/pnas.2436255100; PMID: 14657337
- Bonuccelli G, Casimiro MC, Sotgia F, Wang C, Liu M, Katiyar S, et al. Caveolin-1 (P132L), a common breast cancer mutation, confers mammary cell invasiveness and defines a novel stem cell/metastasis-associated gene signature. Am J Pathol 2009; 174:1650 - 62; http://dx.doi.org/10.2353/ajpath.2009.080648; PMID: 19395651