Abstract
The majority of human tumors express mutant forms of p53 at high levels, promoting gain of oncogenic functions and correlating with disease progression, resistance to therapy and unfavorable prognosis. p53 mutant accumulation in tumors is attributed to the ability to evade degradation by the proteasome, the only currently recognized machinery for p53 disruption. We report here that glucose restriction (GR) induces p53 mutant deacetylation, routing it for degradation via autophagy. Depletion of p53 leads, in turn, to robust autophagic activation and to cell death, while expression of degradation-defective mutant p53 blocks autophagy and enables survival to GR. Furthermore, we found that a carbohydrate-free dietetic regimen that lowers the fasting glucose levels blunts p53 mutant expression and oncogenic activity relative to a normal diet in several animal model systems. These findings indicate that the stability of mutant forms of p53 is influenced by the levels of glucose and by dietetic habits. They also unravel the existence of an inhibitory loop between autophagy and mutant p53 that can be exploited therapeutically.
Introduction
One of the most important driving forces for malignant transformation of epithelial tissues consists in the elimination of the activity of the p53 tumor suppressor via missense mutations of the gene. It is now widely accepted that p53 mutants acquire novel oncogenic functions (GOF) relative to the wild-type protein.Citation1,Citation2 This gain of activity was first appreciated in knock-in mice, where tumor-derived p53 mutant , equivalent to the human R175H and R273H, replaced one or both of the endogenous p53 alleles, leading to a change of the tumor spectrum compared with a p53-null background.Citation3,Citation4 An important addition to the GOF hypothesis came from subsequent evidence demonstrating the importance of p53 mutant stabilization for tumor progression. In its wild-type conformation, p53 is expressed at low levels due to proteasome- and ubiquitin-dependent degradation, which is, in turn, controlled by the E3-ubiquitin ligase MDM2 and by other ubiquitin-conjugating enzymes.Citation2,Citation5,Citation6 The majority of established tumors express mutant p53 at high levels, due to their ability to evade proteolysis. This phenomenon has been attributed to lack of induction of MDM2 transcription, to altered interaction with MDM2 and to the activity of chaperones. However, in knock-in animal models, p53 mutant levels are low in most normal tissues and also in some tumors, unless the dosage of the MDM2 gene is reduced.Citation5 In these conditions, p53 mutants accumulate, correlating with an accelerated onset of tumors and with the appearance of metastatic behavior, which is otherwise rarely seen in a p53-null background. In human tumors the presence of high expression levels of mutant p53 is a negative prognostic factor predictive of relapse and of poor therapeutic responses.Citation6 Thus, understanding and manipulating the mechanisms involved in p53 mutant destabilization is of the utmost importance for cancer therapy and prevention. The only known pathway for p53 degradation, in either a wild-type or mutant conformation, is the proteasome. We and others have previously shown that in the case of wild-type p53, various post-translational modifications, including acetylation and ubiquitination, interfere with its proteasome-dependent clearance, leading to stabilization.Citation7,Citation8 How post-translational modifications affect the activity of mutant p53 is incompletely defined.
Autophagy plays complex and conflicting activities in cancer.Citation9,Citation10 Autophagy is a degradative process through which damaged organelles and abnormally folded proteins are targeted for disruption via the lysosomes. In tumor cells, autophagy promotes survival by extracting energy during nutritional stress and aids in the elimination of potentially toxic products that are generated as a consequence of high metabolic rates. By virtue of these activities, autophagy is envisioned as necessary for cancer proliferation. However, autophagic activation, if uncontrolled and when proceeding to completion, can also lead to cell death, likely due to degradation of cell constituents and organelles required for cellular homeostasis. Furthermore, inhibition of autophagy enhances the production of radical oxygen species (ROS), induces DNA damage and leads to genomic instability, suggesting that loss of autophagy generates an environment that acts instead in favor of tumor progression.Citation11 Indeed, several lines of evidence indicate that autophagy acts as a tumor barrier. Mono-allelic deletion of the Beclin-1 and of other autophagy genes in mice increases tumor propensity, and these genes are frequently lost in human tumors.Citation11,Citation12 There are also noticeable examples whereby activation of autophagy has synthetic lethal effects that result in cell death in defined tumor types, for example, in renal cancers lacking functional VHL.Citation13 Therefore, the outcome of autophagy is likely dependent upon tumor-specific genetic characteristics and needs to be assessed within the context of specific oncogenic signal pathways. Previous studies showed that in the wild-type form, p53 can either inhibit or activate autophagy, leading to cell death or survival depending upon the type of stress and the cellular context.Citation14-Citation16 One recent work has also shown that the anti-autophagic function might be shared by some of the p53 mutants found in tumors.Citation17 The significance of this inhibitory effect of p53 mutants on autophagy is incompletely defined at this point.
In this study we describe a negative feedback loop between some of the 53 mutations found in human tumors and autophagy, which affects p53 stability and oncogenic capacity. We first demonstrate that the expression of mutant p53 is influenced by the levels of glucose, such that high glucose levels stabilize p53, while glucose restriction leads to its depletion via autophagy by implementing de-acetylation and, likely, ubiquitination. Mutant p53 depletion, in turn, renders cells hypersensitive to autophagic activation leading to cell death. Collectively, our experiments identify a new pathway for degradation of mutant p53 and provide evidence that its accumulation and gain of oncogenic function can be manipulated in vivo with dietetic adjustments. These results might foster the rationalization of dietetic interventions in individuals harboring germ-line (Li-Fraumeni syndrome) or somatic p53 mutations.
Results
Glucose restriction leads to p53 mutant deacetylation and degradation
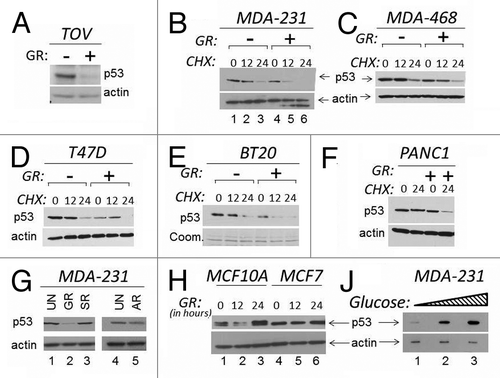
The stability of wild-type p53 is regulated by many signals, such as DNA damage, oxidative or metabolic stress. However, little is known on the pathways that control p53 mutant stability. In normal cells, glucose starvation promotes stabilization of wild-type p53.Citation18 Thus, we asked whether glucose restriction affects p53 mutant levels. In the ovarian cancer cell line TOV, and in breast carcinoma MDA-231 cells that express endogenous p53 mutations at position 175 (p53R175H) and 280 (R280K), respectively, glucose restriction led to depletion of p53 mutant levels (). This p53 loss was observed in the presence of the translation inhibitor cycloheximide (CHX), demonstrating that is due at least in part to degradation (, compare lanes 4–6 with lanes 1–3). To determine whether GR-induced p53 mutant depletion is a generalized phenomenon, we expanded these experiments to various human cancer cell lines of different tissue origin and harboring p53 mutations at various positions. As established with CHX chase experiments, GR shortened the half-life of p53 in breast cancer cells MDA-468 (R273H) (), T47D (L194F) (), BT20 (K132Q) () and in pancreatic cancer cells PANC1 (R280T) (). Thus, GR induces p53 mutant depletion in several cell lines, although we cannot exclude that cells resistant to GR-dependent depletion also exist. We then asked whether p53 mutant degradation occurs in other conditions of metabolic stress. In MDA-231 cells, neither serum nor amino acid starvation affected p53 mutant levels (, compare lanes 3 and 5 with lane 2). By contrast, the expression of wild-type p53 in MCF7 or in MFC10A cells was not significantly affected by GR (). To then determine whether changes in the concentration of glucose influence p53 mutant levels, cells were incubated in 0 mM, 5 mM or 25 mM glucose. It is noteworthy that 5 mM glucose is the physiological concentration inside the cells, while levels above 10 mM reflect a pre-diabetic or diabetic condition within the cell culture system. We observed that progressively higher glucose levels were mirrored by an increase in mutant p53 expression (), indicating that the glucose concentration is an important determinant of its stability.
Figure 1. The levels of glucose affect mutant p53 expression. (A and B). TOV or MDA-231 cells were grown in regular media or in media lacking glucose (indicated as GR, - or +). In the experiment shown in (A), cells were harvested 16 h after GR, and cell extracts were subjected to immunoblot with anti-p53 or anti-actin antibodies. In (B), a time course in the presence of the translation inhibitor cycloheximide (CHX) is shown. Untreated (lanes 1–3) or GR-treated (lanes 4–6) cells were incubated with 40 μM CHX for the time indicated at the top of the panel (in hours). (C–F) The expression levels of p53 were assessed with experiments similar to those shown in (B) in the cell lines indicated at the top of each panel in the presence of GR and of CHX. All panels show p53 and actin levels. (E) shows Coomassie staining of the membrane (abbreviated Coom.). (G) MDA-231 were grown in regular media (25 mM, lanes 1 and 4); or in GR media (lane 2); or in media containing glucose (25 mM) but lacking serum (SR, lane 3); or lacking amino acids (AR, lane 5). (H) MCF10A (lanes 1–3) or MCF7 (lanes 4–6) cells were grown in regular media or in GR media for the time indicated at the top of the panel (in hours). (J) MDA-231 cells were incubated in 0 mM (lane 1), 5 mM (lane 2), 25 mM (lane 3) glucose for 12 h, followed by assessment of p53 and actin levels.
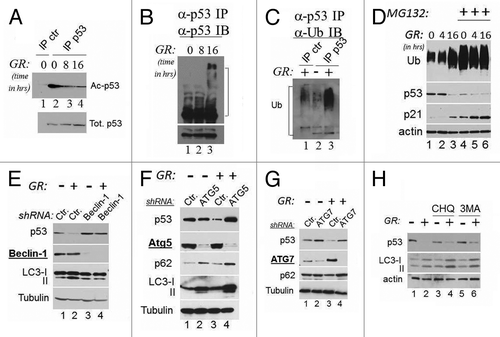
Depletion of glucose reduces the intracellular pools of acetyl-coenzyme A, leading to changes in the acetylation status of several proteins.Citation19 In the case of wild-type p53, acetylation competes with ubiquitination leading to stabilization.Citation7 Therefore, we next investigated whether GR affects mutant p53 acetylation and ubiquitination status. We found that in both TOV (not shown) and MDA-231 cells, GR induced rapid p53 deacetylation (), which coincided with an increase in ubiquitination, as demonstrated by the appearance of high molecular weight isoforms that reacted with antibodies directed against p53 or ubiquitin (). These findings suggested that glucose-induced deacetylation and ubiquitination are a pre-requisite for mutant p53 degradation.
Figure 2. Mutant p53 is deacetylated during GR, and its degradation is regulated by autophagy. (A) Cell extracts derived from MDA-231 cells grown in the presence (lanes 1 and 2) or absence of glucose for 8 h (lane 3) or for 16 h (lane 4) were first equalized for p53 content with a first immunoblot of total p53, then were immunoprecipitated with a mixture of anti-acetyl-p53 antibody (K373 and K320, lanes 1–3), followed by immunoblot with the monoclonal anti-p53 antibody. Lane 1 shows a control immunoprecipitation reaction from untreated cell extracts. The lower panel shows the total p53 inputs used for the immunoprecipitation reactions. (B) Cell extracts as in (A) were immunoprecipitated with agarose-conjugated anti-p53 monoclonal antibody (DO1). The products of these immunoprecipitation reactions were then probed with an anti-goat anti-p53 polyclonal antibody. The lower panel shows a short exposure of the anti-p53 immunoblot. In (C), extracts derived from untreated (lane 2) or GR-treated cells (lanes 1 and 3) were immunoprecipitated with agarose-conjugated anti-p53 antibody (lane 2 and 3), or with a control isotope-matched antibody (lane 1, indicated ctr. IP) and probed in immunoblot with anti-ubiquitin antibodies. Brackets in (B and C) indicate the position of high molecular weight isoforms that react with both anti-p53 and anti-ubiquitin antibodies. (D) MDA-231 cells were grown in regular or GR media for the indicated time, and in the absence (lanes 1–3) or presence (lanes 4–6) of 20 μM MG132 for 16 h. Immunoblots with anti-p53, anti-ubiquitin (Ub), anti-actin or anti-p21 antibodies are shown. (E) MDA-231 cells were transfected with 50 nM of control siRNA (lanes 1 and 2) or with 50 nM of the siRNA specific for Beclin-1 (lane 3 and 4) in the presence (lanes 1 and 3) or absence (lanes 2 and 4) of glucose. Cell extracts were probed with the antibodies indicated at the left of each panel. The two LC3 isoforms, microtubule- (LC3-I; 18 kDa) and autophagosome associated (LC3-II, 16 kDa), are indicated. (F–G) Untreated (lanes 1 and 2 of each panel) or GR-treated (lanes 3 and 4) MDA-231 cells were transfected with control shRNAs (lanes1–3) or with the shRNAs (lanes 2–4, each panel) specific for ATG5 (F) or ATG7 (G). The expression levels of p53, ATG5, ATG7, p62 or LC3 are shown. (H) Cells were cultured in regular media (lanes 1, 3 and 5) or in GR media (lanes 2, 4 and 6) for 16 h. Cells received either chloroquine (20 μM; lanes 3 and 4) or 3MA (5 mM, lanes 5 and 6) for the last 4 h of incubation. p53, LC3 and actin levels were assessed.
GR leads to proteasome-independent but autophagy-dependent degradation of p53
All cells utilize two major systems for protein degradation, the proteasome and autophagy, in turn, consisting of micro-autophagy, chaperone-mediated autophagy and macro-autophagy (from here forth referred to as autophagy). However, the only recognized machinery for degradation of p53 is the proteasome. Therefore, we performed experiments similar to those shown before in the presence of the proteasome inhibitor MG132, with the expectation that if GR routes p53 mutant for disruption by the proteasome, its inhibition should have abolished this effect. To our surprise, MG132 did not rescue p53 degradation (, compare lanes 1–3 with lanes 4–6). Ubiquitinated proteins and p21 accumulated with MG132 treatment, demonstrating that this compound was effective in blocking degradation of proteasomal substrates, yet did not lead to p53 mutant stabilization. Noticeably, the total ubiquitination signal as well as p21 levels increased during GR (, compare lane 1 with lane 3), suggesting that depletion of glucose leads to inhibition of proteasomal activity, in turn, resulting in mutant p53 degradation via another pathway. Thus, we asked whether GR-mediated depletion of p53 is dependent upon autophagy. To monitor autophagy, we assessed the conversion of LC3-I into the lipidated, activated form LC3-II and/or the expression levels of p62, a bona fide autophagic substrate. First, autophagy was induced in GR-treated cells, as demonstrated by increased LC3 conversion and decreased p62 levels (, lane 2 and ). We then studied the effects of the knockdown of various members of the ATG family whose gene products are required for various steps of autophagy. The individual knockdown of ATG6/Beclin-1, of ATG5 or of ATG7, with RNA interference completely rescued p53 mutant loss (). The chemical inhibitors of autophagic/lysosomal degradation chloroquine (CHQ) and 3-methyl-adenine (3MA) also partially prevented GR-induced degradation ().
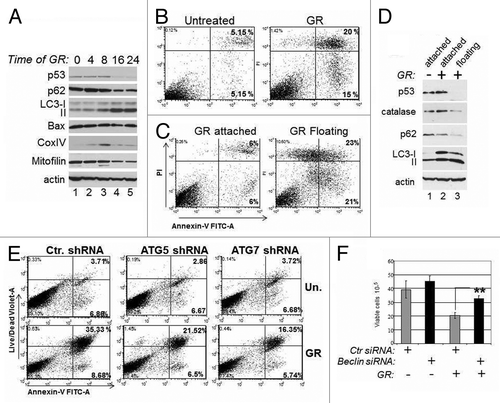
Figure 4. Mutant p53 depletion is accompanied by autophagic cell death. (A) MDA-231 cells were untreated (lane 1) or grown in GR media for the time points indicated at the top of the panel (in hours). At each point, cell extracts were prepared and probed in immunoblot with the indicated antibodies. (B and C). Flow cytometric analysis with Annexin-V vs. propidium iodide (PI) staining of live MDA-231 cells either untreated or glucose-restricted. Cells were analyzed as whole populations (B), or they were separated as attached and floating (C). The percentage of early (right lower panel) and late (right upper panel) apoptotic cells is indicated. (D) Untreated (lane 1) or GR-treated cells for 36 h (lanes 2 and 3) were separated as attached (lane 2) or floating (lane 3) cells. Cell extracts were probed with the indicated antibodies. (E) MDA-231 cells transfected with control shRNA or with the shRNA for ATG5 or ATG7 were selected with puromycin for 1 wk and then left untreated or GR-treated. Flow cytometry was performed as described in (B and C), except the live/dead violet staining was used in place of PI. (F) TOV cells were transfected with control siRNA (gray bars) or with the Beclin siRNA (black bars), as indicated at the bottom of the panel. Thirty-six hours after transfection, cells were subjected to glucose restriction for 8–12 h, then the media was replaced with regular media. Cell viability was assessed 4 d after with trypan blue exclusion. The p values between control and Beclin-siRNA-transfected cells in the GR-treated group are indicated.
Glucose restriction induces the association of mutant p53 with the autophagic machinery and with MDM2
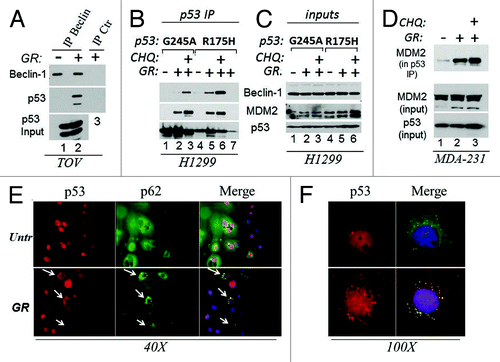
The above results predicted that p53 mutant physically associates with the autophagic apparatus. To test this possibility, we performed immunoprecipitation and immunofluorescence experiments. First, an interaction of p53 with Beclin-1 was detected in cell lysates derived from GR-treated but not from untreated TOV cells (). To rule out cell type-specific effects, we expanded these experiments to various other cell lines including the p53-null, H1299 cells ectopically expressing two different p53 mutants, p53G245A or p53R175H, via a tetracycline inducible system. Beclin-1 was specifically detected in the anti-p53 immunoprecipitation reactions derived from GR-treated cells, and such association was increased by chloroquine, arguing that blocking autophagic flux traps p53 complexes with the autophagic machinery (Fig. Citation2B and C, compare lanes 2–3 and 5–6 with lanes 1 and 4). Consistent with this idea, GR and chloroquine enhanced the co-localization of p53 with cytoplasmic autophagic structures and with lysosomes identified by the presence of p62 and of the lysosomal marker LAMP-1, respectively (; Fig. S1A and B). MDM2 is an important determinant of p53 mutant ubiquitination and stability.Citation2,Citation5,Citation20 Since we found previously that GR induces p53 mutant ubiquitination (), we asked whether GR modifies the affinity of mutant p53 for MDM2. We determined that complex formation between p53 and MDM2 was increased during GR as well as by chloroquine in both H1299 and MDA-231 cells (). Furthermore, treatment with the MDM2/MDMX-inhibitor Nutlin-3a abolished GR-induced p53 mutant loss (Fig. S1C).
Figure 3. GR promotes the interaction of mutant p53 with the autophagic machinery and with MDM2. (A) Cell extracts prepared from untreated or GR-treated TOV cells for 4 h (approximately 5 mg) were immunoprecipitated with the anti-Beclin-1 monoclonal antibody, followed by immunoblot with the anti-p53 (FL393) or anti-Beclin polyclonal (lanes 1 and 2). Similar amounts of cell extracts derived from GR-treated cells were immunoprecipitated with control isotope matched antibody (indicated as IP ctr. lane 3). The bottom panel shows input levels of p53. (B) H1299 cells expressing p53G245A (lanes 1–3) or R175H (lanes 4–7) under the control of a tetracycline inducible promoter were induced with doxycycline for 12 h, then grown in regular media (lanes 1 and 4) or in GR media in the absence (lanes 2, 5 and 7) or presence of chloroquine (CHQ, lanes 3 and 6) for 4 h. Cell extracts (0.5–1 mg) were immunoprecipitated with agarose conjugated anti-p53 antibody, then probed in immunoblot with anti-Beclin-1, anti -MDM2 or anti-p53 antibodies. Lane 7 contains a control immunoprecipitation from p53R175H cell extracts with an isotype matched antibody. A small amount of p53 is detected in this control IP. (C) Input levels of the proteins as in (B) (20 μg). (D) Cell extracts derived from MDA-231 cells untreated (lane 1) or GR-treated (lanes 2 and 3) for 4 h, in the absence (lanes 1 and 2) or presence of CHQ (40 μM, lane 3) were immunoprecipitated with the agarose conjugated FL393 antibody and probed with the MDM2 monoclonal antibody. The panel shows MDM2 in the p53 IP, and input levels of MDM2 or p53. (E) BT20 cells plated on glass coverslips were grown in regular (Unt) or GR media for 16 h, subsequently stained with anti-p53 (red) or anti-p62 (green) antibodies and immunofluorescence images were captured at 40x magnification. The merge images show the red and green channels merged with DAPI. Arrows indicate cells were p62 and p53 co-localize in the cytoplasm. (F) Shows a 100x magnification of p53 staining in untreated or GR-treated cells (red), and the merged image with p62 and DAPI. Note the strong punctate pattern of p53 staining in GR-treated cells. These experiments are quantified in Fig. S1B.
Based on these results, we conclude that autophagy is responsible for p53 mutant destabilization during glucose restriction. The data also reveal the potential importance of MDM2 in this newly identified degradation pathway.
p53 mutant loss during GR is accompanied by activation of autophagy and cell death
Autophagy can be organelle/substrate selective, or proceed as a bulk degradation process of multiple cellular compartments, including the mitochondria.Citation21 While autophagy may defend from stress insults in some instances, it can also lead to cell death. It was previously shown that various forms of p53 mutants are able to block autophagy.Citation17 Therefore, in subsequent experiments, we asked whether by inducing degradation, GR alleviates p53-dependent inhibition of autophagy and, if so, how this impinges upon proliferation. A kinetic analysis of the molecular changes occurring during GR revealed that autophagy was maximally induced concomitantly with p53 mutant loss, as judged based upon the pattern of LC3 conversion and the decreased levels of p62 (). The levels of Bax were only modestly changed, while the mitochondrial markers Mitofilin and Cox IV were reduced, indicative of autophagic degradation of mitochondria. Consistent with this latter idea, at a late time after GR the mitochondria were engulfed with lysosomes (Fig. S2A). Furthermore, a significant percentage of GR-treated cells underwent apoptosis ().
To tackle the molecular events occurring in the cells that survived glucose deprivation, we separated the cell populations as attached or floating. Approximately 40% of apoptosis occurred in the floating cells (), coinciding with a nearly complete loss of p53 expression therein (). p53 levels were instead still high in the attached surviving cells (, compare lanes 2 and 3). The floating cells displayed lower levels of p62 and increased LC3 conversion, strongly arguing that loss of p53 and autophagy are responsible for cell death. An important substrate of autophagic degradation, the radical oxygen species (ROS) scavenger catalase was also decreased.Citation22 Consistent with this observation, GR-induced cell death was accompanied by an increase in ROS levels (Fig. S2B). Since it was shown previously that autophagic degradation of catalase leads to oxidative stress accompanied by autophagic cell death,Citation22 we investigated whether catalase rescues these phenotypes. Incubation of GR-treated cells with catalase reversed ROS production as well as apoptosis (Fig. S2B and C). To confirm the role of autophagy in cell death, we knocked-down various autophagic gene products. The ATG5 or ATG7 shRNAs or the Beclin-1 siRNA largely prevented GR-induced apoptosis (). Thus, GR-induced cell death is associated with p53 depletion and is sustained at least in part by autophagy and consequent depletion of factors, such as catalase, with protective activity against excessive ROS production.
Expression of degradation-insensitive p53 mutant blocks autophagy and confers resistance to GR
The above results correlatively linked p53 mutant depletion with activation of autophagy and cell death. To then examine the relationship between p53 and autophagy further, additional approaches were undertaken. First, we asked whether manipulation of p53 mutant expression levels affects autophagy and influences survival. To this end we employed the p53-null ovarian or lung cancer cell lines, SKOV or H1299, respectively, where the expression of various p53 mutants was ectopically restored, as well as MDA-231 or T47D cell lines harboring a doxycycline (Dox)-inducible p53 shRNA (MDA-231.shp53 and T47D.shp53, respectively).Citation23 We also constructed p53 mutants mimicking acetylation, which, based on our previous results (), were predicted to be insensitive to GR-dependent degradation. To generate the acetylation mutant, the six acetylated carboxyl-terminal lysine residues of p53G245A (K319, K320, K321 and K370, K372, K373) were replaced with glutamine, an acetyl-mimicking residue (from here forth referred to as p53G245A-Δ6K).Citation8 In H1299 cells, induction of p53G245A or p53R175H led to a time-dependent LC3 and p62 accumulation (), which was insensitive to inhibition of autophagy with Bafilomycin A treatment (not shown), demonstrating that it reflects autophagic block. Vice versa, the knockdown of p53R280K in MDA-231.shp53 cells resulted in depletion of p62 and increased LC3 conversion only after 4 h of either glucose or amino acid restriction (). In H1299 cells expressing p53G245A, p62 levels declined concomitantly with GR-induced p53 mutant depletion (, compare lane 3 with lane 1). By contrast, p53G245A-Δ6K was more resistant to degradation and remained capable of maintaining its inhibitory effect on autophagy, as judged based on the pattern of p62 expression and LC3 conversion (, lanes 6 and 4). Thus, the data reveal that several p53 mutations inhibit autophagy in a manner largely dependent upon acetylation and p53 stability.
Figure 5. Mutant p53 blocks autophagy and promotes resistance to metabolic stress. (A) H1299 cells expressing p53G245A (lanes 1–4) or p53R175H (lanes 5–8) were grown without Dox (lanes 1 and 5), or with Dox for the indicated time (in hours). (B) MDA-231.shp53 cells were left untreated (lanes 1 and 4), or grown for 4 h in media lacking amino acids (AR, lanes 2 and 5), or lacking glucose (lanes 3 and 6), in the absence (lanes 1–3) or presence (lanes 4–6) of Dox. In all the experiments performed with these cells, pre-treatment with Dox was performed for at least 7 days before starvation. The levels of p53, LC3, p62 and actin were assessed in both panels. (C) H1299 cells expressing p53G245A (lanes 1–3) or p53G245A-Δ6K (lanes 4–6) were induced with Dox for 24 h. Doxycyxline was then washed out the media, and cells received either complete media (lanes 1 and 4) or GR media for 12 h (lanes 2 and 5) or 24 h (lanes 3 and 6). Cell extracts were probed in immunoblot with anti-p53, anti-p62, anti LC3 antibodies or anti-actin antibodies. The image shown is captured from the same autoradiogram at identical exposure times, and lines in between were cut. (D–E) Cell proliferation of T47D.shRNA and MDA-231.shp53 cells was monitored dynamical and in real time by using the x-CELLigence RT-CA system (see Materials and Methods). Cells were pre-treated with Dox or vehicle control, then plated in the presence or absence of glucose in duplicate wells and cell proliferation was assessed for 96 h. The p values between different samples are shown. (F) Naïve H1299 (lanes 1–3), or H1299 expressing p53G245A (lanes 4–6) or p53G245A-Δ6K (lanes 7–9) were Dox-treated as described previously, and then received GR media (lanes 2, 5 and 8), or rapamycin (Rap.; 20 μg/ml; lanes 3,6,9) for 3 d. Cell viability was assessed with trypan blue exclusion. Double asterisks refer to comparison of p values between p53G245A-Δ6K-expressing cells vs. cells expressing p53G245A.
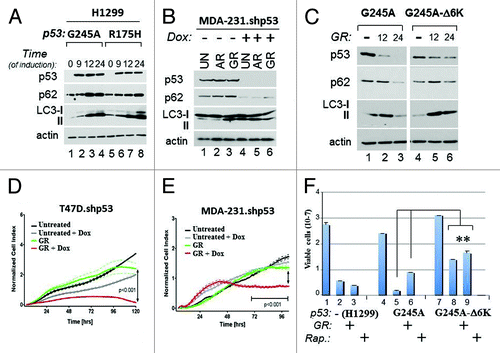
We then examined how mutant p53 affects survival to glucose restriction. Depletion of p53 from T47D.shp53 or MDA-231.shp53 cells with Dox prior to GR significantly accelerated loss of viability (). Vice versa, in the p53-null ovarian cancer cell line SKOV, restriction of glucose led to loss of cell viability and to a change in morphology, which were nearly entirely rescued when p53R175H was expressed with an adenoviral vector (Ad.p53R175H) (Fig. S3A and B). Thus, depletion of p53 enhances, while its overexpression bypasses, loss of viability due to GR also highlighting the vulnerability of p53-null cells to nutritional stress. Similarly to SKOV cells, glucose restriction impaired survival in naïve H1299 cells as well as in cells harboring p53G245A, but the expression of p53G245A-Δ6K conferred partial resistance to GR treatment (, compare lane 8 with lane 2 and 5). This protective activity of p53G245A-Δ6K was also seen in cells treated with the mTOR inhibitor, rapamycin, which mimics a global metabolic stress responseCitation24 (, lane 9).
Based on these results, we conclude that sustained p53 mutant expression and inhibition of autophagy confer resistance to metabolic stress. Therefore, viewed together with previous experiments (), they support the concept that the anti-proliferative activity due to glucose restriction relies upon p53 mutant degradation, which, in turn, leads to autophagic activation.
p53 mutant levels are influenced by the dietary carbohydrate content
How restriction of glucose affects tumor progression in vivo has been investigated only in few instances. Most published studies have explored the effects of either starvation or caloric restriction, this latter achieved with a reduction of 30–40% caloric intake.Citation25 Here we employed a standard laboratory chow diet (indicated as CD or C), a low carbohydrate diet (LCD or LC) or a third diet similar to the CD but with slightly higher carbohydrate content (HC). As shown in Table S1, these diets have nearly identical energetic caloric power of approximately 3.8–4 Kcal/gr. However, in the CD and HC, the majority of percent energy is derived from carbohydrates, while in the case of LCD, calories are mostly derived from proteins. The percent content of other nutrients, including minerals, vitamins and fibers, is otherwise identical.
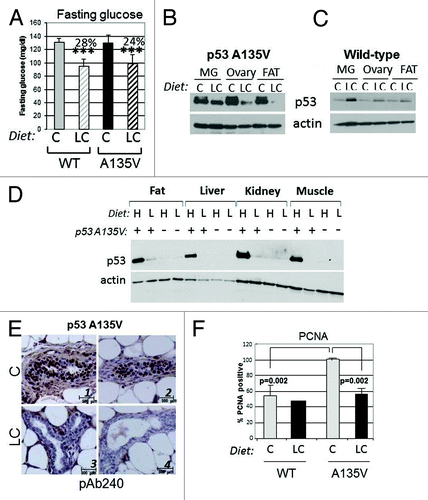
To explore the impact of dietetic interventions on p53 mutant levels, we employed mice harboring a p53 mutant transgene, p53A135V, a transforming p53 mutation.Citation26,Citation27 p53A135V transgenic- or wild-type animals were randomized after weaning so to receive the chow or the LC diet for several months. Assessment of blood glucose levels showed a highly significant reduction of the fasting glycemic values in the animals fed with the LC compared with the CD diet (). Strikingly, the LC diet led to depletion of mutant but not wild-type p53 in the ovary, fat and mammary glands (MG) of female mice (IHC) (). Similarly, the levels of p53A135V were lower in various tissues derived from male mice fed with the LC diet relatively to the HC diet (). The reduction of p53 mutant levels was also appreciable in immunohistochemistry ( and quantified in Fig. S3C). Although at 5 months of age p53A135V do not develop tumors, immunostaining with antibodies recognizing the proliferation marker, PCNA, showed an increased positivity in the epithelial cells of the mammary gland of p53A135V relative to control wild-type animals, which was abrogated by the LC diet (; Fig. S3D). Thus, in this transgenic model system, dietary restriction of carbohydrates prevents accumulation of p53 mutant and reduces proliferation rates.
Figure 6. A carbohydrate-restricted diet lowers fasting glucose levels and blunts p53 mutant expression in a transgenic murine model. (A) Fasting glucose measurements in wild-type or transgenic female mice expressing the p53A135V transgene matched for age and litter after 4 mo of dietetic regimen with either the chow diet (C) or low carbohydrate diet (LC). Average percent reductions between different groups are shown at the top of the histograms and are indicated by brackets. Double asterisks (**) refer to p < 0.01; triple asterisk (***) indicate p < 0.001. (B and C). Female mice fed as described in (A) were sacrificed and cell extracts derived from the mammary gland, ovaries and adipose tissues were probed in immunoblot with anti-p53 or anti-actin antibodies. These experiments were performed three times on different litters. (D) p53A135V positive (+) or wild-type (negative, -) males were sacrificed after 2 mo of feeding with the high carbohydrates (H) or LC (L) diet. Cell extracts were prepared from fat, liver, kidney and muscle, and then probed with the anti-p53 antibody (pAb240) or with the anti-actin antibody. (E) Immunostaining with the antibody pAb240, of the mammary gland derived from p53A135V transgenic mice fed with the chow diet (C, panels 1 and 2) or LC diet (panels 3 and 4). Sections were counterstained with H&E. Experiments are quantified in Figure S3C. (F) Proliferating cell nuclear antigen (PCNA) immunostaining of wild-type and p53A135V transgenic animals fed as indicated. Proliferation rates in the mammary epithelium were assessed by determining the percentage of PCNA-positive cells per total cells in each field examined. Statistical significance (p values) refers to the comparison between p53A135V mice fed with the LC diet, relatively to mice fed with C diet; or to comparison of values between p53A135V mice vs. wild-type control, as indicated by the brackets.
p53 mutant resistant to degradation accelerates oncogenesis in the face of carbohydrate restriction
We next asked how carbohydrate restriction influences p53 levels in developing tumors. To this end, we performed tumor transplantation experiments in nude mice by employing three syngenic mammary cancer lines, possessing a wild-type p53 allele, or expressing the murine counterpart of human p53G245A (p53G242A), or lacking p53.Citation28 In agreement with our previous results, the LC diet quite significantly reduced p53 mutant accumulation, while it stabilized wild-type p53 (, compare lanes 5 and 6 with lanes 1 and 2). Compared to p53-null or wild-type cells, cells expressing p53G242A developed highly aggressive and larger tumors, consistent with the GOF of this mutant, but tumor-forming ability was entirely reverted by the LC diet (). Not unexpectedly, considering previous data showing that p53-null cells are hypersensitive to nutritional stress, the growth of tumors lacking p53 was also inhibited by the LC diet, while significantly less inhibition was seen in wild-type p53-expressing cells.
Figure 7. Effects of the carbohydrate-restricted diet on tumor development and relatively to p53 status. (A) Expression levels of p53 in wild-type, null or G242A murine syngenic cell lines relative to the dietetic regimen as indicated at the top of panels. Athymic nude female mice were injected subcutaneously in the flank. Prior to the injection, animals were randomized to receive the C or LC diet for 2 wk. Tumors were excised and analyzed for p53 or actin content. (B) Tumor volumes in these mice were assessed 3–4 wk after injection. The differences in p values for each group of animals are indicated by brackets. These experiments were performed twice and data were analyzed from 4–8 tumors, depending upon the genotype and conditions of treatment. (C and D) Tumor-forming capacity of H1299 cells expressing p53G245A or p53G245A-Δ6K, after implantation in nude mice and fed as described in (A). Mice received doxycycline once a week. In (C), tumor volumes derived from two independent experiments are shown as averages. Error bars represent standard deviations. The p values between different treatment groups are indicated by brackets. Mice derived from one set of experiments are shown in (D).
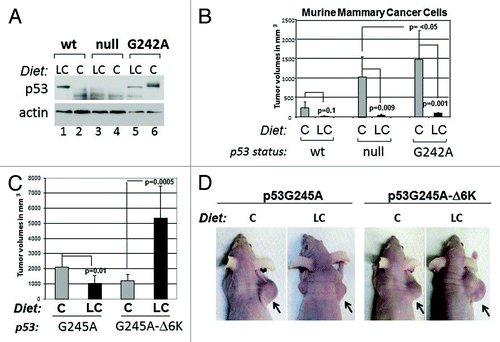
The implication of these results is that a carbohydrate-restricted dietetic regimen abrogates gain of oncogenic function of p53 mutant, and this is the direct result of GR-induced degragation. To test this possibility more directly, we next compared the tumor-forming capacity of H1299 cells harboring p53G245A or p53G245A-ΔK6. With these experiments, we also interrogated the impact of acetylation on tumor inhibition. We found that the LC diet led to tumor suppression in p53G245A-expressing cells, but such inhibition was entirely reverted by expression of p53G245A-ΔK6 (). Surprisingly, the LC diet actually enhanced the oncogenic capacity of p53G24A-ΔK6, indicating that this mutant might lead to the acquisition of novel gain of function activities during GR.
These data not only lend support to the previously proposed concept that p53 mutant stabilization is linked to oncogenic capacity, but also enlighten the importance of the anti-autophagic activities of p53 in this respect, given that the p53G24A-ΔK6 mutant is more efficient in blocking autophagy relative to its native counterpart.
Discussion
It is widely accepted that high levels of p53 mutants in tumors drive tumor progression, metastasis and resistance to therapy.Citation1,Citation2 Therefore, preventing p53 mutant accumulation and gain-of-function activity provides an important chemo-preventive and chemo-therapeutic strategy. Our study shows that in vivo a carbohydrate-free dietetic regimen that lowers the fasting glucose depletes p53 mutant levels in both transgenic and xenograft model systems and abrogates gain-of-function activity. Considering that at least in vitro, other forms of starvation did not prevent p53 mutant accumulation (), it is likely that by promoting p53 mutant depletion, restriction of glucose might be more effective in blunting oncogenic activity relatively to other dietetic interventions. Given the complexity of the p53 mutation spectrum in human tumors, it will be very important to expand our observations to determine whether destabilization of p53 mutants via glucose restriction is a generalized phenomenon. If so, an implication of our findings consists in the rationalization of dietetic interventions specifically aimed at lowering the fasting glucose in individuals harboring germ-line (Li-Fraumeni syndrome) or somatic p53 mutations as a strategy to prevent accumulation and gain of oncogenic functions. We have also shown that, at least in the case of p53G245A, while the LC diet limits its oncogenic activity, the deacetylation-insensitive counterpart, p53G245A-ΔK6, now accelerates tumorigenesis in the face of carbohydrate restriction (). When taken with the molecular characterization of p53G245A-ΔK6, the data argue that high levels of mutant p53 and inhibition of autophagy enable adaptation to metabolic stress. It is further possible that p53G245A-ΔK6 rewires the metabolism of tumor cells during glucose restriction. For example, a recent study showed that several p53 mutations maintain a malignant phenotype by promoting the mevalonate and sterol biosynthetic pathway.Citation23 Other activities of mutant p53 on various branches of the metabolism are rapidly emerging.Citation29,Citation30 Thus, p53 mutants also acquire novel “metabolic” gain of function(s) that could possibly contribute to the resistance of p53G245A-ΔK6 to GR. In essence, our findings reveal that p53 mutant acetylation-deacetylation acts as a nodal switch in affecting the sensitivity of tumor cells to GR.
An important question posed by our and many other studies is how autophagic activation influences cell survival, especially in relationship to p53 status. This is a complex issue, because the ultimate outcome of autophagy is likely dictated by the cell type and by the type of stress.Citation14,Citation31 Data presented here demonstrate that genetic or pharmacologic inhibition of autophagy or superimposing p53 mutant expression in the context of glucose restriction provides a survival advantage, while depletion of mutant p53 accelerates autophagy and GR-induced cell death ( and ). These results indicate that activation of autophagy is detrimental in a p53-null background and point to a protective role of mutant p53 in this respect. Intriguingly, a similar protective activity was previously attributed to wild-type p53.Citation14,Citation29 There are many plausible explanations as to why mutant p53 possesses such protective activity. For example, mitochondrial autophagy, when proceeding to completion, and if not balanced by appropriate rates of biogenesis, leads to cell death.Citation31,Citation32 We have recently identified the mitochondrial citrate transporter CIC as an inhibitor of mitochondrial autophagy in tumor cells.Citation33 Our results further show that CIC is directly activated at a transcriptional level by several forms of mutant p53 (manuscript in preparation). Therefore, in an exciting case scenario, p53 mutant might control the expression of factors, including CIC, that prevent mitophagy, while its depletion induced by GR leads to loss of such protective function and, thus, induces cell death. Wild-type p53 displays a pro-survival non-apoptotic activity during metabolic stress, in large part owing to a metabolic transcriptional program.Citation14,Citation29 It has emerged very recently that such a program is distinguishable from the transcriptional response elicited during DNA damage and specifically relies upon acetylation.Citation34 Our own previous studies demonstrated that acetylation can rescue DNA binding activity of several mutant forms of p53.Citation35 These observations raise the intriguing possibility that via acetylation, wild-type and mutant p53 share discrete pro-survival transcriptional activities, particularly those involved in autophagic and metabolic regulation. Further studies will obviously be necessary to corroborate these provocative hypotheses.
The proteasome is considered the master regulator of p53 stability in both its wild-type and mutant conformation. However, we found that depletion of glucose leads to p53 mutant degradation via the autophagic machinery, demonstrating that alternative pathways for p53 mutant disruption exist. The current model envisions that the proteasome predominantly degrades short-lived and soluble proteins, while autophagy provides an important route for disruption of long-lived and misfolded proteins that tend to be resistant to proteosomal clearance.Citation36 Many, if not all, of the tumor-derived p53 mutations acquire a misfolded configuration and are resistant to proteasome-mediated clearance unless the activity of chaperones is blocked.Citation2,Citation37 Wild-type p53 also possesses an intrinsically disordered and misfolded structure, which is ameliorated by acetylation.Citation38 Moreover, nuclear acetylation prevents misfolding of tumor-derived mutant forms of androgen receptor and restores a DNA-binding competent conformation of mutant p53 itself.Citation35,Citation39 These independent observations suggest that acetylation plays an important role in preventing misfolding of many proteins, including mutant p53. This interpretation could explain why glucose-induced deacetylation might render mutant p53 a suitable substrate for disruption by the autophagic machinery.
In conclusion, our results shed a new light on the molecular mechanisms by which p53 mutants stability is regulated and provide a potential framework for exploiting chemo-preventive dietary interventions as a strategy to control their expression levels and gain of oncogenic activity.
Materials and Methods
Cells, antibodies and reagents
The cell lines employed in this study were obtained from the tissue culture core facility at LCCC.
Cell lines, transfections and generation of the H1299 tetracycline-inducible cells
The generation of tetracycline inducible cells expressing p53R175H or p53G245A was described before (Knights et al. 2006).
Animal models, diets, assessment of body weights and of fasting glucose level
The Tg(Trp53A135V)2Ber/J strain was purchased from Jackson Laboratory and was maintained and genotyped with the protocol provided by JL.
Immunohistochemical staining of the mammary glands
Immunohistochemical staining of mammary gland sections was performed by using the fourth inguinal gland from all mice examined and is described in detail in the supplemental file.
Preparation of tissue samples from mice
Organs were excissed post-mortem; extracts were prepared by pulverizing the tissue mechanically using pestles chilled on dry ice. The tissue powder was subsequently dissolved in RIPA buffer and analyzed in immunoblot.
Tumor transplantation experiments
To produce tumor xenografts 5 × 106 cells were resuspended in PBS and injected subcutaneously in the flanks of female nude mice and kept for 2 wk before the injections under the indicated dietetic regimen.
Statistical analysis
Data are expressed as means ± standard deviations (SD). The two-tailed Student’s t-test was used for all statistical analysis, and Excel was used for statistical calculations. Significant differences are indicated using the standard Michelin Guide scale [p < 0.05, significant (*); p < 0.01, highly significant (**); p < 0.001, extremely significant (***)].
Additional material
Download Zip (892.5 KB)Acknowledgments
This work was supported by NIH-R01 CA102746 grants to M.L.A; C.A. was supported by NIH-RO1 CA129003; A.W. by NIH-R01 CA71508. Funds from The Breast Cancer Research Foundation to J.B. partially supported this work. We are very grateful to Dr. Amato Giaccia who provided important inputs and suggestions during the preparation of this manuscript. This project was in part supported by Award Number P30CA051008 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev 2012; 26:1268 - 86; http://dx.doi.org/10.1101/gad.190678.112; PMID: 22713868
- Prives C, White E. Does control of mutant p53 by Mdm2 complicate cancer therapy?. Genes Dev 2008; 22:1259 - 64; http://dx.doi.org/10.1101/gad.1680508; PMID: 18483214
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004; 119:861 - 72; http://dx.doi.org/10.1016/j.cell.2004.11.006; PMID: 15607981
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004; 119:847 - 60; http://dx.doi.org/10.1016/j.cell.2004.11.004; PMID: 15607980
- Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev 2008; 22:1337 - 44; http://dx.doi.org/10.1101/gad.1662908; PMID: 18483220
- Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 2009; 9:701 - 13; PMID: 19693097
- Kruse JP, Gu W. Modes of p53 regulation. Cell 2009; 137:609 - 22; http://dx.doi.org/10.1016/j.cell.2009.04.050; PMID: 19450511
- Knights CD, Catania J, Di Giovanni S, Muratoglu S, Perez R, Swartzbeck A, et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol 2006; 173:533 - 44; http://dx.doi.org/10.1083/jcb.200512059; PMID: 16717128
- Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab 2011; 13:495 - 504; http://dx.doi.org/10.1016/j.cmet.2011.04.004; PMID: 21531332
- Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev 2011; 25:1999 - 2010; http://dx.doi.org/10.1101/gad.17558811; PMID: 21979913
- Liu EY, Ryan KM. Autophagy and cancer--issues we need to digest. J Cell Sci 2012; 125:2349 - 58; http://dx.doi.org/10.1242/jcs.093708; PMID: 22641689
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672 - 6; http://dx.doi.org/10.1038/45257; PMID: 10604474
- Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, Giaccia AJ. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell 2008; 14:90 - 102; http://dx.doi.org/10.1016/j.ccr.2008.06.004; PMID: 18598947
- Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci USA 2010; 107:18511 - 6; http://dx.doi.org/10.1073/pnas.1006124107; PMID: 20937856
- Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126:121 - 34; http://dx.doi.org/10.1016/j.cell.2006.05.034; PMID: 16839881
- Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D’Amelio M, Djavaheri-Mergny M, et al. A dual role of p53 in the control of autophagy. Autophagy 2008; 4:810 - 4; PMID: 18604159
- Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A, et al. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008; 7:3056 - 61; http://dx.doi.org/10.4161/cc.7.19.6751; PMID: 18818522
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005; 18:283 - 93; http://dx.doi.org/10.1016/j.molcel.2005.03.027; PMID: 15866171
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009; 324:1076 - 80; http://dx.doi.org/10.1126/science.1164097; PMID: 19461003
- Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res 2003; 1:1001 - 8; PMID: 14707283
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12:401 - 10; http://dx.doi.org/10.1038/nrc3262; PMID: 22534666
- Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA 2006; 103:4952 - 7; http://dx.doi.org/10.1073/pnas.0511288103; PMID: 16547133
- Freed-Pastor WA, Mizuno H, Zhao X, Langerød A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012; 148:244 - 58; http://dx.doi.org/10.1016/j.cell.2011.12.017; PMID: 22265415
- Peng T, Golub TR, Sabatini DM. The immunosuppressant rapamycin mimics a starvation-like signal distinct from amino acid and glucose deprivation. Mol Cell Biol 2002; 22:5575 - 84; http://dx.doi.org/10.1128/MCB.22.15.5575-5584.2002; PMID: 12101249
- Lee C, Longo VD. Fasting vs dietary restriction in cellular protection and cancer treatment: from model organisms to patients. Oncogene 2011; 30:3305 - 16; http://dx.doi.org/10.1038/onc.2011.91; PMID: 21516129
- Donehower LA, Lozano G. 20 years studying p53 functions in genetically engineered mice. Nat Rev Cancer 2009; 9:831 - 41; http://dx.doi.org/10.1038/nrc2731; PMID: 19776746
- Harvey M, Vogel H, Morris D, Bradley A, Bernstein A, Donehower LA. A mutant p53 transgene accelerates tumour development in heterozygous but not nullizygous p53-deficient mice. Nat Genet 1995; 9:305 - 11; http://dx.doi.org/10.1038/ng0395-305; PMID: 7773294
- Tzeng YJ, Zimmermann C, Guhl E, Berg B, Avantaggiati ML, Graessmann A. SV40 T/t-antigen induces premature mammary gland involution by apoptosis and selects for p53 missense mutation in mammary tumors. Oncogene 1998; 16:2103 - 14; http://dx.doi.org/10.1038/sj.onc.1201733; PMID: 9572491
- Maddocks OD, Vousden KH. Metabolic regulation by p53. J Mol Med (Berl) 2011; 89:237 - 45; http://dx.doi.org/10.1007/s00109-011-0735-5; PMID: 21340684
- Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer 2009; 9:691 - 700; http://dx.doi.org/10.1038/nrc2715; PMID: 19759539
- Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov 2007; 6:304 - 12; http://dx.doi.org/10.1038/nrd2272; PMID: 17396135
- Gusdon AM, Chu CT. To eat or not to eat: neuronal metabolism, mitophagy, and Parkinson’s disease. Antioxid Redox Signal 2011; 14:1979 - 87; http://dx.doi.org/10.1089/ars.2010.3763; PMID: 21126205
- Catalina-Rodriguez O, Kolukula VK, Tomita Y, Preet A, Palmieri F, Wellstein A, et al. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 2012; In press PMID: 23100451
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012; 149:1269 - 83; http://dx.doi.org/10.1016/j.cell.2012.04.026; PMID: 22682249
- Perez RE, Knights CD, Sahu G, Catania J, Kolukula VK, Stoler D, et al. Restoration of DNA-binding and growth-suppressive activity of mutant forms of p53 via a PCAF-mediated acetylation pathway. J Cell Physiol 2010; 225:394 - 405; http://dx.doi.org/10.1002/jcp.22285; PMID: 20589832
- Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 2008; 4:141 - 50; PMID: 17986870
- Dey A, Lane DP, Verma CS. Modulating the p53 pathway. Semin Cancer Biol 2010; 20:3 - 9; http://dx.doi.org/10.1016/j.semcancer.2010.02.004; PMID: 20193765
- Stanga S, Lanni C, Govoni S, Uberti D, D’Orazi G, Racchi M. Unfolded p53 in the pathogenesis of Alzheimer’s disease: is HIPK2 the link?. Aging (Albany NY) 2010; 2:545 - 54; PMID: 20876941
- Thomas M, Dadgar N, Aphale A, Harrell JM, Kunkel R, Pratt WB, et al. Androgen receptor acetylation site mutations cause trafficking defects, misfolding, and aggregation similar to expanded glutamine tracts. J Biol Chem 2004; 279:8389 - 95; http://dx.doi.org/10.1074/jbc.M311761200; PMID: 14670946