Abstract
The p53 gene has been implicated in many cancers due to its frequent mutations as well as mutations in other genes whose proteins directly affect p53’s functions. In addition, high expression of p53 [wild-type (WT) or mutant] has been found in the cytoplasm of many tumor cells, and studies have associated these observations with more aggressive tumors and poor prognosis. Cytoplasmic mis-localization of p53 subsequently reduced its transcriptional activity and this loss-of-function (LOF) was used to explain the lack of response to chemotherapeutic agents. However, this hypothesis seemed inadequate in explaining the apparent selection for tumor cells with high levels of p53 protein, a phenomenon that suggests a gain-of-function (GOF) of these mis-localized p53 proteins. In this study, we explored whether the direct involvement of p53 in the apoptotic response is via regulation of the caspase pathway in the cytoplasm. We demonstrate that p53, when present at high levels in the cytoplasm, has an inhibitory effect on caspase-9. Concurrently, knockdown of endogenous p53 caused an increase in the activity of caspase-9. p53 was found to interact with the p35 fragment of caspase-9, and this interaction inhibits the caspase-9 activity. In a p53-null background, the high-level expression of both exogenous WT and mutant p53 increased the resistance of these cells to cisplatin, and the data showed a correlation between high p53 expression and caspase-9 inhibition. These results suggest the inhibition of caspase-9 as a potential mechanism in evading apoptosis in tumors with high-level p53 expression that is cytoplasmically localized.
Introduction
Since the discovery of p53,Citation1-Citation5 50% of human cancers have been found to have mutations in the TP53 gene, which directly compromise its functions, while in a large proportion of the remaining tumors, mutations or altered expression of other genes occur, which affect the functions of this important protein.Citation6 Across a wide-range of cancers, p53 expression is associated with malignant progression, metastasis and poor prognosis,Citation7-Citation9and similar observations were made in mouse models.Citation10 Such observations had led to numerous studies investigating the relationship between p53 status and clinical response.
In tumors bearing a p53 mutation, a large percentage is missense mutations, i.e., full-length p53 proteins with a single amino-acid change.Citation11 Among the mutations found, certain residues such as 175, 248 and 273 are hot spots for mutations. These residues are within the DNA-binding domain of p53 and have been found to be critical for p53’s binding to DNA and the induction of p53 target genes such as CDKN1A (p21).Citation12,Citation13 In addition, cytoplasmic localization of p53 was noted in varied tumor backgrounds.Citation14-Citation17 Hence, a common hypothesis used to explain the possible cause of these cancers is the inability of p53 (through mutation and/or cytoplasmic mislocalization) to maintain homeostasis due to the impaired transactivation of target genes involved in important cell processes, such as cell cycle arrest and apoptosis.
Although the above hypothesis is supported by studies showing the relationship between p53 cytoplasmic localization and tumor metastasis and poor prognosis,Citation18,Citation19 the LOF hypothesis seemed inadequate in explaining the concurrent findings of high expression of both WT and mutant p53 in the cytoplasm of tumors.Citation20-Citation22 These data suggest a possible GOF of p53 in eliciting oncogenic transformation, which is specific to the cytoplasmic milieu. Many mechanisms have been suggested, including the ability of mutant p53 to bind and inactivate WTp53, p63 and p73 and the inhibition of autophagy.Citation23,Citation24 In other studies, mutant p53 has been found to acquire other GOF by interacting with p63 to activate genes involved in metastasis and chemoresistance.Citation25,Citation26 Recently, mutant p53 was found to interact and inhibit mitochondrial caspase-3.Citation27 Nevertheless, WT and mutant p53 proteins are observed in remarkably high levels throughout the entire cytoplasm in tumor cells, suggesting that the global oncogenic role of mis-localized p53 may not be restricted to the mitochondria.
Caspases are a distinct, highly conserved class of intracellular cysteine proteases expressed as inactive proenzymes. Upon activation by receiving death stimuli, caspases are proteolytically cleaved to generate heterotetramers that are enzymatically active. Activated caspases then initiate a cascade of reactions, leading to distinct morphological characteristics, such as chromatin condensation, apoptotic body formation and, ultimately, cell death.Citation28 Caspases are normally found in the cytoplasm, with traces of localization (caspase-2, caspase-3 and caspase-9) in the mitochondria.Citation29 Based on the report by FrankCitation27 and the observation of overexpressed p53 in the cytoplasm of tumors, it is possible that p53 could be more intimately involved with the caspase family then initially thought. The GOF of p53 could extend to this family of proteins, which is critical for apoptosis.
To explore the role of p53 on caspase activation in the cytoplasm, we emulated the high concentration of p53 in the cytoplasm in tumor cells through the addition of recombinant p53 to cytoplasmic extracts. We found that high levels of cytoplasmic p53 inhibited the cleavage of caspase-9 in the presence of the chemotherapeutic drug, cisplatin. We showed that p53 interacted with caspase-9 directly and the re-expression of p53 in p53-null cells increased the resistance of these cells to cisplatin. In addition, a correlation between p53 overexpression and caspase-9 inhibition was observed in these experiments. The data suggest that the inhibition of caspase-9 may be a mechanism of apoptosis-evasion in tumors overexpressing cytoplasmically localized p53.
Results
p53 confers resistance to cisplatin in a p53-null background
Several studies have documented the increased resistance of tumor cells overexpressing p53 to chemotherapeutic drugs.Citation7-Citation9,Citation30 In our investigation, we first verified if a similar phenomenon could be observed in our cell lines. We used a H1299 cell line with inducible expression of p53R175H [ecdysone-inducible (EI)-R175H]. Cells were seeded at low density and treated with the inducing agent (ponasterone A) to express p53 for 24 h before subsequent exposure to increasing doses of cisplatin for 48 h. At 208 µM cisplatin concentration, EI-R175H cells exhibited an 8-fold increase in cell viability compared to the EI-Vector control (). Analysis of the cells by annexin V staining also showed that EI-R175H cells expressing the mutant p53 had a significant reduction in the population of annexin V-stained cells, indicating reduced level of apoptosis (). These results indicated a marked increase in chemoresistance in cells expressing the mutant p53R175H in conditions of acute exposure to cisplatin.
Figure 1. Presence of p53 confers resistance to cisplatin. (A) EI-H1299 cells were induced with ponasterone A to express p53 prior to treatment with increasing dosage of cisplatin. Cell viability was determined through 7-ADD staining, flow cytometry and analyzed using FlowJo software. (B) EI-H1299 cells were induced with ponasterone A to express p53 prior to treatment with 208 µM of cisplatin. Apoptosis was measured by detecting annexin V staining using flow cytometry and the data was analyzed using FlowJo software. (C) H1299 cells were induced with ponasterone A to express p53 prior to treatment with 25 µM cisplatin for 48 h. The cells were then allowed to recover in the absence of cisplatin over 12 days and were stained with crystal violet to visualize cell growth. Cytosolic extracts of the various cell lines were probed for expression of p53 (bottom panel). The area of cell growth was measured using ImageJ, and the graph (top panel) shows the change in cell growth area of the three cell lines between the induced and non-induced cells. (D) HCT-116 p53-/- cells were transfected with the various GFP-tagged p53 before 0.5 mM of cisplatin was added. Cells were counted at every 24-h period post administration of cisplatin. The percentages of transfected cells were calculated and the ratios of transfected cells in cisplatin to those in PBS were obtained. Un-normalized data used to calculate the fold change is presented in Figure S2.
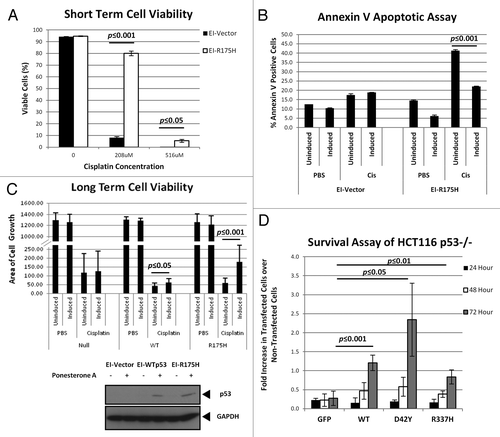
We next investigated if the inducible cell lines were able to exhibit chemoresistance to cisplatin at low dosage of 25 µM. Similar to the earlier experiment, cells (with the inclusion of the cell line expressing WTp53 from a ponasterone A-inducible promoter; EI-WTp53) were induced with ponasterone A to express p53 for 24 h to cisplatin administration, after which, cells were allowed to recover in a medium without cisplatin but still containing ponasterone A (to maintain p53 expression) for 12 d. Cells were then stained with crystal violet to determine the density of cell growth. The images were scanned and analyzed using ImageJ to determine the area of cell growth. In the presence of cisplatin, EI-WTp53 and EI-R175H cells showed more robust cell growth under the induction of ponasterone A as compared to their counterparts with no expression of p53. While EI-WTp53-expressing p53 was able to recover slightly better (approximately 50% more) than its non-induced counterpart, EI-R175H showed a significantly higher density of cell growth (about 3-fold) when p53 expression was induced (, top panel). The expression of p53 in the cell lines was verified using immunoblots for p53 on the cytoplasmic extracts of the cell lines (, bottom panel).
To explore if p53 with mutations at other sites could also confer such levels of resistance, we chose to work with two other p53 mutants, namely p53D42Y and p53R337H. The D42Y mutation is within the N terminus of the protein, while R337H resides in the tetramerization domain of p53. The use of these two mutants was intended to complement the results of the R175H mutation (which resides within the DNA-binding domain of p53). The HCT116 p53-/- cell line was used as a p53-null background to re-express either the p53D42Y and p53R337H mutants or WTp53 as a control. Cells were transfected with plasmids containing the various GFP-tagged p53 genes before cisplatin administration. As these cells only have transient expression of p53, we evaluated the changes in the population of cells with p53 expression over a 72-h period. Cells expressing the GFP-tagged p53 can be easily counted under the fluorescence microscope. In the presence of cisplatin, the percentage of cells with p53 expression (expressed over total number of cells counted with Hoechst 33342 stain) increased steadily over the 72-h experimental period. At the end of the 72 h, cells expressing p53D42Y showed the highest resistance to cisplatin (approximately 9-fold increase over GFP control), followed by WTp53 and p53R337H, both with 4-fold and 3-fold increase over the GFP control, respectively (). Taken together, these findings demonstrate a global role for both WT and mutant p53 to drive chemoresistance in cancer cell lines.
Inhibition of caspase-9 is specific to p53
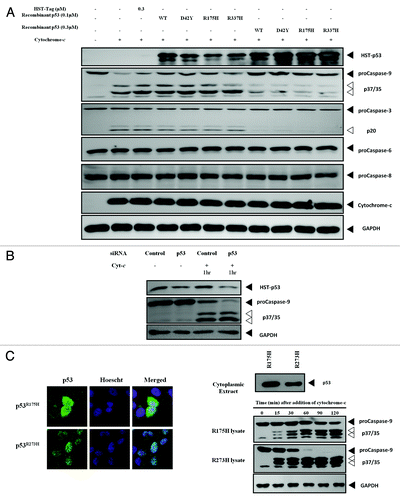
From the recent work done on the GOF of p53 and that by Frank, we hypothesized that p53’s GOF could be acting on the caspase family of proteins in the cytoplasm, particularly the caspase pathways downstream of the mitochondria. To test this hypothesis, bacterially produced and purified recombinant p53 (both WT and mutant) was added to S100 cytosolic extracts from HCT116 p53-null cells. Apoptosis was induced with the addition of recombinant cytochrome-c (rCyt-c) to study the transcription-independent effects of p53. The addition of rWTp53 and of mutant p53 proteins led to a decrease in the active, cleaved p37/p35 bands of caspase-9, with the concurrent restoration of the p46 proform. Similarly, the downstream caspase-3 also showed a reduction in the intensity of the cleaved band p20, with the restoration of the procaspase-3 band of p34. Both caspase-6 and caspase-8, which are further downstream of caspase-3, did not show any change in the cleavage profiles (). As the inhibition was observed on caspase-9, the apical caspase of the mitochondrial apoptotic pathway, caspase-9 was assumed to be the target of p53’s inhibitory action. To confirm this inhibitory effect, we performed a knockdown of endogenous p53 using siRNA in HEK293 cells, a cell line with high cytoplasmic p53 levels. The knockdown of p53 resulted in an increase of caspase-9 cleavage when apoptosis was induced with rCyt-c (), confirming what we observed with recombinant p53 proteins’ effect on caspase-9. We then isolated the cytosolic extracts of H1299 cells stably expressing either p53R175H or p53R273H and induced caspase-9 cleavage through the addition of rCyt-c. Lysates were harvested at various time points to analyze the caspase-9 profile. Cells expressing p53R175H had a higher level of cytoplasmic p53 than those expressing p53R273H, and this correlated with accelerated caspase-9 cleavage in the latter (). The data obtained suggests that p53 does possess a GOF for the caspase family, in particular caspase-9. In addition, the data showed the correlation between cytoplasmic p53 levels and the extent of caspase-9 inhibition.
Figure 2. Different cytoplasmic levels of p53 affect cleavage of caspase-9. (A) Recombinant WT and mutant p53 were added to cytochrome-c challenged S100 lysates of HCT116 p53-/- cells and the caspase cleavage profiles were probed. (B) Endogenous p53 in HEK293 cells was knocked-down using siRNA and the S100 cytosolic extracts of the cells were induced with recombinant cytochrome-c to initiate apoptosis. (C) Immunocytochemistry showing the localization of p53R175H and p53R273H in H1299 cells stably expressing these mutants (left panel). Immunoblots of p53 and caspase-9 of the cytochrome-c challenged S100 lysates from each cell type at different time points were probed (right panel).
p53 inhibits the activity of caspase-9 directly in an in vitro enzyme assay
To study if the inhibition of caspase-9 by p53 is mediated through direct interaction, a recombinant system consisting of the various p53 proteins and recombinant active caspase-9 was developed. Caspase-9 activity was measured using colorimetric assays, and significant inhibition (more than 50%) of caspase-9 activity was observed in the presence of p53 (, left panel; see also Fig. S2A). In addition, a dose-dependent inhibition of caspase-9 activity by the recombinant p53 proteins was evident (, right panel; see also Fig. S2B).
Figure 3. WT and mutant p53 interacts directly with caspase-9.(A) One µM of recombinant p53 was incubated with recombinant active caspase-9 and the caspase-9 activity was measured after 1 h incubation (left panel). Increasing concentrations of the various recombinant p53 used in (A) were incubated with recombinant active caspase-9, and the caspase-9 activity was measured after 1 h incubation (right panel). (B) Procaspase-3 was expressed using an IVT system and the recombinant active caspase-9-induced cleavage was probed in the presence of control proteins and recombinant WTp53 after 2 h incubation. (C) One µM of recombinant p53 was added to recombinant active caspase-3 (left panel) or caspase-6 (right panel) and the activities of the respective caspases were measured after 1 h incubation.
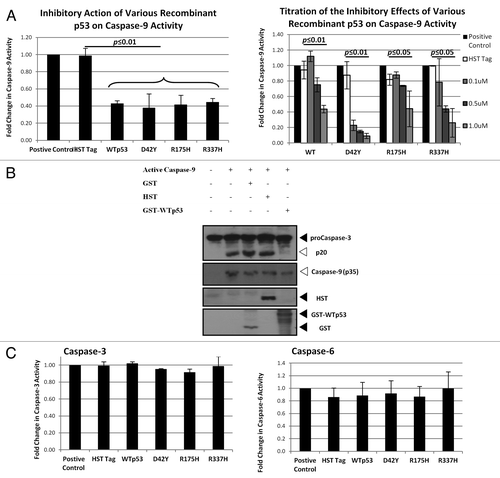
To further test this phenomenon, caspase-3 was expressed using an in vitro translation (IVT) system, and activation of this caspase was induced by the addition of recombinant active caspase-9. In this system, the presence of recombinant WTp53 resulted in a reduction of caspase-3 cleavage, indicating the inhibition of caspase-9 by WTp53 (). We tested if the p53-dependent inhibition of caspase-9 can be observed on other caspases. Our data confirmed that this inhibition was specific to caspase-9 as no significant reduction in activities was observed against pure recombinant active caspase-3 or caspase-6 using the colorimetric assay system ().
p53 interacts with p35 of caspase-9
To further demonstrate the direct interaction of p53 and caspase-9, co-immunoprecipitation (co-IP) experiments were performed using whole-cell lysates and S100 cytosolic extracts of cells that have been induced to undergo apoptosis. However, an endogenous interaction between p53 and caspase-9 could not be detected under these experimental conditions (data not shown). It appears that in normal physiological conditions, the concentration of cytoplasmically localized p53 is too low to allow for the detection of this interaction. Instead, recombinant p53 was expressed using IVT to achieve a sufficiently high concentration of the protein. Recombinant active caspase-9 was then added to the protein and co-IP was performed. Mdm2, a well-known negative regulator of p53 known to bind strongly to the proteinCitation31 was also expressed using IVT system and included as a positive control. Here, the co-IP of both p53 and the p35 fragment of caspase-9 were observed, verifying the direct in vitro interaction between the two proteins ().
Figure 4. WTp53 interacts with the p35 fragment caspase-9.(A) p53 was expressed using an IVT system, and immunoprecipitation was performed in the presence of IVT-expressed MDM2 or recombinant active caspase-9. (B) Apoptosis was induced with dATP and rCyt-c in S100 lysates of H1299 cells stably expressing p53R175H. Antibody pairs between DO-1 (p53) and the above was added to the lysates and anti-mouse coated (donor ) and anti-rabbit (acceptor) beads were used to detect interactions between the proteins (top panel). Fluorescence signal were measured for lysates induced for apoptosis and expressed as a ratio over signal obtained for uninduced lysates for each antibody pair (bottom left panel). Immunoblots for p53 and caspase-9 was done for the lysates used for this assay (bottom right panel).
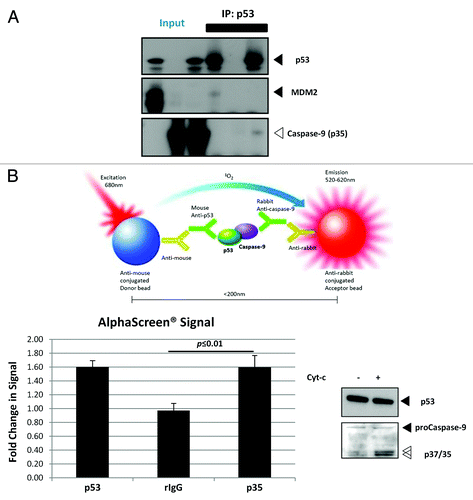
The observation that the detection of an endogenous p53-caspase-9 interaction falls below the limits of sensitivity of traditional co-IP protocols suggests that the interaction between the two endogenous proteins could also be transient. Here, the AlphaScreen® assay was used to detect the direct interaction between p53 and caspase-9 by using the H1299 cells stably expressing p53R175H, shown to harbor high levels of p53 in the cytoplasm (). In , p53 and active caspase-9 antibody pair gave almost similar levels of fluorescence as compared to the positive control between the two different antibodies targeting p53 itself. These data indicated that there is a direct interaction between p53 and the p35 fragment of active caspase-9, and that the interaction between the two proteins requires p53 to be in sufficiently high concentrations.
Inhibition of caspase-9 activities correlated with expression of WT and mutant p53
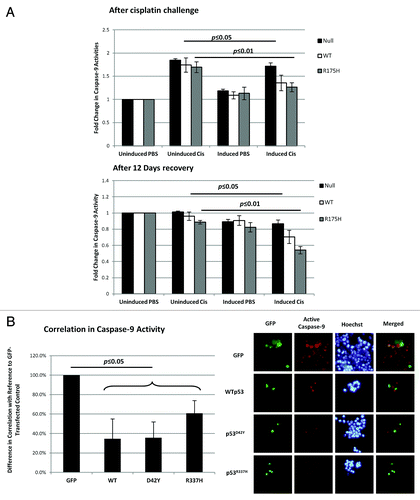
The results obtained in this study so far suggest that the inhibition of caspase-9 by cytoplasmically localized p53 can be a mechanism in evading cell death. As such, we would expect that the increased cell survival observed in both the H1299-inducible cell lines and the HCT116 p53-null with transient p53 expression was due to impeded caspase-9 activity. To test this hypothesis, the experiments shown in were repeated to measure the caspase-9 activity. In the H1299 with inducible p53 expression, caspase-9 activity of those cells expressing WTp53 and p53R175H was significantly lower compared to those without p53 expression (). The measured caspase-9 activity immediately after the 48-h challenge with cisplatin (, top panel) and after the 12-d cisplatin-free recovery period (, bottom panel) showed the same expected results. For the HCT116 p53-null cells with transient p53 expression, a caspase-9-specific substrate, which yields a fluorescence product upon cleavage by active endogenous caspase-9, was added to the cells after the 72-h cisplatin challenge. Cells were imaged, and the correlation between caspase-9 activity and p53-positive cells was assessed. The cellular expression of p53 correlated with lowered caspase-9 activity (). Together with the results in , the data indicated a suppression of caspase-9 activity, suggesting the increased chemoresistance to cisplatin was through the inhibition of caspase-9.
Figure 5. Resistance to cisplatin in the presence of p53 correlated with the inhibition of caspase-9 activity. (A) H1299 cells were induced with ponasterone A to express p53 prior to treatment with 25 µM cisplatin for 48 h. After 48 h with cisplatin, caspase-9 activity was measured and normalized to cell density (top panel). The cells were then allowed to recover in the absence of cisplatin over 12 d. Caspase-9 activity after the 12-d recovery period was also measured (bottom panel). (B) HCT-116 p53-/- cells were transfected with the various GFP-tagged p53 before 0.5 mM of cisplatin was added. At 72 h, substrates that are specific to the action of active caspase-9 were added to the cells to yield a fluorescent product 1 h prior to imaging. Cells positive for both GFP fluorescence and caspase-9 activity were counted and expressed as a ratio over the total number of GFP-fluorescence-positive cells (left panel). The right panel shows the representative images used to calculate the correlation percentages.
Discussion
The data from this study demonstrated p53’s ability to inhibit caspase-9 activity through direct interaction with the p35 fragment. Previous studies have shown p53’s ability to influence the expression levels of caspases, such as caspase-3 and caspase-6.Citation32,Citation33 However, our study is one of the first to show a direct interaction of p53 with a member of the caspase family of proteins. The use of a cell-free system derived primarily from cytosol devoid of nuclear content has allowed a focus on the transcription-independent influence of p53 in the caspase activation cascade. Also, the lack of mitochondria in the cell-free system, as well as the manual addition of rCyt-c to the lysates, limited the influence of p53 on the Bax/Bak-mediated cytochrome-c release from the mitochondria, events which are upstream of caspase activation in cells,Citation34,Citation35 underscoring the importance of the use of the cell-free system in uncovering this hitherto unknown oncogenic facet of p53 that is unrelated to its well-studied transactivation function.
In this study, p53 was found to interact with the p35 fragment of caspase-9. The idea of an inhibitor that targets the active form of caspases is not unprecedented in the field of apoptosis study. Protein families, such as those of IAP (inhibitor-of-apoptosis) proteins, have the ability to regulate both initiator and effector caspases by binding to and inhibiting the actions of cleaved caspases, such as caspase-9 and caspase-3.Citation36,Citation37 p53’s interaction and inhibition of caspase-9 at the p35 fragment is analogous to that of the x-linked inhibitor of apoptosis protein (XIAP), where XIAP was found to bind to monomeric caspase-9.Citation38 As the activity of caspase-9 is amplified manifold when present as a heterodimer of p35 and p12 subunits, the binding action of XIAP to monomeric caspase-9 retards the ability of caspase-9 to form highly active heterodimer molecules.Citation37,Citation39 Although there is no evidence in this study to determine the mechanism of p53’s inhibition on active caspase-9, it is plausible that p53 might act and inhibit caspase-9 in a similar fashion as XIAP in its binding mechanism.
The inhibition of caspase-9, the most apical caspase of the mitochondrial apoptotic pathway, can have a profound effect on cell fate. When activated, downstream targets such as caspase-3 and PARP are cleaved to enable apoptosis to take place.Citation40 In addition, a small amount of caspase-3 is usually shunted into the caspase amplification loop for rapid signal amplification. In this amplification loop, activated caspase-3 cleaves and activates caspase-6,Citation41 then caspase-8 by caspase-6,Citation42 and subsequently, activated caspase-8 can back cleave caspase-3 to complete the amplification loop.Citation43 The activation of this caspase amplification loop can lead to an escalation of caspase activities, resulting in a swift death response. The ability of p53 to inhibit caspase-9 can potentially thwart the initiation of this pathway. Together with other known inhibitors of caspases, such as XIAP, Bcl2L12Citation44,Citation45 and nucleophosmin,Citation46 the threshold for apoptosis in cells with sufficiently high levels of cytoplasmic p53 could be raised to a level where chemotherapeutic drugs could no longer elicit cell death.
In this study, the mutant p53 conferred different levels of chemoresistance to cisplatin, with p53D42Y showing the highest level of protection against cisplatin, followed by p53R337H. The D42Y mutation is located within the transactivation domain of p53 and characterized by an amino acid substitution from a positive electrically charged side group to one with a hydrophobic side group. This change in side chain properties could accentuated the cisplatin-induced cell death resistance, probably through a structural change in the protein, which retards p53’s transactivation property, presumably by inhibiting the phosphorylation of serine 46, which is essential for the transactivation of the pro-apoptotic gene, p53AIP1.Citation47 p53R337H, on the other hand, has a less drastic change in the side chain properties (the substitution involved amino acid side chains, which are both positively charged). The mutation at amino acid residue 337 (located within the tetramerization domain) lowers the ability of p53 to tetramerize, an essential step for the transactivation of p53 target genes.Citation48 Coupled to caspase-9 inhibition, the lack of transactivation by the mutant p53 could increase the threshold of cells to cytotoxin-induced apoptosis. More studies will have to be done to determine the exact mechanisms involved in the different levels of cytoprotection of the mutant p53.
It is interesting to note that from our study, WTp53 (like the mutants used in this study) can also inhibit active caspase-9. It appears that this inhibitory role is an inherent function of WTp53, which has probably eluded detection as WTp53 is usually very unstable (and therefore present in very low amounts) in the cytoplasm due to the rapid degradation by the 26S proteosome.Citation49 However, the choice of HEK293 cells in this study has enabled this phenomenon to be observed. HEK293 cells were immortalized using adenovirus type 5.Citation50 Since E1B-55K proteins of adenoviruses has been shown to bind p53 and sequester it in the cytoplasm,Citation51,Citation52 the high levels of cytoplasmic p53 in these cell lines can then be manipulated to show the inhibitory effect on caspase-9.
Although this study demonstrated the inhibitory effect of p53 (both WT and mutant) on caspase-9, p53 has to be in sufficiently high amounts for this effect to be observed. Also, since caspase-9 is predominantly in the cytoplasm, the subcellular localization of p53 also plays an important role for the inhibition to be pronounced. This suggests that in normal physiological conditions, the inhibition of caspase-9 by p53 could be very low. This could also explain our unsuccessful co-IP attempts to demonstrate the interaction between endogenous p53 and caspase-9. However, in tumor cells overexpressing cytoplasmically localized p53, the impact of such inhibition becomes significant. Data from this study shows the increased chemoresistance to cisplatin cytotoxicity of cells expressing p53 and implicates the suppression of caspase-9 activity as a mechanism of action.
Besides its transactivation role, p53 have been reported to have transcription-independent roles in the cytosol. These studies claimed that p53 can bind to Bcl-2 and Bcl-xL proteins (both of which are anti-apoptotic) to release pro-apoptotic proteins such as Bax.Citation53 Bax, upon activation by p53, can initiate the permeabilization of the mitochondrial membrane, allowing the release of cytochrome-c, which can then activate caspase-9.Citation35,Citation54 p53 with R175H mutation was found to be defective in its interaction with Bcl-2 and Bcl-xL.Citation53 However, no available data can be found on the ability of mutant p53 to activate Bax. It is possible that the inhibitory effect on caspase-9 by the mislocalized p53 in tumor cells has a cumulative effect with mutant p53’s LOF in transactivation (through mutation and nuclear exclusion) and activation of mitochondrial membrane permeabilization, which may then account for the more aggressive nature of such tumors. To address this question in greater detail, more experiments are necessary to compare the effects of nuclear vs. cytoplasmically localized p53 on caspase-9 activity and cell growth in the presence of chemotherapeutic agents.
The findings from this study adds to the list of new functions that both WT and mutant p53 possess, which together can potentially raise the level of chemoresistance in tumors with high levels of p53 protein. Therefore, it is imperative to look for new chemotherapeutic drugs that can activate the apoptotic pathways regardless of the p53 status. A recent work by MurphyCitation55 presented a new class of transplatinum compound, which could elicit cell death in both chemosensitive (harboring WTp53) and cisplatin- and oxaliplatin-resistant (harboring mutant p53) cells. It will be interesting to see if the use of these transplatinum compounds could restore the ability of cells to overcome the inhibitory action of p53 on caspase-9 shown in our study. However, the application of a single drug could result in the development of new chemoresistance. As such, beyond the quest for new chemotherapeutic drugs, combination drug therapy (as reviewed by BlagosklonnyCitation56) may be one solution to fight these chemoresistant tumors.
Materials and Methods
Generation of EI-H1299 cell lines expressing WT and mutant p53
H1299 cells were maintained in DMEM supplemented with 10% FCS. To establish the EI-H1299 base cell line, pVgRXR was stably transfected into H1299 cells using Lipofectamine 2000 (Invitrogen; #11668) according to the manufacturer’s protocol. Single clones were selected in 100 μg/mL zeocin (Invitrogen; #R-250). The base EI H1299 cell line was selected through screening by transient transfection with pIND-GFP, followed by treatment with 2.5 μg/mL Ponasterone A (Invitrogen; #H101). Inducible p53-expression constructs (p-TK-Hygro-p53WT/MUT) were stably transfected into the EI H1299 base cell line. Clones were selected at limiting dilutions in 600 μg/mL Hygromycin B (Sigma Aldrich; #H3274).
Cell lines
Human embryonic kidney (HEK) 293 cells (a generous gift from Dr. Low Boon Chuan, NUS) were cultured in RPMI medium (Sigma-Aldrich; #R8758) supplemented with 10% fetal bovine serum (FBS) (Gibco; #10437028) and 1% penicillin/streptomycin (Gibco; #15070). Human non-small cell lung carcinoma H1299 p53-/- (ATCC), H1299 EI-vector, EI-WTp53 and EI-R175H were cultured in DMEM (Sigma-Aldrich) supplemented with 10% FBS and 1% penicillin-streptomycin. Human colon tumor (HCT)-116 p53-/- were cultured in McCoy 5A medium (Sigma-Aldrich; #M4892) supplemented with 10% FBS and 1% penicillin-streptomycin.
Reagents, antibodies and immunoblotting
Cisplatin [cis-Diamineplatinum(II) dichloride], Hoechst-33342, dATP and rCyt-c were purchased from Sigma-Aldrich (#P4393, #B2261, #D6500, #C6749). Caspase-3, caspase-9, caspase-6 and caspase-8 antibodies were from Cell Signaling Technology (#9662, #9502, #9762, #9746); p53 antibody (DO-1) from Santa Cruz Biotechnology (#sc-126); GAPDH from Ambion (#Am4300); Alexa-488 from Invitrogen (#A21151); S-tag-HRP from Bethyl Laboratories (#A190-134P), and MDM2 antibody (2A10) was a kind gift from Dr. Borek Vojtesek. Immunoblots were developed using enhanced chemiluminescence (Pierce; #34077).
Cytosolic fraction extraction
Cells were harvested, washed twice in PBS and resuspended in ice-cold IDP buffer (20 mM Hepes-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1% protease inhibitor cocktail and 3 mM DTT). After incubation on ice, the cells were disrupted by dounce homogenizing in a Wheaton Dounce Homogenizer (Millville; #357542) with a tight pestle. Cell extracts were directly centrifuged at 21,000 g for 20 min at 4oC. The resulting supernatants (S100 cell-free extracts) were collected.
p53 knockdown
A siRNA duplex targeting p53 (Cell Signaling Technology, #6231S) was transfected into HEK293 cells using the DharmaFECT transfection reagent (Dharmacon Inc; #T-2001) for 48 h before the S100 cytosolic extracts were obtained. As negative control, a control siRNA duplex (Cell Signaling Technology; #6231) was used.
In vitro caspase-9 assays
Recombinant p53 proteins were produced and analyzed using circular dichroism (Fig. S3) as previously described.Citation57 The various p53 proteins were added to S100 lysates of HCT116 p53-/-cells before apoptosis were induced with dATP and rCyt-c. Lysates were subsequently analyzed for caspase-9 profile using immunoblotting. In the recombinant protein enzyme assay experiments, the various p53 proteins were incubated with recombinant active caspase-9 (Calbiochem; #218807) in reaction buffer before caspase-9 activity was measured using 200 µM of LEHD-pNA substrate (BioVision Inc; #K119), and the yield of the colored pNA product was measured at 405 nm.
IVT system and co-IP
WT caspase-3 was expressed using an IVT system (Promega; #L1170). Recombinant GST-WTp53 (Santa Cruz; #sc-4246) and the various tags were added to the protein mixture before caspase-3 cleavage was induced using recombinant active caspase-9 for 2 h at room temperature.
Recombinant WTp53 and MDM-2 were produced using another IVT system (Novagen; #70876). The proteins and recombinant active caspase-9 were incubated in IDP buffer. p53 antibody coupled to Protein-G coated Dyna-beads® (Invitrogen; #100) was used to co-immunoprecipitate p53 and the interacting proteins.
AlphaScreen® assay
H1299 cells stably expressing p53R175H was harvested for the S100 cytosolic fraction as described above. Apoptosis was induced with dATP and rCyt-c. Rabbit antibodies to p53 (CM-1), p35 fragment of caspase-9 and control rabbit IgG were added to acceptor beads coated with anti-rabbit antibodies while mouse DO-1 antibodies was added to donor beads coated with anti-mouse antibodies and incubated for 30 min. The acceptor beads-rabbit antibody mix were added to the lysates and incubated for 30 min before the donor beads-DO-1 mix was added and incubated for another 30 min. All incubation steps were performed at room temperature. Fluorescence at 520‒620 nm was measured to detect close proximity between the protein pairs.
Apoptotic assay
EI-R175H cells were seeded at 1 × 105 cells per 12-well and incubated with either PonA (2.5 μg/mL) or vehicle control. Following 24 h of p53R175H induction, cells were treated with cisplatin (208 μM) or vehicle control as indicated for 20 h. Cells were harvested, washed twice in cold PBS and then resuspended in 100 μL of binding buffer (140 mM NaCl, 2.5 mM CaCl2 and 10 mM HEPES pH 7.4) and incubated with 5 μL of Annexin V-FITC (BD Bioscience; #556570) and 200 ng of 7-AAD viability dye (Invitrogen; #A1310) in the dark for 15 min. Samples were processed using a FACScalibur flow cytometer (BD Bioscience) and analyzed using FlowJo software (Tree Star Inc.).
HCT116 p53-/- cells were transfected with pXJ40-HA-GFP, pXJ40-HA-GFP-WTp53/pXJ40-HA-GFP-p53D42Y/pXJ40-HA-GFP-p53R175H/pXJ40-HA-GFP-p53R337H using Lipofectamine-2000 before cisplatin was administered 6 h post-transfection. At every 24 h, cells were stained with Hoechst-33342 before images were taken. For caspase-9 activity assays, fluorescence caspase-9 substrate, Red-LEHD-FMK from Caspase-9 Detection Kit (Calbiochem; #QIA116) was added to the cells 1 h before images were taken. All images were processed and data was obtained using ImageJ (National Institute of Health). Images for both GFP and caspase-9 red-LEHD-FMK were transformed by ImageJ using a set threshold. Images at each field for both fluorescence were merged. Cells that were positive for both GFP and caspase-9 were counted using the “analyze/analyze particles” function in ImageJ and expressed over GFP-positive cells.
EI-H1299 cells were seeded in 96-well plates, induced for p53 expression with ponasterone A (1 µg/ml) for 24 h prior to treatment with 25 µM cisplatin for 48 h. After the 48-h cisplatin treatment and 12 d of recovery in cisplatin-free medium, caspase-9 activity was measured using the Caspase-Glo®-9 (Promega; #G8211) assay according to manufacturer’s instructions.
Cell viability assay
EI-H1299 cells were seeded at low density in 6-well plates. p53 expression was induced with ponasterone A (1 µg/mL) for 24 h before the indicated dose of cisplatin was administered for a further 48 h. The cell viability was assessed using 7-AAD as previously described.Citation58
Cell growth assay
For long-term growth assays, cells were seeded in 6-well plates, induced for p53 expression and treated with cisplatin as described for cell viability assay. After cisplatin treatment, cells were allowed to recover in DMEM without cisplatin for 12 d. Cells were stained with crystal violet to visualize cell growth density. Cell growth density was determined using ImageJ (NIH). All images were transformed using a set threshold. As the cell growth pattern was often irregularly shaped, the freehand tool was used to draw around the area with highest cell growth density. The area within was then determined using the “Analyze/Measure” function.
Additional material
Download Zip (447.1 KB)Acknowledgements
The authors would like to thank Yan T., Hayford C. and Siau J.W. for resource management; Tan B.X. and Ahmad B. for their kind assistance and materials provided.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note
This work was supported by Ministry of Education (Singapore) Tier 2 grant (AcRF grant T208B3112) and the Agency of Science, Technology and Research (Singapore).
References
- Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979; 17:43 - 52; http://dx.doi.org/10.1016/0092-8674(79)90293-9; PMID: 222475
- Kress M, May E, Cassingena R, May P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol 1979; 31:472 - 83; PMID: 225566
- Melero JA, Stitt DT, Mangel WF, Carroll RB. Identification of new polypeptide species (48-55K) immunoprecipitable by antiserum to purified large T antigen and present in SV40-infected and -transformed cells. Virology 1979; 93:466 - 80; http://dx.doi.org/10.1016/0042-6822(79)90250-2; PMID: 222051
- Smith AE, Smith R, Paucha E. Characterization of different tumor antigens present in cells transformed by simian virus 40. Cell 1979; 18:335 - 46; http://dx.doi.org/10.1016/0092-8674(79)90053-9; PMID: 227604
- Lane DR, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells.. Nature 1979; 278:261 - 3; PMID: 218111
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer 2006; 6:909 - 23; http://dx.doi.org/10.1038/nrc2012; PMID: 17128209
- Crook T, Vousden KH. Properties of p53 mutations detected in primary and secondary cervical cancers suggest mechanisms of metastasis and involvement of environmental carcinogens. EMBO J 1992; 11:3935 - 40; PMID: 1327751
- Allred DC, Clark GM, Elledge R, Fuqua SA, Brown RW, Chamness GC, et al. Association of p53 protein expression with tumor cell proliferation rate and clinical outcome in node-negative breast cancer. J Natl Cancer Inst 1993; 85:200 - 6; http://dx.doi.org/10.1093/jnci/85.3.200; PMID: 8423624
- Levesque MA, Katsaros D, Yu H, Zola P, Sismondi P, Giardina G, et al. Mutant p53 protein overexpression is associated with poor outcome in patients with well or moderately differentiated ovarian carcinoma. Cancer 1995; 75:1327 - 38; http://dx.doi.org/10.1002/1097-0142(19950315)75:6<1327::AID-CNCR2820750615>3.0.CO;2-P; PMID: 7882283
- Goh AM, Coffill CR, Lane DP. The role of mutant p53 in human cancer. J Pathol 2011; 223:116 - 26; http://dx.doi.org/10.1002/path.2784; PMID: 21125670
- IARC TP53 Mutation Database [Internet]. Available from: http://www-p53.iarc.fr/Statistics.html
- Watcharasit P, Bijur GN, Zmijewski JW, Song L, Zmijewska A, Chen X, et al. Direct, activating interaction between glycogen synthase kinase-3beta and p53 after DNA damage. Proc Natl Acad Sci USA 2002; 99:7951 - 5; http://dx.doi.org/10.1073/pnas.122062299; PMID: 12048243
- Thukral SK, Blain GC, Chang KK, Fields S. Distinct residues of human p53 implicated in binding to DNA, simian virus 40 large T antigen, 53BP1, and 53BP2. Mol Cell Biol 1994; 14:8315 - 21; PMID: 7969167
- Schlamp CL, Poulsen GL, Nork TM, Nickells RW. Nuclear exclusion of wild-type p53 in immortalized human retinoblastoma cells. J Natl Cancer Inst 1997; 89:1530 - 6; http://dx.doi.org/10.1093/jnci/89.20.1530; PMID: 9337350
- Bosari S, Viale G, Roncalli M, Graziani D, Borsani G, Lee AK, et al. p53 gene mutations, p53 protein accumulation and compartmentalization in colorectal adenocarcinoma. Am J Pathol 1995; 147:790 - 8; PMID: 7677190
- Moll UM, Riou G, Levine AJ. Two distinct mechanisms alter p53 in breast cancer: mutation and nuclear exclusion. Proc Natl Acad Sci USA 1992; 89:7262 - 6; http://dx.doi.org/10.1073/pnas.89.15.7262; PMID: 1353891
- Moll UM, LaQuaglia M, Bénard J, Riou G. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc Natl Acad Sci USA 1995; 92:4407 - 11; http://dx.doi.org/10.1073/pnas.92.10.4407; PMID: 7753819
- Stenmark-Askmalm M, Stål O, Sullivan S, Ferraud L, Sun XF, Carstensen J, et al. Cellular accumulation of p53 protein: an independent prognostic factor in stage II breast cancer. Eur J Cancer 1994; 30A:175 - 80; http://dx.doi.org/10.1016/0959-8049(94)90082-5; PMID: 7908819
- Sun XF, Carstensen JM, Zhang H, Stål O, Wingren S, Hatschek T, et al. Prognostic significance of cytoplasmic p53 oncoprotein in colorectal adenocarcinoma. Lancet 1992; 340:1369 - 73; http://dx.doi.org/10.1016/0140-6736(92)92558-W; PMID: 1360088
- Høgdall EVS, Christensen L, Høgdall CK, Frederiksen K, Gayther S, Blaakaer J, et al. Distribution of p53 expression in tissue from 774 Danish ovarian tumour patients and its prognostic significance in ovarian carcinomas. APMIS 2008; 116:400 - 9; http://dx.doi.org/10.1111/j.1600-0463.2008.00917.x; PMID: 18452430
- Ghavam-Nasiri MR, Rezaei E, Ghafarzadegan K, Seilanian-Toosi M, Malekifard H. Expression of p53 in colorectal carcinoma: correlation with clinicopathologic features. Arch Iran Med 2007; 10:38 - 42; PMID: 17198452
- Guillou L, Estreicher A, Chaubert P, Hurlimann J, Kurt AM, Metthez G, et al. Germ cell tumors of the testis overexpress wild-type p53. Am J Pathol 1996; 149:1221 - 8; PMID: 8863671
- Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A, et al. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008; 7:3056 - 61; http://dx.doi.org/10.4161/cc.7.19.6751; PMID: 18818522
- Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol 2011; 7:285 - 95; http://dx.doi.org/10.1038/nchembio.546; PMID: 21445056
- Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011; 20:79 - 91; http://dx.doi.org/10.1016/j.ccr.2011.06.004; PMID: 21741598
- Martynova E, Pozzi S, Basile V, Dolfini D, Zambelli F, Imbriano C, et al. Gain-of-function p53 mutants have widespread genomic locations partially overlapping with p63. Oncotarget 2012; 3:132 - 43; PMID: 22361592
- Frank AK, Pietsch EC, Dumont P, Tao J, Murphy ME. Wild-type and mutant p53 proteins interact with mitochondrial caspase-3. Cancer Biol Ther 2011; 11:740 - 5; http://dx.doi.org/10.4161/cbt.11.8.14906; PMID: 21307660
- Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem 1999; 68:383 - 424; http://dx.doi.org/10.1146/annurev.biochem.68.1.383; PMID: 10872455
- Zhivotovsky B, Samali A, Gahm A, Orrenius S. Caspases: their intracellular localization and translocation during apoptosis. Cell Death Differ 1999; 6:644 - 51; http://dx.doi.org/10.1038/sj.cdd.4400536; PMID: 10453075
- Jaros E, Perry RH, Adam L, Kelly PJ, Crawford PJ, Kalbag RM, et al. Prognostic implications of p53 protein, epidermal growth factor receptor, and Ki-67 labelling in brain tumours. Br J Cancer 1992; 66:373 - 85; http://dx.doi.org/10.1038/bjc.1992.273; PMID: 1503912
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992; 69:1237 - 45; http://dx.doi.org/10.1016/0092-8674(92)90644-R; PMID: 1535557
- MacLachlan TK, El-Deiry WS. Apoptotic threshold is lowered by p53 transactivation of caspase-6. Proc Natl Acad Sci USA 2002; 99:9492 - 7; http://dx.doi.org/10.1073/pnas.132241599; PMID: 12089322
- Wong RPC, Tsang WP, Chau PY, Co NN, Tsang TY, Kwok TT. p53-R273H gains new function in induction of drug resistance through down-regulation of procaspase-3. Mol Cancer Ther 2007; 6:1054 - 61; http://dx.doi.org/10.1158/1535-7163.MCT-06-0336; PMID: 17363498
- Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev 2000; 14:2060 - 71; PMID: 10950869
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303:1010 - 4; http://dx.doi.org/10.1126/science.1092734; PMID: 14963330
- Kasof GM, Gomes BC. Livin, a novel inhibitor of apoptosis protein family member. J Biol Chem 2001; 276:3238 - 46; http://dx.doi.org/10.1074/jbc.M003670200; PMID: 11024045
- Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, et al. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell 2003; 11:519 - 27; http://dx.doi.org/10.1016/S1097-2765(03)00054-6; PMID: 12620238
- Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997; 388:300 - 4; http://dx.doi.org/10.1038/40901; PMID: 9230442
- Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 2001; 410:112 - 6; http://dx.doi.org/10.1038/35065125; PMID: 11242052
- Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell 1998; 1:949 - 57; http://dx.doi.org/10.1016/S1097-2765(00)80095-7; PMID: 9651578
- Hirata H, Takahashi A, Kobayashi S, Yonehara S, Sawai H, Okazaki T, et al. Caspases are activated in a branched protease cascade and control distinct downstream processes in Fas-induced apoptosis. J Exp Med 1998; 187:587 - 600; http://dx.doi.org/10.1084/jem.187.4.587; PMID: 9463409
- Cowling V, Downward J. Caspase-6 is the direct activator of caspase-8 in the cytochrome c-induced apoptosis pathway: absolute requirement for removal of caspase-6 prodomain. Cell Death Differ 2002; 9:1046 - 56; http://dx.doi.org/10.1038/sj.cdd.4401065; PMID: 12232792
- Stennicke HR, Jürgensmeier JM, Shin H, Deveraux Q, Wolf BB, Yang X, et al. Pro-caspase-3 is a major physiologic target of caspase-8. J Biol Chem 1998; 273:27084 - 90; http://dx.doi.org/10.1074/jbc.273.42.27084; PMID: 9765224
- Stegh AH, Kesari S, Mahoney JE, Jenq HT, Forloney KL, Protopopov A, et al. Bcl2L12-mediated inhibition of effector caspase-3 and caspase-7 via distinct mechanisms in glioblastoma. Proc Natl Acad Sci USA 2008; 105:10703 - 8; http://dx.doi.org/10.1073/pnas.0712034105; PMID: 18669646
- Stegh AH, DePinho RA. Beyond effector caspase inhibition: Bcl2L12 neutralizes p53 signaling in glioblastoma. Cell Cycle 2011; 10:33 - 8; http://dx.doi.org/10.4161/cc.10.1.14365; PMID: 21200141
- Leong SM, Tan BX, Bte Ahmad B, Yan T, Chee LY, Ang ST, et al. Mutant nucleophosmin deregulates cell death and myeloid differentiation through excessive caspase-6 and -8 inhibition. Blood 2010; 116:3286 - 96; http://dx.doi.org/10.1182/blood-2009-12-256149; PMID: 20606168
- Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 2000; 102:849 - 62; http://dx.doi.org/10.1016/S0092-8674(00)00073-8; PMID: 11030628
- Stenger JE, Tegtmeyer P, Mayr GA, Reed M, Wang Y, Wang P, et al. p53 oligomerization and DNA looping are linked with transcriptional activation. EMBO J 1994; 13:6011 - 20; PMID: 7813439
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997; 387:299 - 303; http://dx.doi.org/10.1038/387299a0; PMID: 9153396
- Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 1977; 36:59 - 74; http://dx.doi.org/10.1099/0022-1317-36-1-59; PMID: 886304
- König C, Roth J, Dobbelstein M. Adenovirus type 5 E4orf3 protein relieves p53 inhibition by E1B-55-kilodalton protein. J Virol 1999; 73:2253 - 62; PMID: 9971808
- Blair Zajdel ME, Blair GE. The intracellular distribution of the transformation-associated protein p53 in adenovirus-transformed rodent cells. Oncogene 1988; 2:579 - 84; PMID: 3290806
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11:577 - 90; http://dx.doi.org/10.1016/S1097-2765(03)00050-9; PMID: 12667443
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997; 91:479 - 89; http://dx.doi.org/10.1016/S0092-8674(00)80434-1; PMID: 9390557
- Murphy RF, Komlodi-Pasztor E, Robey R, Balis FM, Farrell NP, Fojo T. Retained platinum uptake and indifference to p53 status make novel transplatinum agents active in platinum-resistant cells compared to cisplatin and oxaliplatin. Cell Cycle 2012; 11:963 - 73; http://dx.doi.org/10.4161/cc.11.5.19447; PMID: 22333583
- Blagosklonny MV. Wt p53 impairs response to chemotherapy: make lemonade to spare normal cells. Oncotarget 2012; 3:601 - 7; PMID: 22802145
- Bell S, Hansen S, Buchner J. Refolding and structural characterization of the human p53 tumor suppressor protein. Biophys Chem 2002; 96:243 - 57; http://dx.doi.org/10.1016/S0301-4622(02)00011-X; PMID: 12034444
- Pishas KI, Al-Ejeh F, Zinonos I, Kumar R, Evdokiou A, Brown MP, et al. Nutlin-3a is a potential therapeutic for ewing sarcoma. Clin Cancer Res 2011; 17:494 - 504; http://dx.doi.org/10.1158/1078-0432.CCR-10-1587; PMID: 21098696