Abstract
Subunit 6 of the COP9 signalosome complex, CSN6, is known to be critical to the regulation of the MDM2-p53 axis for cell proliferation and anti-apoptosis, but its many targets remain unclear. Here we show that p57Kip2 is a target of CSN6, and that CSN6 is a negative regulator of p57Kip2. CSN6 associates with p57Kip2, and its overexpression can decrease the steady-state expression of p57Kip2; accordingly, CSN6 deficiency leads to p57Kip2 stabilization. Mechanistic studies show that CSN6 associates with p57Kip2 and Skp2, a component of the E3 ligase, which, in turn, facilitates Skp2-mediated protein ubiquitination of p57Kip2. Loss of Skp2 compromised CSN6-mediated p57Kip2 destabilization, suggesting collaboration between Skp2 and CSN6 in degradation of p57Kip2. CSN6’s negative impact on p57Kip2 elevation translates into cell growth promotion, cell cycle deregulation and potentiated transformational activity. Significantly, univariate Kaplan-Meier analysis of tumor samples demonstrates that high CSN6 expression or low p57 expression is associated with poor overall survival. These data suggest that CSN6 is an important negative regulator of p57Kip2, and that overexpression of CSN6 in many types of cancer could lead to decreased expression of p57Kip2 and result in promoted cancer cell growth.
Keywords: :
Introduction
The COP9 signalosome (CSN) protein complex is implicated in cell cycle regulation. It has been shown that CSN is involved in the degredation of proteins that regulate the cell cycle, such as p27,Citation1 c-Jun,Citation2 p53,Citation3-Citation5 COP1Citation6 and 14-3-3σ Citation6 via the ubiquitin-proteasome pathway. CSN is a protein complex, which consists of eight subunits (CSN1 to CSN8). First characterized as a repressor of plant photomorphogenesis, this evolutionarily conserved multiprotein complex has been found in both plants and animals. While the detailed biochemical function of the CSN in animal cells remains uncharacterized, the complex and its individual subunits have been implicated in a wide variety of regulatory processes, including cell cycle control, signal transduction, transcriptional activation and tumorigenesis.Citation7,Citation8 Interestingly, the eight subunits of the COP9 signalosome are each paralogous to one of the eight subunits that form the lid complex (19S) of the 26S proteasome.Citation9,Citation10 The lid complex can recognize ubiquitinated substrates and then funnel them into the proteolytic core complex for degradation. The homology between the COP9 signalosome and the 19S lid complex implies that the COP9 signalosome plays a role in protein degradation by functioning as a mediator of the cross talk between substrates and the 26 proteasome. At issue then is the characterization of COP9 signalosome’s many targets.
The p57Kip2 protein (abbreviated as p57) is a cyclin-dependent kinase inhibitor, sharing p27Kip1 homology,Citation11 also known as CDKN1c. It is a maternally expressed, paternally imprinted gene located on chromosome 11p15.5.Citation12,Citation13 p57-knockout mice have altered cell proliferation, differentiation and a variety of other abnormalities.Citation14,Citation15 Many of these defective phenotypes are also observed in patients with Beckwith-Wiedemann syndrome, a childhood overgrowth syndrome, suggesting that the loss of p57 has a role in the syndrome.Citation14,Citation15 As a negative regulator of cell cycle, p57 is a potential tumor-suppressor gene.Citation16-Citation18 Although decreased expression of p57 has been found in many types of cancer, including bladder carcinoma, gastric cancer and pancreatic cancer,Citation13 p57-associated mutations are rarely found.Citation17,Citation19 It is possible that post-transcriptional deregulation of p57 is the cause of decreased p57 expression in cancers, but it remains to be seen how an oncogenic signal can downregulate p57 expression at the protein level.
It is well known that ubiquitination is critical to the regulation of p57 activity through Skp2,Citation20 an important component of the E3 ligase. Recently, we have identified CSN6 of the COP9 complex as playing a pivotal role in the regulation of E3 ligases, such as MDM2 and COP1, by destabilizing their respective targets.Citation4,Citation6 Importantly, CSN6 overexpression is found in several types of cancer,Citation4,Citation8 linking it to oncogenic activity. It is not clear whether or not CSN6 has any link to Skp2 for ubiquitin regulation. In this study, we demonstrate that CSN6 and p57 interact. Furthermore, we indicate that CSN6 is involved in the negative regulation of p57 via the Skp2-mediated ubiquitination pathway. We characterize that CSN6 is involved in p57 destabilization, which, in turn, has an impact on cell proliferation and transformation. Significantly, human tumor sample studies demonstrate that high CSN6 and low p57 correlate with poor overall survival. Our studies provide important insights into the oncogenic activity of CSN6 during tumorigenesis.
Results
CSN6 is involved in HER2 signaling-mediated p57 regulation
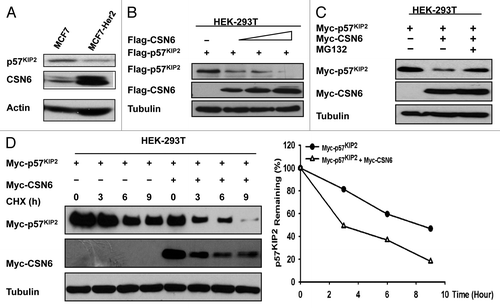
HER2 signaling can positively promote the cell cycle.Citation21 Recently, we demonstrated that HER2-Akt signaling positively regulates CSN6 stability.Citation22 Given that CSN6 overexpression can promote cell proliferation,Citation4 it is possible that CSN6 can control the cell cycle by regulating the CDK inhibitor, which affects cylin-Cdk activity. We determined the expression level of CSN6 in isogenic cell lines with different HER2 statuses. CSN6 protein levels are higher in HER2-overexpressing cells (MCF7 overexpressed with HER2) than in non-HER2-overexpressing cells (MCF7) (). Importantly, while CSN6 is high in HER2-overexpressing cells, p57 CDK inhibitor is low in such a context (). We then explored whether or not CSN6 is involved in p57 downregulation. Cells’ CDK inhibitor p57 levels decreased in a dose-dependent manner when transfected with increasing amounts of CSN6 (). CSN6-mediated p57 downregulation was suppressed by MG132, a proteasome inhibitor, suggesting the involvement of the 26S proteasome (). To investigate if CSN6 downregulates p57 at the post-transcriptional level, we examined the turnover rate of p57 in the presence of the de novo protein synthesis inhibitor, cycloheximide (CHX) (). Indeed, overexpression of CSN6 increased the turnover rate of p57 when compared with the non-overexpression control group (). This result suggests that CSN6 is a negative regulator of p57.
Figure 1. CSN6 negatively regulates p57 stability. (A) HER2 and CSN6 negatively regulate the steady-state expression of p57. Equal amounts of cell lysates were immunoblotted with indicated antibodies. (B) CSN6 decreases the steady-state expression of p57 in a dose-dependent manner. 293T cells were co-transfected with indicated plasmids and increasing amounts of CSN6. Equal amounts of cell lysates were immunoblotted with indicated antibodies. (C) CSN6-mediated degradation of p57 is proteasome-dependent. 293T cells were co-transfected with the indicated plasmids. Cells were treated with MG132 for 6 h before harvesting. Equal amounts of cell lysates were immunoblotted with the indicated antibodies. (D) p57 turnover rate is decreased in CSN6-overexpressing cells. 293T cells transfected with the indicated plasmids were treated with cycloheximide (CHX) (100 µg/ml) for the indicated times. Cell lysates were immunoblotted with the indicated antibodies. Integrated OD values of p57 bands at each time point were measured using a densitometer. Levels of p57 at time zero were set at 100%. Remaining p57 is indicated graphically (right).
CSN6 regulates p57 protein stability by enhancing the ubiquitination level of p57
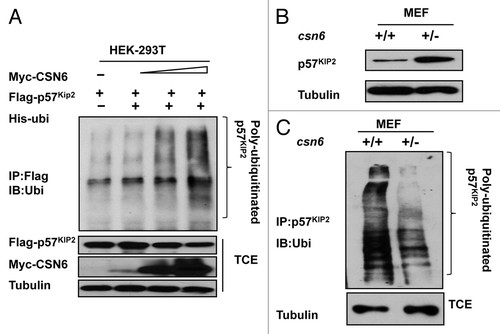
To further investigate the impact of CSN6 on p57 stability, we examined the ubiquitination of p57, and found that CSN6 increased the ubiquitination level of p57 in a dose-dependent manner (). We previously obtained csn6+/− mouse embryonic fibroblast (MEF)Citation4 through CSN6 knockout. We found that csn6+/− MEF had an accumulation of p57 when compared with wild-type MEF, consistent with the idea that CSN6 can cause p57 downregulation (). Consistently, CSN6 haploinsufficiency, as seen in csn6+/− MEFs, diminished the levels of p57 polyubiquitination when compared with WT MEF (). These results suggest that CSN6 negatively regulates p57 stability via increased levels of p57 ubiquitination, thereby reducing p57’s steady-state expression.
Figure 2. CSN6 is critical in affecting p57 ubiquitination. (A) CSN6 increases p57 ubiquitination in a dose-dependent manner. 293T cells were transfected with the indicated plasmids and increasing CSN6 expressing plasmids. The proteasome inhibitor MG132 was added 6 h prior to cell harvesting. The amount of ubiquitinated p57 was analyzed by immunoprecipitation (IP) with anti-Flag followed by immunoblotting (IB) with anti-ubi. TCE, total cell extracts. (B) CSN6 haplo-insufficiency leads to p57 stabilization. Indicated WT and csn6+/− MEF cells were immunoblotted with anti-p57 or tubulin antibodies. (C) CSN6 haplo-insufficiency leads to decreased p57 ubiquitination. Indicated MEFs were treated with proteasome inhibitor MG132 for 6 h prior to cell harvesting. The amount of ubiquitinated p57 was analyzed by immunoprecipitation (IP) with anti-p57 followed by immunoblotting (IB) with anti-ubi. TCE, total cell extracts.
CSN6 interacts with p57 and Skp2
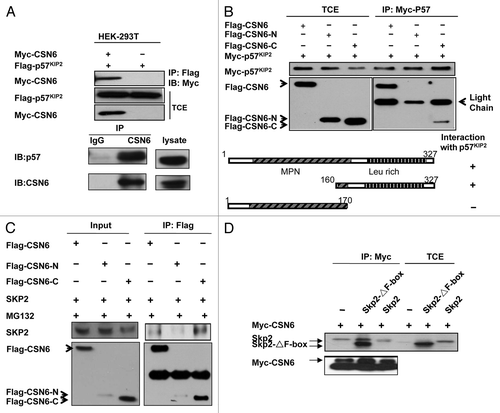
Although CSN6 is involved in the destabilization of p57, it is not clear whether or not there is an interaction between CSN6 and p57. To investigate this possible interaction, we analyzed cell lysates co-transfected with CSN6 and p57. Indeed, CSN6 associated with p57 as assayed by Co-IP (). Endogenous interaction between these two proteins was also observed (). To map the interaction domain, we co-transfected CSN6 domains with p57. We found that full-length Flag-tagged CSN6 co-immunoprecipitated with p57 from cell lysates. Interestingly, we showed that the C-terminal CSN6 domain (aa 150–327) interacted with p57 but not the N-terminal region containing MPN domain (). Also, we showed that full-length CSN6 associated with Skp2, an important component of the E3 ubiquitin ligase (). Interestingly, we also showed that the C-terminal CSN6 domain (aa 150–327) interacted with Skp2 efficiently (). We further mapped the CSN6 binding region on Skp2, and found that CSN6 interacted with the Skp2 F-box deletion mutant efficiently (), suggesting that CSN6 interacts with Skp2 not through the F-box domain. These results indicate that CSN6 associates with p57 and Skp2 using its C-terminal domain to form a complex that critically affects the stability of p57.
Figure 3. CSN6 interacts with p57 through C-terminal domain. (A) Association between CSN6 and p57. HEK-293T cells transfected with indicated plasmids were immunoprecipitated with anti-Flag antibodies and immunoblotted with anti-Myc. TCE, total cell extracts. Cell lysates of HEK293 cells were immunoprecipitated with anti-CSN6 and then immunoblotted with anti-p57 for endogenous binding. (B) Mapping of p57 binding domains on CSN6. Indicated Flag-tagged CSN6 constructs were co-transfected with p57 into 293T cells. Cell lysates were immunoprecipitated with anti-Myc and immunoblotted with anti-Flag. Schematic representation of the mutants is shown. TCE, total cell extracts. (C) Mapping of Skp2 binding sites on CSN6. Indicated Flag-tagged CSN6 constructs were co-transfected with Skp2 into 293T cells. Cell lysates were immunoprecipitated with anti-Flag and immunoblotted with the indicated antibodies. (D) CSN6 does not interact with Skp2 on F-box domain. Indicated Flag-tagged Skp2 constructs were cotransfected with CSN6 into 293T cells. Cell lysates were immunoprecipitated with anti-Myc and immunoblotted with the indicated antibodies. TCE, total cell extracts.
Skp2 is required for CSN6-mediated p57 downregulation
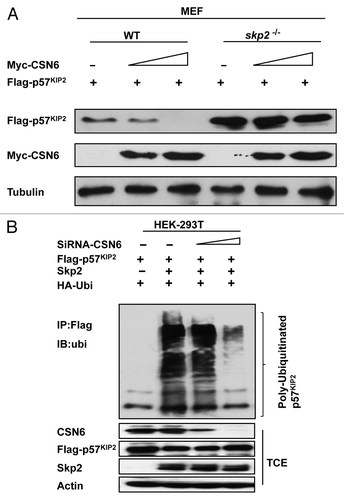
To determine the role of Skp2 in CSN6-mediated p57 downregulation, we employed skp2 MEF−/− cells in the studies. We analyzed WT and skp2−/− MEF cell lysates and found that skp2−/− MEF cells had elevated p57 when compared with wt MEF cells (). Importantly, we examined the steady-state expression of p57 in the presence of increasing amounts of CSN6 in these cells and found that in constrast to wt MEF, CSN6 does not lead to a reduction in the steady-state expression of p57 in skp2−/− MEF, i.e., Skp2 deficiency compromised the negative impact of CSN6 on p57 (). We further investigated the ubiquitination process, and found that Skp2, as expected, increased the level of p57 ubiquitination (). Surprisingly, the level of p57 ubiquitination is reduced when CSN6 is depleted by siRNA, even in the presence of Skp2 (). These results indicate that CSN6 and Skp2 collaborate to enhance p57 ubiquitination, thereby decreasing the steady-state expression of p57.
Figure 4. CSN6-mediated p57 downregulation is Skp2-dependent. (A) Skp2 deficiency diminished CSN6’s capability to reduce steady-state expression of p57. Indicated WT and Skp2−/− MEF cells after transfection with indicated plasmids were immunoblotted with indicated antibodies. (B) Skp2-mediated p57 ubiquitination requires the presence of CSN6. 293T cells were transfected with the indicated plasmids. The proteasome inhibitor MG132 was added 6 h prior to cell harvesting. The amount of ubiquitinated p57 was analyzed by immunoprecipitation (IP) with anti-Flag followed by immunoblotting (IB) with anti-Ubi. TCE, total cell extracts.
HER2-CSN6 regulates cell growth by antagonizing p57 activity
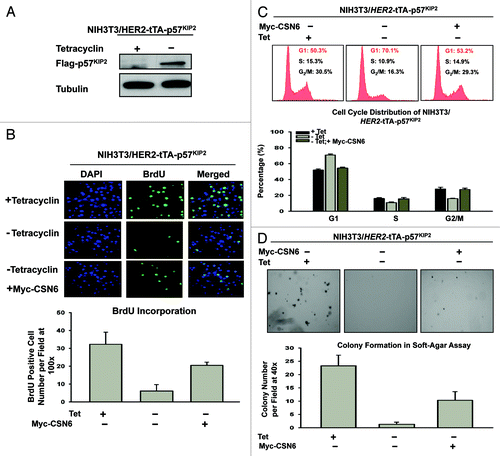
Since CSN6 downregulates p57, we reasoned that CSN6 would have a role in cell proliferation. Because p57 inhibits CDK activity, we first tested whether p57 could block cell proliferation activity using a BrdU incorporation assay in HER2-overexpressing cells. We established a cell line, NIH3T3/HER2/tTA-p57, to perform a p57 induction experiment in the absence of tet () and then determined the BrdU incorporation rate in these p57-expressing cells. The data showed that the BrdU incorporation rate of p57-expressing cells (-tet treatment) decreased drastically after the p57 induction (). Since CSN6 can facilitate p57 degradation, we sought to examine the impact of CSN6 expression on cell growth in this system. We found that CSN6 overexpression in NIH3T3/HER2/tTA-p57 (when p57 is induced) antagonized p57-mediated reduced BrdU incorporation (), suggesting that overexpression of CSN6 can reverse the cell inhibition caused by p57. Consistently, DNA histogram analysis by FACS showed that induced expression of p57 led to an increased G1 DNA content that was well above that seen in non-induced cells (i.e., G1 arrest), while CSN6 overexpression in the presence of p57 induction reduced G1 arrest (). We sought to further examine the effect of CSN6 on p57-mediated inhibition of cell transformation. We found that NIH3T3/HER2/tTA-p57 cells with p57 induction showed a decrease in cell colonies with anchorage-independent growth when compared with non-induced cells (), while CSN6 overexpression in the presence of p57 induction prevented such an inhibition from occurring (). These data demonstrate the biological consequences of CSN6’s negative impact on p57 activity.
Figure 5. CSN6 impacts on p57 to regulate HER2-mediated cell proliferation, cell cycle progression and cell transformation. (A) p57 is induced in NIH3T3/HER2/tTA-p57 cell in the absence of tet. NIH3T3/HER2/tTA-p57 cells were treated with (+) or without (-) tet and the cell lysates were immunoblotted with indicated antibodies. (B) CSN6 expression antagonizes p57-mediated reduction of BrdU incorporation. NIH3T3/HER2/tTA-p57 cells were transfected with CSN6 and then treated without (-) tet. The cells were fixed for anti-BrdU staining. BrdU-positive cells were counted and presented as a bar graph. Error bars represent 95% confidence interval. (C) CSN6 expression hinders p57-mediated G1 arrest. NIH3T3/HER2/tTA-p57 cells were transfected with CSN6 and then treated without (-) tet. The cells were analyzed by FACS for cell cycle distribution. The percentages of each cell cycle phase are presented as a bar graph. Error bars represent 95% confidence interval. (D) CSN6 expression compromises p57-mediated inhibition of cell transformation. NIH3T3/HER2/tTA-p57 cells were transfected with CSN6 and then treated without (-) tet. The cells were analyzed for anchorage-independent growth by soft agar colony formation assay. The numbers of colony formation are presented as a bar graph.
Overexpression of CSN6 and downregulation of p57 in cancer
To understand the relationship between CSN6 and p57 in cancer, we examined the expression of CSN6 and p57 by western blotting a collection of 25 primary human breast cancer samples. We noticed that CSN6 is elevated in a high percentage of cancer samples (19 out of 25) with concurrent p57 downregulation (). In this set of 19 samples with CSN6 overexpression, 11 had low levels of p57 expression (). These results suggest that CSN6 overexpression correlates with the p57 downregulation in cancers. We further quantitated the expression of CSN6 and p57 using immunohistochemical staining and, in another cohort of breast cancer patients with clinical records, correlated expression levels with the overall survival. We found a strong inverse relationship between CSN6 and p57 expression (). In tumors where CSN6 expression is low, p57 expression is high; whereas in tumors where CSN6 expression is high, p57 expression is low. These results indicate that downregulation of p57 correlates very well with the overexpression of CSN6 in tumor tissue, which suggests that downregulation of p57 plays an important role in CSN6-mediated tumorigenesis of breast cancer. Univariate Kaplan-Meier analysis showed that high expression levels of CSN6 were associated with poor overall survival (log rank test, p = 0.025) (). Accordingly, low expression levels of p57 were associated with poor overall survival (log rank test, p < 0.001) (). CSN6 and p57 were significant (p < 0.05) predictive factors in separate multivariate Cox regression models for both overall survival and progression-free survival in this cohort after controlling for age, stage, ER, PR and HER2 status. Therefore, the protein expression levels of CSN6 and p57 are significant prognostic factors for breast cancer.
Figure 6. Expression levels of CSN6 and p57 in breast cancer samples and their correlations with survival. (A) A large percentage of breast cancer samples have high CSN6 and low p57. Cell lysates from 25 primary human breast cancer samples were immunoblotted with indicated antibodies. (B) Kaplan-Meier analysis showed that high expression of CSN6 and low expression of p57 were associated with poor overall survival. The expression levels of CSN6 and p57 were investigated by immunohistochemical staining, and were correlated with the overall survival rate. (C) Model of how CSN6-mediated p57 degradation affects cell proliferation, cell cycle progression and cell transformation.
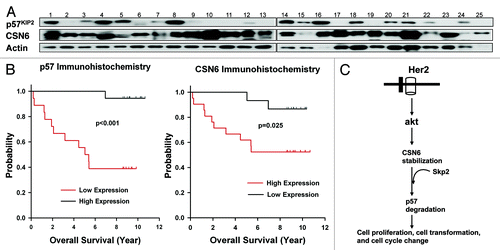
Table 1. Expression of CSN6 and p57 of breast cancer tissue samples
Discussion
The COP9 complex and its individual subunits have been implicated in cell cycle regulation.Citation7,Citation8 We previously showed that CSN6 is overexpressed in many types of cancers.Citation4,Citation8 Here, we discover that CSN6 has a critical role in controlling the homeostasis of the p57 CDK inhibitor by enhancing ubiquitin-proteasomal degradation of p57. Our results provide invaluable insight into the effects of CSN6 overexpression on p57 expression during cancer cell growth.
The F-box protein of the SCF (SKP1–CUL1–F-box protein) ubiquitin ligases provides the substrate targeting specificity of the complex.Citation23 Our interaction studies showing that CSN6 uses its C-terminal domains to interact with Skp2 (F-box protein) and p57 () provide critical insights into the role of CSN6 in the coordination of SCFSkp2 and its target. These data suggest that CSN6 may function as an adaptor during p57 ubiquitination and degradation. Indeed, overexpression of CSN6 accelerates the p57 ubiquitination and degradation process (), while loss of CSN6 compromises the degradation event (). So far, the domain functions of CSN6 remain largely unknown. Structurally, the CSN6 protein has a MPN (Mpr1p and Pad1p N-terminal) domain, which is found in the N terminus of yeast Mpr1 and Pad1 proteins.Citation24-Citation26 The MPN domain consists of polar residues that resemble the active site residues of metalloproteasesCitation27 and is involved in proteasome-associated deneddylation activity.Citation28 In contrast, the C-terminal domain of CSN6 is less characterized, and its biological function remains undetermined. Our data show that p57 does not interact with the MPN domain of CSN6, and that both p57 and Skp2 strongly interact with the C-terminal domain of CSN6. How this interaction participates in the activities of Skp2 in relation to p57 ubiquitination remains to be determined. It is important to point out that Skp2 is involved in p57 degradation.Citation20 Additionally, FBL 12, another F-box protein, is also involved in p57 ubiquitin-mediated degradation.Citation29 CSN6-mediated p57 degradation may involve FBL12; however, we showed that CSN6-mediated p57 degradation is compromised in skp2−/− cells (), suggesting that Skp2, not FBL 12, is the major player. Nevertheless, it is important to point out that Skp2 is induced by serum, while FBL12 is upregulated by TGFβ,Citation29 and it remains to be determined whether FBL12 and CSN6 collaborate to degrade p57 under TGFβ signaling. Recently, missense mutations located in the PCNA binding motif of p57 were found to cause decreased ubiquitination of p57.Citation30,Citation31 These mutants are associated with IMAGe syndrome (intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita and genital anomalies) and have the characteristics of gain-of-function, i.e., these mutants greatly inhibit cell growth. It remains to be determined if these mutants have lost their bindings to CSN6 or Skp2, decreasing their ubiquitination and allowing them a more active role in the control of cell growth. Interestingly, the p57 mutants, which are associated with IMAGe syndrome, are less ubiquitinated and can effectively restrict eye growth in Drosophila.Citation30 In our study, we show that the absence of CSN6 overexpression leads to less ubiquitination of p57, causing it to inhibit cell growth (), confirming the significance of less-ubiquitinated p57 in the inhibition of cell growth. In contrast, p57-mediated cell growth inhibition is compromised when CSN6 is overexpressed (); this supposedly is a manifestation of CSN6-enhanced p57 ubiquitination and degradation (). Similar effects are observed in cell cycle distribution: p57-mediated G1 arrest is alleviated by the expression of CSN6 (). These data indicate that p57 ubiquitination is critically regulated during cell growth.
It has been indicated that Akt is the downstream regulator of the HER2 signaling that facilitates cell growth. We have recently shown that Akt can mediate CSN6 stabilization.Citation22,Citation32 With this in mind, our study clearly indicates that CSN6-mediated p57 degradation is an extension of the HER2-Akt-CSN6 axis. This revelation fills the gap in knowledge about this axis’ regulation of cell cycle growth via CDK inhibitor p57 (). This signal direction can cause cell proliferation, compromise G1 arrest and potentiate cell transformation as depicted in our working model (). Our mechanistic studies of CSN6-mediated p57 degradation explain how the observed overexpression of CSN6 in cancers can lead to the downregulation of p57 and cause a reduction in overall survival (). Clearly, the CSN6-p57 link will be an important molecular target for rational cancer therapy. Enhancing the p57 stabilization, CDK inhibition or CSN6 inhibition may be useful therapeutic strategy for future CSN6-overexpressing cancer interventions.
Materials and Methods
Cell lines and reagents
HEK-293T (human embryonic kidney cell line), MCF7 and HER18 (human breast cancer cell line) as previously described.Citation22 Wt MEF, csn6+/− MEF, skp2−/− MEF as described previously.Citation4 NIH3T3/HER2/tTA-cells as described previously.Citation33 Flag-tagged p57 DNA fragment was cloned into modified pUHD10–3 hygromycin vectorCitation34 and confirmed by sequencing. BrdU, MG132 and cycloheximide were obtained from Sigma. SiCSN6 RNA as previously described.Citation4 Antibodies: CSN6 (BIOMOL international), Flag (M2 monoclonal antibody, Sigma), tubulin (Sigma), p57 (sigma), ubiquitin (Zymed Laboratories, Inc.), Skp2 (abcam). Skp2ΔFbox, Flag-CSN6, Myc-CSN6, Flag-p57 were previously described.Citation4,Citation12,Citation35 N-CSN6 (1–170 aa) and C-CSN6 (160–327aa) mutants were constructed by PCR cloning.
Immunoblotting and immunoprecipitation
Total-cell lysates were processed as previously described.Citation36 Cell lysates for western blot or immunoprecipitation were lysed with 100–300 μl 1x lysis buffer [0.5 L batch: 7.5 g 1 M Tris (Fisher), 15 ml 5M NaCl (Fisher), 0.5 ml NP-40 (USB Corp.), 0.5 ml Triton X-100 (Sigma) and 1 ml 0.5 M EDTA (Fisher)] for 20 min at 4°C. Lysates were immunoblotted with indicated antibodies. For immunoprecipitation, cell lysates were prepared and standardized as before and 1 mg protein immunoprecipitated with appropriately diluted antibody in lysis buffer overnight. Antibody was pulled down with 50 μl of either Protein A or G beads. Beads were centrifuged at a low speed for 10 min and supernatant was discarded. Dried beads were mixed with 2x loading dye and boiled for 5 min. Lysate samples were loaded onto gels and SDS-PAGE was performed as before.
Protein turnover assay
The cells were transfected with the indicated plasmids and incubated at 37°C with 5% (vol/vol) CO2 for 24 h. Then cycloheximide was added into the media to a final concentration of 100 μg/ml. The cells were harvested at the indicated times after CHX treatment. The protein levels were analyzed by immunoblotting.
Ubiquitination assay
csn6+/− MEFs and the 293T cells cotransfected with indicated plasmids or were used for the experiments. At 24 h post-transfection, cells were treated with 50 μg/ml of MG132 for 6 h. The p57 proteins were immunoprecipitated by the anti-Flag or anti-p57 antibody. The protein complexes were then resolved by SDS-polyacrylamide gel and probed with anti-ubiquitin or Anti-HA to observe the ubiquitinated p57.
BrdU incorporations immunofluorescence
Incorporation of BrdU was examined under a fluorescence microscope with fluorescein isothiocyanate-conjugated anti-BrdU as previously described.Citation33 At least 300 cells were counted for BrdU staining. The percentage of BrdU-positive cells was measured. Data shown are from a typical experiment performed in triplicate.
FACS analysis of DNA contents
NIH3T3/HER2/tTA-p57 cells were transfected with CSN6 and then treated with 2 mg/ml tet or left untreated and were subjected to FACS analysis. Cells were incubated at 37°C for 30 min in a buffer containing 50 μg/ml propidium iodine, 5 mM MgCl2, 10 mM TRIS-HCl pH 7.0, 25 μg/ml RNaseA. This was followed by FACS analysis to measure the DNA contents.
Anchorage-independent soft-agar colony formation assay
NIH3T3/HER2/tTA-p57 cells were transfected with CSN6 and then treated with 2 mg/ml tet or left untreated and were then subjected to the assays. These assays were performed as described.Citation37
Human tumor samples
Thirty-five samples of primary breast tumors were provided by the First Affiliated Hospital and China Medical University. Collection and usage of all patient materials and information were conducted according the institutional guidelines and the Declaration of Helsinki Principles. Immunohistochemical staining was performed to evaluate the expression of CSN6 and p57. The age, pathological stage, ER, PR and HER2 status, overall survival and recurrence-free survival were obtained by retrospective chart review. Primary breast tumor samples were obtained from patients who had undergone operations to treat breast cancer. They were collected as freshly frozen tissues and were stored in the tissue bank of the University of Texas MD Anderson Cancer Center. Research using all human specimens and data was conducted under protocols approved by Institutional Review Board of UT MD Anderson Cancer Center. Informed consent was obtained for use of these pathologic samples for research.
Acknowledgments
This work was supported by grants in part from the National Institutes of Health (NIH) (R01CA089266), Directed Medical Research Programs (DOD SIDA BC062166 to S.J.Y. and M.H.L.) and Susan G. Komen Breast Cancer Foundation (KG081048). The University of Texas M.D. Anderson Cancer Center is supported by NIH core grant CA16672. B.C. is supported by National Natural Science Foundation of China, 30972939. We thank Stephen Skerl for critical reading.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Tomoda K, Kubota Y, Kato J. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature 1999; 398:160 - 5; http://dx.doi.org/10.1038/18230; PMID: 10086358
- Naumann M, Bech-Otschir D, Huang X, Ferrell K, Dubiel W. COP9 signalosome-directed c-Jun activation/stabilization is independent of JNK. J Biol Chem 1999; 274:35297 - 300; http://dx.doi.org/10.1074/jbc.274.50.35297; PMID: 10585392
- Bech-Otschir D, Kraft R, Huang X, Henklein P, Kapelari B, Pollmann C, et al. COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J 2001; 20:1630 - 9; http://dx.doi.org/10.1093/emboj/20.7.1630; PMID: 11285227
- Zhao R, Yeung SC, Chen J, Iwakuma T, Su CH, Chen B, et al. Subunit 6 of the COP9 signalosome promotes tumorigenesis in mice through stabilization of MDM2 and is upregulated in human cancers. J Clin Invest 2011; 121:851 - 65; http://dx.doi.org/10.1172/JCI44111; PMID: 21317535
- Zhang XC, Chen J, Su CH, Yang HY, Lee MH. Roles for CSN5 in control of p53/MDM2 activities. J Cell Biochem 2008; 103:1219 - 30; http://dx.doi.org/10.1002/jcb.21504; PMID: 17879958
- Choi HH, Gully C, Su CH, Velazquez-Torres G, Chou PC, Tseng C, et al. COP9 signalosome subunit 6 stabilizes COP1, which functions as an E3 ubiquitin ligase for 14-3-3σ. Oncogene 2011; 30:4791 - 801; http://dx.doi.org/10.1038/onc.2011.192; PMID: 21625211
- Wei N, Deng XW. The COP9 signalosome. Annu Rev Cell Dev Biol 2003; 19:261 - 86; http://dx.doi.org/10.1146/annurev.cellbio.19.111301.112449; PMID: 14570571
- Lee MH, Zhao R, Phan L, Yeung SC. Roles of COP9 signalosome in cancer. Cell Cycle 2011; 10:3057 - 66; http://dx.doi.org/10.4161/cc.10.18.17320; PMID: 21876386
- Karniol B, Chamovitz DA. The COP9 signalosome: from light signaling to general developmental regulation and back. Curr Opin Plant Biol 2000; 3:387 - 93; http://dx.doi.org/10.1016/S1369-5266(00)00101-1; PMID: 11019806
- Li L, Deng XW. The COP9 signalosome: an alternative lid for the 26S proteasome?. Trends Cell Biol 2003; 13:507 - 9; http://dx.doi.org/10.1016/j.tcb.2003.08.002; PMID: 14507477
- Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994; 78:59 - 66; http://dx.doi.org/10.1016/0092-8674(94)90572-X; PMID: 8033212
- Lee MH, Reynisdóttir I, Massagué J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev 1995; 9:639 - 49; http://dx.doi.org/10.1101/gad.9.6.639; PMID: 7729683
- Lee MH, Yang HY. Negative regulators of cyclin-dependent kinases and their roles in cancers. Cell Mol Life Sci 2001; 58:1907 - 22; http://dx.doi.org/10.1007/PL00000826; PMID: 11766887
- Yan Y, Frisén J, Lee MH, Massagué J, Barbacid M. Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev 1997; 11:973 - 83; http://dx.doi.org/10.1101/gad.11.8.973; PMID: 9136926
- Zhang P, Liégeois NJ, Wong C, Finegold M, Hou H, Thompson JC, et al. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 1997; 387:151 - 8; http://dx.doi.org/10.1038/387151a0; PMID: 9144284
- Lee MH, Yang HY. Regulators of G1 cyclin-dependent kinases and cancers. Cancer Metastasis Rev 2003; 22:435 - 49; http://dx.doi.org/10.1023/A:1023785332315; PMID: 12884917
- Kavanagh E, Joseph B. The hallmarks of CDKN1C (p57, KIP2) in cancer. Biochim Biophys Acta 2011; 1816:50 - 6; PMID: 21447370
- Guo H, Tian T, Nan KJ, Wang W. p57: A multifunctional protein in cancer (Review). [Review] Int J Oncol 2010; 36:1321 - 9; PMID: 20428755
- Borriello A, Caldarelli I, Bencivenga D, Criscuolo M, Cucciolla V, Tramontano A, et al. p57(Kip2) and cancer: time for a critical appraisal. Mol Cancer Res 2011; 9:1269 - 84; http://dx.doi.org/10.1158/1541-7786.MCR-11-0220; PMID: 21816904
- Kamura T, Hara T, Kotoshiba S, Yada M, Ishida N, Imaki H, et al. Degradation of p57Kip2 mediated by SCFSkp2-dependent ubiquitylation. Proc Natl Acad Sci USA 2003; 100:10231 - 6; http://dx.doi.org/10.1073/pnas.1831009100; PMID: 12925736
- Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol 2001; 3:973 - 82; http://dx.doi.org/10.1038/ncb1101-973; PMID: 11715018
- Xue Y, Chen J, Choi HH, Phan L, Chou PC, Zhao R, et al. HER2-Akt signaling in regulating COP9 signalsome subunit 6 and p53. Cell Cycle 2012; 11:4181 - 90; http://dx.doi.org/10.4161/cc.22413; PMID: 23095642
- Jin J, Ang XL, Shirogane T, Wade Harper J. Identification of substrates for F-box proteins. Methods Enzymol 2005; 399:287 - 309; http://dx.doi.org/10.1016/S0076-6879(05)99020-4; PMID: 16338364
- Rinaldi T, Bolotin-Fukuhara M, Frontali L. A Saccharomyces cerevisiae gene essential for viability has been conserved in evolution. Gene 1995; 160:135 - 6; http://dx.doi.org/10.1016/0378-1119(95)00212-O; PMID: 7628709
- Penney M, Wilkinson C, Wallace M, Javerzat JP, Ferrell K, Seeger M, et al. The Pad1+ gene encodes a subunit of the 26 S proteasome in fission yeast. J Biol Chem 1998; 273:23938 - 45; http://dx.doi.org/10.1074/jbc.273.37.23938; PMID: 9727008
- Wei N, Deng XW. Making sense of the COP9 signalosome. A regulatory protein complex conserved from Arabidopsis to human. Trends Genet 1999; 15:98 - 103; http://dx.doi.org/10.1016/S0168-9525(98)01670-9; PMID: 10203806
- Maytal-Kivity V, Reis N, Hofmann K, Glickman MH. MPN+, a putative catalytic motif found in a subset of MPN domain proteins from eukaryotes and prokaryotes, is critical for Rpn11 function. BMC Biochem 2002; 3:28; http://dx.doi.org/10.1186/1471-2091-3-28; PMID: 12370088
- Lyapina S, Cope G, Shevchenko A, Serino G, Tsuge T, Zhou C, et al. Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science 2001; 292:1382 - 5; http://dx.doi.org/10.1126/science.1059780; PMID: 11337588
- Kim M, Nakamoto T, Nishimori S, Tanaka K, Chiba T. A new ubiquitin ligase involved in p57KIP2 proteolysis regulates osteoblast cell differentiation. EMBO Rep 2008; 9:878 - 84; http://dx.doi.org/10.1038/embor.2008.125; PMID: 18660753
- Arboleda VA, Lee H, Parnaik R, Fleming A, Banerjee A, Ferraz-de-Souza B, et al. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat Genet 2012; 44:788 - 92; http://dx.doi.org/10.1038/ng.2275; PMID: 22634751
- Riccio A, Cubellis MV. Gain of function in CDKN1C. Nat Genet 2012; 44:737 - 8; http://dx.doi.org/10.1038/ng.2336; PMID: 22735584
- Iyer SV, Iwakuma T. A novel link between the HER2-Akt and MDM2-p53 pathways via CSN6. Cell Cycle 2012; 11:4112; http://dx.doi.org/10.4161/cc.22606; PMID: 23099920
- Yang H, Zhao R, Yang HY, Lee MH. Constitutively active FOXO4 inhibits Akt activity, regulates p27 Kip1 stability, and suppresses HER2-mediated tumorigenicity. Oncogene 2005; 24:1924 - 35; http://dx.doi.org/10.1038/sj.onc.1208352; PMID: 15688030
- Reynisdóttir I, Polyak K, Iavarone A, Massagué J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev 1995; 9:1831 - 45; http://dx.doi.org/10.1101/gad.9.15.1831; PMID: 7649471
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 2004; 428:190 - 3; http://dx.doi.org/10.1038/nature02330; PMID: 15014502
- Laronga C, Yang HY, Neal C, Lee MH. Association of the cyclin-dependent kinases and 14-3-3 sigma negatively regulates cell cycle progression. J Biol Chem 2000; 275:23106 - 12; http://dx.doi.org/10.1074/jbc.M905616199; PMID: 10767298
- Yang HY, Shao R, Hung MC, Lee MH. p27 Kip1 inhibits HER2/neu-mediated cell growth and tumorigenesis. Oncogene 2001; 20:3695 - 702; http://dx.doi.org/10.1038/sj.onc.1204472; PMID: 11439332