Abstract
The ATR-dependent intra-S checkpoint protects DNA replication forks undergoing replication stress. The checkpoint is enforced by ATR-dependent phosphorylation of CHK1, which is mediated by the TIMELESS-TIPIN complex and CLASPIN. Although loss of checkpoint proteins is associated with spontaneous chromosomal instability, few studies have examined the contribution of these proteins to unchallenged DNA metabolism in human cells that have not undergone carcinogenesis or crisis. Furthermore, the TIMELESS-TIPIN complex and CLASPIN may promote replication fork protection independently of CHK1 activation. Normal human fibroblasts (NHF) were depleted of ATR, CHK1, TIMELESS, TIPIN or CLASPIN and chromosomal aberrations, DNA synthesis, activation of the DNA damage response (DDR) and clonogenic survival were evaluated. This work demonstrates in NHF lines from two individuals that ATR and CHK1 promote chromosomal stability by different mechanisms that depletion of CHK1 produces phenotypes that resemble more closely the depletion of TIPIN or CLASPIN than the depletion of ATR, and that TIMELESS has a distinct contribution to suppression of chromosomal instability that is independent of its heterodimeric partner, TIPIN. Therefore, ATR, CHK1, TIMELESS-TIPIN and CLASPIN have functions for preservation of intrinsic chromosomal stability that are separate from their cooperation for activation of the intra-S checkpoint response to experimentally induced replication stress. These data reveal a complex and coordinated program of genome maintenance enforced by proteins known for their intra-S checkpoint function.
Introduction
A function of the ATR (ataxia-telangiectasia mutated and Rad3-related) kinase is to activate the intra-S checkpoint in response to replication stress (reviewed in refs. Citation1–Citation3). Replication stress occurs when movement of the replication fork is impaired. Replication forks are stalled when the CDC45/MCM2–7/GINS (CMG) helicase complex encounters a lesion or a natural configuration of DNA that impedes strand separation. Replication forks are uncoupled when polymerization is stalled by certain modifications of the DNA template that can arise from endogenous or exogenous sources. Chemicals such as hydroxyurea, which reduces nucleotide pools, or aphidicolin, which inhibits polymerases, can also uncouple replication forks. Two key structural changes occur at uncoupled replication forks. One is the generation of excess RPA-coated single-stranded DNA (ssDNA) and another is the generation of additional 5′-double-stranded DNA (dsDNA) with a free 5′ end in juxtaposition to ssDNA via new primer synthesis.Citation4 These alterations of the replication fork are physiological processes that convert specific impediments to replication into sites for the recruitment of checkpoint proteins. Briefly, activation of the ATR-dependent intra-S checkpoint involves the following events. ATR is recruited to uncoupled replication forks through the interaction of its binding partner, ATRIP, with RPA-coated ssDNA.Citation5 The RAD9-RAD1-HUS1 (9-1-1) complex is loaded at dsDNA-ssDNA junctions by the RAD17-RFC clamp loader for recruitment of TOPBP1, which stimulates ATR kinase activity.Citation6-Citation9 Phosphorylation of the signal transducer kinase CHK1 by the sensor kinase ATR is mediated by CLASPIN and the TIMELESS-TIPIN (TIM-TIPIN) heterodimer, and phosphorylated CHK1 (P-CHK1) diffuses through the nucleus to enforce the intra-S checkpoint.Citation10-Citation14 CLASPIN interacts with TIMELESS and TIPINCitation15-Citation17 and biochemical studies suggest that TIPIN may bridge interactions between CLASPIN and RPA-coated ssDNA to promote association of CLASPIN-CHK1 with sites of altered replication forks.Citation16 Therefore, the activation of the ATR-dependent intra-S checkpoint in response to uncoupled replication forks requires specific structural alterations of replication forks and a coordinated assembly of sensor, activator, mediator and transducer proteins.
Activation of the ATR-dependent intra-S checkpoint in response to experimentally induced replication stress protects uncoupled replication forks, slows the rate of replication at other forks, inhibits origin firing and delays entry into mitosis,Citation2,Citation18 illustrating the influence of this pathway on replication dynamics and cell cycle progression. Importantly, whether in the presence or absence of experimentally induced replication stress, checkpoint proteins contribute to cell viability and maintenance of genomic stability. Mice knocked-out for Atr, Chk1 or Timeless are embryonic lethal,Citation19-Citation21 and mammalian cells deficient for ATR, CHK1, TIMELESS, TIPIN or CLASPIN exhibit chromosomal instability.Citation10,Citation11,Citation19,Citation22-Citation26 Chromosomal breaks have been linked to fragile sites in cells deficient for ATR, CHK1 or CLASPIN in the presence or absence of aphidicolin.Citation22,Citation23,Citation25 Likewise, S. cerevisiae require MEC1 and RAD53 (functional orthologs of ATR and CHK1, respectively) for survival [the genetic study of their functions is possible by co-deletion of suppressor of mec1 lethality (SML1)]Citation27 and Mec1 and Rad53, as well as other proteins that contribute to S-phase checkpoint activation, suppress gross chromosomal rearrangements in the absence of experimentally induced replication stress.Citation28
The mechanisms by which checkpoint proteins suppress spontaneous chromosomal breakage are largely unknown. Much of what is known about the ATR-CHK1 signaling pathway has been derived from studies of replication fork uncoupling induced by hydroxyurea, aphidicolin or DNA-damaging agents. As an extension of this knowledge, it has been proposed that the pathway preserves inherent genomic stability via CHK1 signaling in response to replication fork uncoupling caused by products of cellular metabolism and environmental sources that modify DNA.Citation29,Citation30 However, in addition to DNA template lesions, evolutionarily acquired natural barriers also can impair replication fork progression. How each type of natural barrier alters the structure of a replication fork or its replisome components is relatively unknown. Recent studies have shown that TIMELESS and TIPIN and their yeast counterparts are important for replication past certain DNA sequences, structures and protein-DNA barriers.Citation31-Citation36 TIMELESS, TIPIN and CLASPIN orthologs travel with the replication forkCitation26,Citation37,Citation38 and interact with replisome components (including MCM helicase subunits, replicative polymerases, AND1, PCNA and RPA).Citation15,Citation39,Citation40 Although TIM-TIPIN mediates ATR-CHK1 signaling in response to experimentally induced replication stress, loss of TIM-TIPIN is associated with increased ssDNA and activation of CHK1,Citation41 suggesting that TIMELESS and TIPIN have functions for preservation of replication fork structure that are independent of ATR-CHK1 signaling. Also, several contributions of Mrc1 (yeast analog of CLASPIN) to unperturbed DNA replication are independent of its role(s) in intra-S checkpoint regulation.Citation42 Models have been proposed in which TIM-TIPIN coordinates helicase and polymerase activities to create a replisome-pausing complex to protect forks that have encountered challenges to processive DNA replication.Citation15,Citation37,Citation41,Citation43-Citation45 Taken together, these observations suggest that components of the ATR-CHK1 signaling pathway may have separable contributions to maintenance of intrinsic genomic stability.
This work examined whether the “sensor-mediator-transducer” mechanism of ATR-dependent intra-S checkpoint activation is engaged to promote normal DNA metabolism, thereby suppressing spontaneous chromosomal aberrations. Very few studies of the main contributions of checkpoint proteins to DNA metabolism have been performed in diploid human cells that have not undergone carcinogenesis or viral transformation with crisis. Furthermore, direct comparisons of checkpoint protein depletions rarely have been made in the same experimental system. We performed a comprehensive analysis of the contributions of ATR-dependent intra-S checkpoint proteins to unchallenged DNA replication in normal human fibroblasts (NHF) expressing the catalytic domain of telomerase (hTERT). These lines provide a useful in vitro model of human somatic cell division with high fidelity replication and segregation of the genome. Chromosomal aberrations, CHK1 phosphorylation, DNA synthesis, activation of the DNA damage response (DDR) and clonogenic expansion were evaluated in cultures with siRNA-mediated depletion of ATR, CHK1, TIMELESS, TIPIN or CLASPIN. Our results indicate that ATR, CHK1 and TIMELESS have separable contributions to DNA replication and chromosomal stability.
Results
Depletion of intra-S checkpoint proteins generated different levels of chromosomal instability
NHF1-hTERT were transiently depleted of intra-S checkpoint sensor, mediator and transducer proteins by siRNA and various phenotypic outcomes related to DNA replication and chromosomal stability were evaluated. The degree of protein depletion routinely was ≥ 95% at 24 or 48 h after introduction of siRNA (). Note that targeting of TIMELESS or TIPIN, which form a heterodimer, reduces expression of the binding partner. Also, depletion of CHK1 or TIPIN, but not TIMELESS, transiently reduced CLASPIN levels by ~50% at the 24 h time point. The effect of CHK1 depletion on reduction of CLASPIN expression has been reported previously.Citation46 Chromosomal aberrations (breaks, gaps and exchanges) were examined in Giemsa-stained metaphases depleted of ATR, CHK1, TIMELESS, TIPIN or CLASPIN (). The chromosomal aberration frequencies of NHF1-hTERT electroporated with non-targeting control (NTC) siRNA were 0.045 ± 0.01 and 0.026 ± 0.01 at 24 and 48 h, respectively. At 24 h, the incidence and frequency of chromosomal aberrations were statistically greater than control for all depletions except TIPIN (). Chromosomal aberrations primarily were sister chromatid breaks (), but also included gaps () and exchanges (), indicative of breaks that took place during S, G2 or M, as opposed to chromosome breaks that are derived from breaks that take place in G1. At 48 h, chromosomal aberrations were nearly baseline in cells depleted of CHK1, TIPIN or CLASPIN, whereas they had increased over time in cells depleted of ATR or TIMELESS (~40-fold and ~15-fold increases in chromosomal aberration frequency over the control, respectively) (). The chromosomal aberration frequency of ATR-depleted cells was significantly greater than that of TIMELESS-depleted cells (p < 0.01), and depletion of ATR or TIMELESS produced chromosomal aberration frequencies that were significantly greater than CHK1 depletion (p < 0.0001 and p < 0.01, respectively) (). Co-targeting of TIMELESS with TIPIN, CLASPIN or ATR appeared to produce greater-than-additive increases in chromosomal aberration frequency (). The frequencies of chromosomal aberrations for each of the depletions were reflected by the incidence of metaphases with at least one aberration ().
Figure 1. Representative western blots from a single experiment showing ≥ 95% depletion of checkpoint proteins from NHF1-hTERT at 24 or 48 h after electroporation of siRNAs.
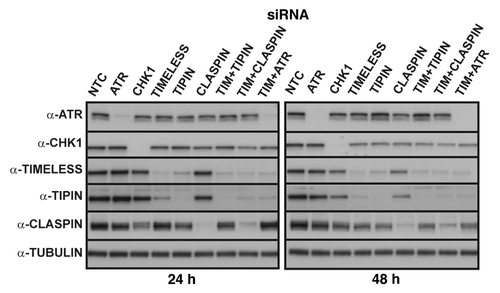
Figure 2. Chromosomal aberrations observed in Giemsa-stained metaphases produced from NHF1-hTERT depleted of checkpoint proteins. Frequency of breaks, gaps and exchanges at 24h (A) or 48 h (B) after introduction of siRNAs (error bars show + SEM for overall frequency of aberrations). Average frequency = total number of aberrations divided by the total number of metaphases evaluated per independent experiment, then averaged across independent experiments. Incidence of metaphases with at least one chromosomal aberration (C). Graphs represent averages of three or more experiments (+ S.D.). Markers indicate degree of statistical difference from NTC siRNA control: # p < 0.01, * p < 0.005, ** p < 0.0005, *** p < 0.0001. Representative pictures of chromosomal aberrations: chromatid break (D) and gap (E) from NHF1-hTERT electroporated with ATR siRNA; an incomplete, complex exchange (F) and a radial exchange (G) from NHF1-hTERT electroporated with TIMELESS and TIPIN siRNAs.
Overall, similar results were obtained from a second NHF line from a different donor (Fig. S1A–E). The basal chromosomal aberration frequencies for NHF10-hTERT electroporated with NTC siRNA were 0.07 ± 0.02 and 0.05 ± 0.01 at 24 and 48 h, respectively. In NHF10-hTERT, the chromosomal aberration frequency and incidence increased between 24 and 48 h in cells depleted of ATR or TIMELESS, decreased in cells depleted of CHK1 and did not increase significantly in cells depleted of TIPIN (Fig. S1A–C). Chromosomal aberrations were low in NHF1-hTERT or NHF10-hTERT depleted of CLASPIN, and chromosomal aberration frequency and incidence were not statistically different from the control in CLASPIN-depleted NHF10-hTERT. Co-depletion of TIMELESS with CLASPIN or ATR produced increases that appeared to be additive in the incidence of metaphases with chromosomal aberrations and greater-than-additive increases in the frequency of chromosomal aberrations. The chromosomal aberration frequency, but not the incidence of metaphases with chromosomal aberrations, decreased considerably over time in NHF10-hTERT co-depleted of ATR and TIMELESS. However, in such cells, 7% and 34% of metaphases at the 24 h and 48 h time points, respectively, exhibited chromosomal shattering, depicted in Figure S1D and E, which excluded such metaphases from accurate counts of chromosomal aberrations. Therefore, the chromosomal aberration frequency of NHF10-hTERTco-depleted of ATR and TIMELESS is underestimated. Furthermore, co-depletion of ATR and TIMELESS appeared to produce metaphase spreads showing a combination of chromosome shattering and discohesion (Fig. S1E), as TIMELESS is required for sister chromatid cohesion (SCC) in human cells.Citation14,Citation26
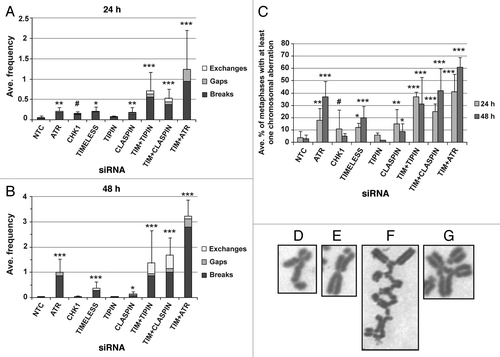
Chromosome exchanges are generated from erroneous rejoining of broken chromosomes. Exchanges were not detected in > 1,400 metaphases from NHF1-hTERT and NHF10-hTERT electroporated with NTC siRNA. Among the single depletions, the exchange frequency ranged from 0.02 ± 0.01 (CLASPIN siRNA) to 0.08 ± 0.05 (ATR siRNA) when data from all time points and cell lines were combined (). Considering the failure to detect exchanges in metaphases obtained from cells that were electroporated with NTC siRNA, it was apparent that checkpoint proteins suppressed formation of these aberrations. Furthermore, exchanges were considerably increased when cells were co-depleted of TIMELESS and TIPIN or TIMELESS and CLASPIN, but not when co-depleted of TIMELESS and ATR. Additionally, the majority of exchanges observed in double depletions were incomplete (not all ends were joined) and complex (involving multiple chromosomes) (). Radial structures also were observed ().
Figure 3. Average frequency of exchanges when data from NHF1-hTERT and NHF10-hTERT were combined. Each bar depicts averages of five or more independent experiments (+ S.D.). The TIMELESS+TIPIN siRNA exchange frequency was statistically different from the exchange frequencies of TIMELESS or TIPIN siRNAs (p < 0.0001 for both comparisons), and the TIMELESS+CLASPIN siRNA exchange frequency was statistically different from the exchange frequencies of TIMELESS or CLASPIN siRNAs (p < 0.0001 for both comparisons). The TIMELESS+ATR siRNA exchange frequency was not statistically different from the ATR siRNA exchange frequency (p = 0.14).
The degree of chromosomal instability was not predicted by the ability or inability to phosphorylate CHK1
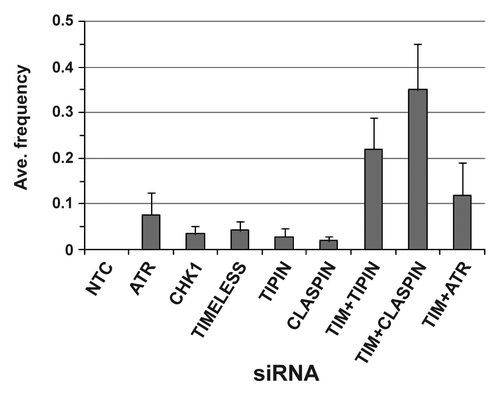
When compared directly, depletion of different components of the ATR-CHK1 signaling pathway produced strikingly different levels of chromatid-type chromosomal aberrations in NHF lines (; Fig. S1A–C). CHK1 phosphorylation is the key event for full deployment of the ATR-dependent checkpoint response to stalled replication forks and is important for prevention of fork collapse that potentially can give rise to chromosomal aberrations. The status of CHK1 phosphorylation was evaluated for each of the depletions (). CHK1 phosphorylation at S345 was not different from the control in cells depleted of ATR or CLASPIN and was not apparent in cells depleted of CHK1 itself. Depletion of TIMELESS or TIPIN produced ~10-fold increases in P-CHK1 S345 at the 24 h time point and 3- to 5-fold increases at the 48 h time point (). Co-targeting of TIMELESS and TIPIN did not appear to produce additive or greater-than-additive increases in P-CHK1 S345. CHK1 phosphorylation in cells depleted of TIMELESS was ATR-dependent and mediated by CLASPIN.
Figure 4. Cells depleted of TIMELESS or TIPIN exhibit ATR-dependent phosphorylation of CHK1 mediated by CLASPIN. Representative western blots from a single experiment depicting phosphorylation of CHK1 at S345 at 24 or 48 h after introduction of siRNAs (A). ImageJ software was used to normalize P-CHK1 S345 to total CHK1 and the results were expressed as average fold change compared with NTC. (B) Graph depicts average of three experiments (+ S. D.).
Global inhibition of DNA synthesis occurred in cells depleted of CHK1, TIMELESS, TIPIN or CLASPIN, but not ATR
As depletion of CHK1, CLASPIN or TIMELESS is known to alter DNA replication dynamics,Citation12,Citation47-Citation50 we examined whether replication fork distress correlated with the chromosomal instability observed in mitosis for the various depletions. It was immediately apparent from the flow cytometry profiles depicted in that disruption of DNA synthesis occurred when NHF1-hTERT was depleted of CHK1, TIMELESS, TIPIN or (to a lesser extent) CLASPIN, but not when cells were depleted of ATR. reports the average percent of cells within the population that had S-phase DNA content, which is further divided into S-phase cells showing normal incorporation of BrdU, reduced incorporation of BrdU, or failure to incorporate BrdU (a, b or c, respectively; see NTC siRNA 24 h profile in ). Compared with the control, S-phase cells showing reduction or failure to incorporate BrdU increased by ~10-fold in cells depleted of CHK1, TIMELESS or TIPIN at 24 h (). By 48 h, DNA synthesis appeared to stop almost completely in affected cells (). The percentage of cells showing control levels of BrdU incorporation was reduced at 48 h after introduction of siRNA to 60%, 50% and 60% of control, respectively, in CHK1-, TIMELESS- or TIPIN-depleted cells (). An experiment in which a 2 h pulse of BrdU was followed at 8 h intervals for 24 h indicated that cells depleted of CHK1, TIMELESS or TIPIN experienced impaired DNA synthesis and were unable to complete replication of their genomes at various stages of S phase (Fig. S2). The degree of impaired DNA synthesis was not as severe in CLASPIN-depleted cells. Co-depletion of TIMELESS and TIPIN did not increase the number of cells showing aberrant replication compared with targeting either protein alone. Co-depletion of TIMELESS with CLASPIN or ATR produced additive increases in cells showing reduction or failure to incorporate BrdU. BrdU incorporation in ATR-depleted cells closely resembled the control at both time points ().
Figure 5. NHF1-hTERT depleted of checkpoint proteins exhibit reduced BrdU incorporation and alterations in cell cycle progression. Bivariate plots from a single experiment show linear propidium iodide signal (DNA content) vs. log signal of anti-BrdU directly conjugated to AlexaFluor 488 at 24 h (A) or 48 h (B) after introduction of siRNAs. Three regions were drawn on the profiles to define G1, G2 and normal S phase (A) populations. Two regions were drawn to define aberrant S phase (B and C). Cells with > 2N and < 4N DNA content above the G1 and G2 regions but below the S phase region defined by the NTC profile showed reduced BrdU incorporation (B). Cells with > 2N and < 4N DNA content that fell below the G1/S and G2/S boundaries showed failure to incorporate BrdU (C). Average percent of cells with S-phase DNA content within the whole population partitioned by those showing normal BrdU incorporation, reduced BrdU incorporation and no BrdU incorporation obtained from four or more experiments (+ S.D.). (C and D) Except for ATR siRNA, the percent of cells showing reduced or no incorporation of BrdU was statistically different (p < 0.0001) from NTC siRNA for all of the depletions at 24 h (C) and 48 h (D) time points. (E and F) Replicate experiments were first normalized to the matched NTC flow cytometry profile before comparison across experiments. Regions drawn on NTC profiles for G1, G2 and all cells with S-phase DNA content were applied to depletion profiles and used to calculate the fold change compared with NTC siRNA. All depletions were statistically different (p < 0.05) from NTC siRNA for G1 except ATR and CHK1 at 24 and 48 h, and TIPIN, CLASPIN and TIMELESS+TIPIN at 48 h. Depletions that were statistically different (p < 0.05) from NTC siRNA for S phase included TIMELESS and TIMELESS+ATR at 24 and 48 h, TIMELESS+CLASPIN at 24 h. All depletions were statistically different (p < 0.05) from NTC siRNA for G2 except ATR at 24 and 48 h, CHK1 and TIMELESS+ATR at 24 h.
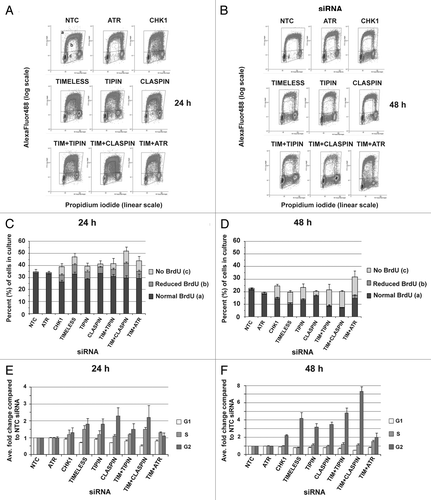
Inhibition of DNA synthesis was accompanied by perturbation of the cell cycle in NHF1-hTERT that were depleted of TIMELESS, TIPIN, CHK1 or CLASPIN, with obvious accumulations in G2, subtle reductions of G1 and subtle increases in S phase (). By 48 h after introduction of siRNA, depletion of CHK1, TIMELESS, TIPIN or CLASPIN resulted in 2- to 4-fold increases in cells with G2 DNA content (). Co-depletion of TIMELESS with ATR attenuated the accumulation of cells in G2 observed with depletion of TIMELESS alone, whereas co-depletion with CLASPIN did not have this effect. Depletion of ATR did not alter the cell cycle. Similar results were obtained from NHF10-hTERT (Fig. S3A–D).
Activation of the DNA damage response (DDR) was associated with global inhibition of DNA synthesis
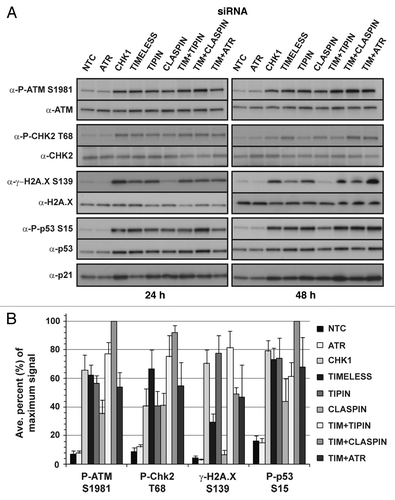
Depletion of checkpoint proteins resulted in different levels of chromosomal aberrations (; Fig. S1A–C), and cells depleted of CHK1, TIMELESS, TIPIN or CLASPIN exhibited replication stress and overall perturbation of the cell cycle, whereas depletion of ATR did not appear to affect cell cycle progression (). The inhibition of DNA replication observed in a subset of cells depleted of checkpoint proteins may have been due to irrecoverable replication fork breakage. Therefore, the activation of the DDR was examined for each of the depletions (). Phosphorylation of ATM was increased by ~10–20-fold over the control in cells depleted of CHK1, TIMELESS or TIPIN and by ~5-fold over the control in cells depleted of CLASPIN. Substrates of ATM were phosphorylated, including CHK2 T68, p53 S15 and H2A.X S139. Phosphorylation of p53 S15 was accompanied by induction of p21Waf1. H2A.X phosphorylation (γ-H2A.X) in TIMELESS-depleted cells was not as robust as that observed in cells depleted of CHK1 or TIPIN. The lower level of γ-H2A.X in CLASPIN-depleted cells correlated with the lower level of P-ATM compared with depletion of CHK1 or TIPIN. Activation of the DDR in cells depleted of ATR was indistinguishable from the control. Similar results were obtained in NHF10-hTERT (Fig. S4). Although ATR-depleted cells did not activate ATM despite considerable increases in chromosomal aberrations (; Fig. S1A–C), ATR-depleted NHF1-hTERT was still capable of activating ATM in response to ionizing radiation (IR) (Fig. S5).
Figure 6. Depletion of checkpoint proteins in NHF1-hTERT activates the DNA-damage response (DDR). Representative western blots from a single experiment showing phosphorylation and/or upregulation of DDR biomarkers at 24 or 48 h after introduction of siRNAs (A). Western blots from and were from the same experiment. ImageJ software was used to normalize phospho-protein levels to total protein levels (B). In order to compare across experiments and different antibodies, the fold change in phosphorylation over the NTC siRNA control of the largest effect was set as the maximal (100%) signal for each experiment and then the results of each experiment were averaged. The data from the two time points were combined as the pattern of induced phosphorylation was not different. Graph depicts average of three experiments (+ S.D.).
Cell viability was impaired, to varying degrees, by different checkpoint protein depletions
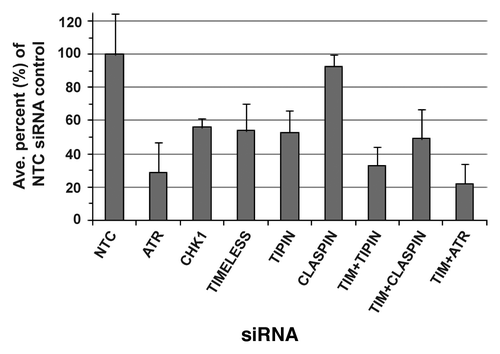
NHF lines depleted of checkpoint proteins exhibited cytotoxic chromosomal aberrations; after some depletion, a fraction of cells appeared to stop in S phase, possibly due to irrecoverable replication fork breakage. The effects of these deleterious events on cell viability were evaluated by a clonal expansion assay (). Cells depleted of CHK1, TIMELESS or TIPIN showed ~50% reduction in clonal expansion, whereas depletion of ATR reduced clonal expansion by 70%. Clonal expansion was reduced by less than 10% with depletion of CLASPIN.
Figure 7. Clonal expansion of NHF1-hTERT depleted of checkpoint proteins in the absence of applied DNA damage. Graph depicts averages of three or more experiments (+ S.D.).
Discussion
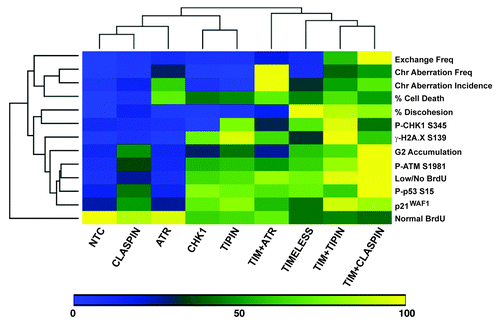
At uncoupled replication forks, efficient ATR-CHK1 signaling is highly dependent on specific protein-DNA assemblies that recruit ATR and CHK1 and promote their interaction. ATR, CHK1 and the mediators TIMELESS, TIPIN and CLASPIN also are important for suppression of chromosomal instability in the absence of experimentally induced replication stress.Citation10,Citation19,Citation20,Citation25,Citation26,Citation51 It has been proposed that ATR-CHK1 signaling during the normal course of DNA replication is necessary to prevent chromosomal damage that could arise from failure to protect uncoupled replication forks, and it also has been proposed that a basal level of ATR-CHK1 signaling is required to prevent excess origin firing that could result in replication fork instability (reviewed in refs. Citation30 and Citation52). Here, in a set of direct comparisons made in two NHF lines from different donors, depletion of the ATR kinase, the CHK1 kinase or mediators of ATR and CHK1 interaction generated different levels of chromosomal instability. DNA synthesis, activation of the DDR and clonal expansion were examined in order to understand these differences, and these measures revealed further separation of the functions of checkpoint proteins in normal DNA metabolism. To provide an overall summary and comparison of the results, an unsupervised, hierarchical heat map was created to cluster the various experimental endpoints obtained from NHF1-hTERT (). When the magnitudes of the responses were scaled and sorted in this manner, two major clades appeared. The clade containing the results for control, ATR, CLASPIN, CHK1 and TIPIN siRNAs was divided into subsets, showing distinct separation of ATR- or CHK1-depletion phenotypes. TIMELESS depletions were separated from the other depletions due to the specific contribution of TIMELESS to sister chromatid cohesion in NHF lines (SCC data obtained from Smith-Roe et al. 2011Citation14). Overall, these results indicate that ATR, CHK1 and TIMELESS support different functions to preserve intrinsic genomic stability, some of which must be independent of ATR-CHK1 signaling.
Figure 8. Unsupervised, hierarchical cluster heat map comparison of various experimental outcomes after checkpoint protein depletion from NHF1-hTERT. In order to make relative comparisons between siRNAs and phenotypic outcomes, the siRNA that produced the highest effect for each independent experiment was set to 100% and all the other values were expressed relative to the highest value for that experiment. Once converted to a scale of 1–100, the converted values were averaged. Determinations are from data obtained at 48 h post-electroporation except for clonogenic expansion data and for western blot data. Western blot data from 24 and 48 h data were combined as the patterns of activation were similar. Scaling of sister chromatid discohesion was generated from Smith-Roe et al. (2011).Citation14 The effect of co-targeting TIMELESS and ATR on SCC was not reported in Smith-Roe et al. (2011), but is reported here as 4.7 ± 2.5% for NHF1-hTERT (scaled into ) and 30.5 ± 12.6% for NHF10-hTERT.
Different degrees of chromosomal instability in NHF depleted of ATR vs. CHK1
Depletion of ATR or CHK1 revealed that these proteins have separable contributions to chromosomal stability in NHF lines. Despite a ~10-fold increase in the incidence of metaphases with chromosomal aberrations compared with the control (; Fig. S1C at 48 h), ATR-depleted cells did not activate the DDR (), exhibited normal BrdU incorporation (; Fig. S3A and B) and did not accumulate in G2 (; Fig. S3C) (the rate of entry into mitosis for ATR-depleted NHF was similar to cells electroporated with NTC siRNA, unpublished observations). Even though the frequency of chromosomal aberrations observed in ATR-depleted cells continued to increase over time (1.0 ± 0.27 at 48 h, vs. 2.7 ± 0.02 at 72 h, Fig. S6A) as did the incidence (37 ± 6.2% at 48 h, vs. 63 ± 2.5% at 72 h, Fig. S6B), only slight alterations in BrdU incorporation or cell cycle distribution were observed (Fig. S6C and D), and the DDR was also only slightly increased above baseline (Fig. S6E and F). ATR-depleted NHF lines showed no signs of distress other than increased chromosomal aberrations, shattered metaphases and reduced clonal expansion (). These results were highly similar to those observed in mouse embryonic fibroblasts (MEFs) that had undergone conditional elimination of ATR.Citation53 Conversely, although CHK1-depleted cells showed a modest increase in chromosomal aberrations at 24 h, they did not differ from the control at 48 h. Furthermore, CHK1-depleted cells exhibited dramatic inhibition of DNA synthesis throughout S phase, accumulation in G2 (CHK1 siRNA reduced the rate of entry into mitosis by 32%, unpublished observations) and robust activation of the DDR.
If activation of CHK1 were a key event for suppression of spontaneous chromosomal aberrations (which are expected to arise from uncoupling of replication forks by DNA template lesions produced by endogenous sources), depletion of ATR or CHK1 would be expected to result in similar degrees of chromosomal instability. Strikingly, instead of sharing phenotypic characteristics of ATR-depleted cells, depletion of CHK1 most closely resembled depletion of TIPIN (). Furthermore, the degree of chromosomal instability of CHK1- or TIPIN-depleted cells was not different from the control (48 h), even though TIPIN-depleted cells activated CHK1. A key difference between depletion of ATR or CHK1 was the severe inhibition of DNA synthesis observed in CHK1-depleted cells. This observation suggests that CHK1 may have an ATR-independent function for the promotion of normal DNA synthesis throughout S phase, implying a regulatory effect of CHK1 through protein-protein interactions as opposed to its kinase activity. CHK1 can associate with chromatin in the absence of applied replication stress and independently of ATR, TOPBP1, HUS1, NBS1 or CLASPIN,Citation54-Citation56 and kinase-independent functions for CHK1 have been reported for replication of damaged DNA via interactions with PCNA.Citation40,Citation57 The activation of the DDR in CHK1-depleted cells and accumulation in G2 may indicate that sufficient activation of the G2 checkpoint took place for successful repair of damaged DNA in cells that were able to complete S phase. However, some CHK1-depleted cells may have had such a high level of DNA damage generated during S phase that they failed to reach mitosis and, hence, the chromosomal instability of CHK1-depleted cells appears to be less than that of ATR-depleted cells. It might be expected that CHK1-depleted cells would accumulate in S phase if they are not able to complete duplication of their genomes. However, not only did CHK1-depleted cells accumulate in G2, they also activated p53 for engagement of the G1 checkpoint, restricting entry into S phase to re-fill the compartment. Cell cycle-specific analysis of broken DNA or DDR markers could distinguish between these alternative explanations for the difference in chromosomal instability observed in metaphases from cells depleted of ATR vs. CHK1.
ATR-depleted cells showed no evidence of impaired DNA replication or response to DNA breaks, yet exhibited the highest degree of chromosomal instability of any single depletion and the most severe impairment of clonal expansion (). The chromatid breaks and gaps observed in ATR-depleted NHF lines may have been due to entry into mitosis with low levels of incompletely replicated DNA. The chromosomal aberration frequency data for ATR depletion in this study was highly similar to that reported by Casper et al. (2002).Citation22 The experiments performed by Casper et al. (2002) revealed that a significant percentage of DNA breaks occur at fragile sites when cells are deficient for ATR function even without exposure to aphidicolin. Fragile sites are rare (~100 identified), and their replication tends to be completed late in S phase.Citation51 Impaired replication of fragile site DNA would not be detected by flow cytometric analysis of BrdU incorporation. Regions of unreplicated DNA, which do not activate ATM, could break during chromatin condensation in mitosis to produce aberrations that resemble chromatid breaks and gaps in metaphase preparations. It may be possible that such breaks could escape activation of ATM. Although the frequencies of chromosomal aberrations in ATR-depleted NHF lines were ~40- to 50-fold greater than control (48 h), the frequencies reflect on average only one to two breaks per metaphase. This level of chromosomal breakage might be insufficient to produce detectable P-ATM and to activate checkpoints in the absence of ATR; however, at 48 h, 34% of metaphases from NHF10-hTERT depleted of ATR had shattered chromosomes (Fig. S1D), yet activation of the DDR was barely distinguishable from the control (Fig. S4B). The absence of ATM activation in response to endogenously generated DNA damage in ATR-depleted cells remains to be fully characterized, and the possibility that ATM cannot respond to frank dsDNA breaks in ATR-depleted NHF1-hTERT has been discounted (Fig. S5). Perhaps DDR signaling pathways are not fully operational in mitotic cells, or this cell cycle compartment is too small for DDR markers to be detected by western blotting. Conversely, activation of ATM in NHF lines depleted of the other checkpoint proteins may have been associated with collapsed replication forks, and, as such, the degree of P-ATM correlated very well with inhibition of DNA synthesis, whereas P-ATM did not correlate well with chromosomal instability (). Although ssDNA is part of fragile site etiology and cells deficient for ATR, CHK1 or CLASPIN express fragile sites,Citation25,Citation51,Citation58 ATR and CHK1 appear to separately govern additional mechanisms for genome-wide completion of DNA replication and prevention of chromosomal aberrations.
Separation-of-function of the TIMELESS-TIPIN heterodimer and genomic stability
The higher level of chromosomal aberrations produced when targeting TIMELESS compared with targeting TIPIN, CHK1 or CLASPIN may have been related to the dramatic sister chromatid cohesion defect observed in TIMELESS-depleted fibroblastsCitation14 (and represented in ). Targeting TIMELESS produced ~15-fold and ~7-fold increases in chromosomal aberration frequency and incidence, respectively, over the control in NHF1-hTERT and NHF10-hTERT, whereas targeting TIPIN did not show a statistical increase in chromosomal aberrations in either cell line (; Fig. S1B and C). Previously, we reported that targeting TIMELESS for depletion produced a 100-fold increase in defective SCC whereas depletion of TIPIN or CLASPIN produced ~10-fold increases and depletion of CHK1 was not statistically different from the control.Citation14 Conversely, targeting TIMELESS or TIPIN for depletion produced similar levels of replication stress (), P-CHK1 (), P-ATM () and accumulation in G2 (), and co-targeting TIMELESS and TIPIN did not result in additive or greater-than-additive outcomes for those measures. These observations illustrate protein complex-dependent functions of TIM-TIPIN and independent functions for TIMELESS, and, thus far, such striking distinctions have not been reported in other experimental models. Considering that TIMELESS-depleted cells did not show greater inhibition of DNA synthesis or greater activation of ATM compared with TIPIN-depleted cells, it was unlikely that TIMELESS-dependent cohesion was rescuing broken replication forks during S phase (e.g. by promoting homologous recombination), even though TIMELESS contributes to establishment of SCC during S phase in the X. laevis egg extract system.Citation59 TIMELESS- or TIPIN-depleted cells also showed equivalent reduction of clonal expansion (), suggesting that discohesion was not a strong contributor to lethality. Even though cells depleted of CHK1, TIMELESS, TIPIN or CLASPIN exhibited an accumulation in G2, only TIMELESS siRNA produced a considerable increase in chromosomal aberrations among these single depletions. Furthermore, co-targeting of TIMELESS with TIPIN, CLASPIN or ATR increased chromosomal aberrations in a greater-than-additive manner, even though on other measures, these co-depletions produced similar or additive results, suggesting that TIMELESS-dependent cohesion was responsible for suppressing the chromosomal aberrations that otherwise would have been present in the single knockdowns. TIMELESS-related cohesion generated in S phase may be important for repair of DNA damage during G2, as cohesion generated during S phase has been shown to be required for post-replicative repair of dsDNA breaks.Citation60 In TIMELESS-depleted MEF, γ-H2A.X appears in cells with S-phase DNA content.Citation41,Citation61 Phosphorylation of H2A.X is an important, early step for recruitment of DNA repair factors at sites of dsDNA breaks. In yeast, Mec1 and Tel1 phosphorylate H2A at sites of dsDNA breaks, promoting loading of cohesin and generation of large cohesin domains at the site of damage.Citation62 Notably, γ-H2A.X in TIMELESS-depleted cells was half of that observed in TIPIN- or CHK1-depleted cells (). Similar levels of ATM activation or ATR activation (as inferred by P-CHK1) occurred in cells depleted of TIMELESS or TIPIN ( and ). Therefore, loss of TIMELESS, but not TIPIN, appeared to impair ATR/ATM phosphorylation of H2A.X. It remains to be determined whether impaired phosphorylation of H2A.X and increased sister chromatid discohesion in TIMELESS-depleted cells are linked or independent, but both could contribute to the reduced repair of chromatid breaks.
Essential functions of the replication fork protection complex
Cessation of DNA replication appeared to occur throughout S phase for cells depleted of TIMELESS, TIPIN or CLASPIN (; Fig. S2). As demonstrated in yeast by analysis of replication pause sites, TIM-TIPIN orthologs and Mrc1 may create a pausing complex that prevents fork collapse when natural barriers are encountered.Citation32 TIM-TIPIN and CLASPIN may have a similar role in mammalian cells. Recent studies have shown that TIMELESS contributes to the replication of difficult-to-replicate DNA, including centromeric DNA, trinucleotide repeats and telomeres.Citation33,Citation35,Citation36 Considering that TIM-TIPIN interacts with CMG helicase components, AND1 and polymerases ε, δ and α, and that TIM-TIPIN-deficient cells showed spontaneous activation of CHK1, TIM-TIPIN may provide a link between DNA unwinding and polymerization to preserve stalled or uncoupled replication forks in a state competent for restart.Citation45 The mechanism(s) by which TIM-TIPIN orthologs might protect/restart replication forks independently of CHK1 activation remains to be fully characterized.
Loss of TIM-TIPIN from mammalian cells reduces the efficiency of DNA replication,Citation12,Citation13,Citation15 but inhibition of DNA synthesis previously has not been identified as a primary cause of lethality. Targeting TIMELESS or TIPIN for depletion dramatically inhibited DNA synthesis, which correlated well with reduction of clonal expansion compared with other measures of distress (). Approximately 40–50% of cells with S-phase DNA content showed aberrant incorporation of BrdU in TIMELESS- or TIPIN-depleted NHF lines (; Fig. S3B), and clonal expansion was suppressed by ~50% (). In MEFs depleted of TIMELESS, only ~2–4% of cells with S-phase DNA content show inhibition of DNA synthesis.Citation41 However, it was demonstrated that ATR-dependent activation of CHK1 rescued inhibition of DNA synthesis in TIMELESS-depleted MEFs.Citation41 Unlike the findings in MEFs, a clear protective effect of P-CHK1 on DNA synthesis in TIMELESS-depleted NHF lines was not observed. Abrogating P-CHK1 by co-targeting TIMELESS with CLASPIN or ATR produced additive, as opposed to synergistic, increases in replication stress at 24 and 48 h, indicating that P-CHK1 was not suppressing a measure of replication stress initiated by loss of TIMELESS. These co-depletions also indicated that the inhibition of DNA synthesis observed in TIM-TIPIN-depleted cells was not enforced by P-CHK1, which is of interest, as activation of CHK1 in response to experimentally induced replication stress is associated with transient inhibition of DNA synthesis by inhibition of origin firing and DNA chain elongation.Citation2 Therefore, fork protection functions associated with CHK1 activation appeared to be ineffective in human fibroblasts depleted of TIMELESS or TIPIN. The cause of embryonic death of Timeless-knockout mice was not fully characterized.Citation21 Whereas the ATR-dependent intra-S checkpoint response is attenuated, but not abrogated, when cells are depleted of TIMELESS or TIPIN,Citation12,Citation14 our findings suggest that TIM-TIPIN may have an essential function in DNA replication, which could underlie the lethality of Timeless-knockout mice.Citation21 Interestingly, although tof1Δ yeast are viable, tof1Δ is synthetic lethal with pfh1Δ, a helicase that is required for fork movement through a variety of natural barriers.Citation63
CLASPIN-depleted cells showed a modest inhibition of DNA synthesis compared with depletion of TIM-TIPIN (). Also, unlike TIM-TIPIN-depleted cells, a dramatic P-CHK1 signal was not observed in CLASPIN-depleted cells (). Combined with the good survival of CLASPIN-depleted cells (), these observations suggest that TIM-TIPIN may have a primary role in replication of natural barriers compared with CLASPIN. With regard to chromosomal instability, CLASPIN depletion produced low frequencies of chromosomal aberrations (0.18 ± 0.04 at 24 h and 0.14 ± 0.03 at 48 h, ), and these frequencies were very similar to the degree of chromosomal instability reported by Focarelli et al. (2009),Citation25 in which the effect of CLASPIN depletion on fragile site expression was examined in primary human fibroblasts. CLASPIN appeared to have an important role for suppression of exchanges when cells were depleted of TIMELESS, as the frequency of exchanges increased by 5-fold over what would be expected if there were no interaction between TIMELESS and CLASPIN for suppression of erroneous chromosomal repair (). Co-targeting of TIMELESS with TIPIN also increased the frequency of exchanges in a similar manner. The increased exchange frequency with co-depletion of TIMELESS with TIPIN or CLASPIN, but not ATR, suggests an ATR-independent function of the fork protection complex for prevention of exchanges. It may be possible that TIM-TIPIN and CLASPIN prevent the formation of certain substrates at replication forks that require repair via non-homologous end-joining pathways.
Loss of CHK1 phosphorylation did not correlate with degree of chromosomal instability in NHF lines
It has been proposed that the ATR-CHK1 signaling pathway prevents replication fork collapse during unperturbed replication and suppresses spontaneous DNA damage by regulating origin firing (reviewed in ref. Citation29). In this model, abrogation of basal P-CHK1 releases CDC25 family members from inhibition, thereby releasing CDK2 from inhibition and allowing targets of CDK2 to enhance basal levels of origin firing. When origin firing is increased, the rate of replication fork displacement is slowed,Citation64 and the excess, slow replication forks are thought to cause imbalances that lead to fork collapse.Citation29 However, our observations that inhibition of DNA synthesis in CHK1-depleted cells was not phenocopied by depletion of ATR, that depletion of TIPIN or CHK1 resulted in equivalent levels of fork collapse even though CHK1 was robustly activated in TIPIN-depleted cells and that CLASPIN depletion did not phenocopy depletion of CHK1 or ATR (), did not support disregulation of Cdc25 and CDK activity as the governing mechanism behind replication fork failure in NHF lines that are deficient for basal ATR-CHK1 signaling. The findings presented here may reflect differences between the DNA metabolism of NHF lines vs. cancer cell lines and the differences in origin regulation between human somatic cells and the Xenopus egg extract system.Citation65,Citation66
Summary
The contributions of ATR, CHK1 and the mediator proteins TIMELESS, TIPIN and CLASPIN to normal DNA metabolism were studied in genetically stable, diploid human fibroblast lines. In two NHF lines, the presence or absence of phosphorylated CHK1 did not correlate with the degree of chromosomal instability, inhibition of DNA synthesis, activation of the DDR or survival. Instead, the components of the intra-S checkpoint activation pathway made different contributions to replication fork stability and chromosomal integrity. ATR may only be absolutely required for replication of DNA at fragile sites, whereas CHK1, TIMELESS, TIPIN and (to a lesser extent) CLASPIN are required for DNA replication throughout S phase. Through its distinct influence on sister chromatid cohesion, TIMELESS provided an additional level of genome maintenance by facilitating repair of broken DNA prior to mitosis. Whereas ATR-dependent intra-S checkpoint signaling is important for responding to unintentional modifications of template DNA, components of the pathway appear to have P-CHK1-independent contributions to replication of inherent barriers to DNA replication that are heterogeneous, numerous and must be duplicated. CHK1, TIMELESS, TIPIN and CLASPIN may have structural roles for replication fork stabilization or restart that differ depending on the nature of the block to replication and whether ssDNA is generated from the block. On the other hand, these proteins do interact and how each one contributes to the stability of the association of the other members of the complex is difficult to determine. The data at hand do not allow the complete exclusion of the possibility that phenotypic endpoints measured when protein abundance was depleted via siRNA may reflect different degrees of reduction of the single protein or protein complexes, and/or their cellular locations, and how much is required to affect a specific biological endpoint. Therefore, how components of canonical checkpoint signaling pathways contribute to unperturbed DNA replication still remains to be fully elucidated.
Materials and Methods
Normal human fibroblast lines and culture
Low-passage foreskin fibroblasts from two different individuals were immortalized by ectopic expression of the catalytic subunit of human telomerase to create NHF1-hTERT and NHF10-hTERT fibroblast lines.Citation67-Citation70 Cell lines were cultured in Dulbecco’s modified Eagle’s medium supplemented with 2 mM L-glutamine and 10% fetal bovine serum (all culture reagents were obtained from Sigma-Aldrich). Cell lines were grown at 37°C in a humidified atmosphere of 5% CO2 and were determined to be free of mycoplasma contamination using the PlasmoTestTM system (Invivogen).
Protein depletion by siRNA
NHF lines were electroporated with siRNAs using the normal human dermal fibroblast nucleofection kit VPD-1001 (Lonzo) and electroporation program U-23. The total amount of siRNA introduced into cells for single vs. double depletions was held constant at 200 pmol siRNA per 1 million cells (100 pmol of targeting siRNA was combined with 100 pmol of NTC siRNA for single depletions). ON-TARGETplus duplex siRNAsCitation71 were purchased from Dharmacon: NTC (D-001210–02), CHK1 (J-003255), CLASPIN (J-005288), TIMELESS (J-019488–05), TIPIN (J-020843). The ON-TARGETplus SMARTpool siRNA for ATR (L-003202) also was purchased from Dharmacon.
Flow cytometry
NHF1-hTERT or NHF10-hTERT were incubated with 10 µM BrdU for 2 h before harvest. Trypsinized cells were fixed overnight at 4°C in 70% ethanol in calcium- and magnesium-free phosphate buffered saline (PBS). Fixative was removed and cells were incubated in 3 ml of 0.08% pepsin in 0.1 N HCl at 37°C for 20 min. Pepsin was removed and nuclei were incubated in 1.5 ml of 2 N HCl at 37°C for 20 min. A volume of 3 ml of 0.1 M sodium borate was added to the nuclei in acid. Nuclei were spun out of neutralized acid and washed with 2 ml HSF-T (10 mM HEPES pH 7.4, 150 mM NaCl, 4% FBS and 0.1% sodium azide with 0.5% Tween-20 added on the day of use) and then incubated at room temperature with anti-BrdU clone MoBU-1 conjugated to AlexaFluor488 (Invitrogen) in HSF-T for 2 h. DNA was stained overnight with a solution of 50 µg/ml propidium iodide (PI) (Sigma-Aldrich) and 5 µg/ml PureLink RNase A (Invitrogen). Nuclei were analyzed for anti-BrdU AF488 and PI fluorescence using a Beckman-Coulter (Dako) CyAn ADP and Summit 5.2 software (Beckman Coulter, Inc.).
Western blotting and antibodies
Cells were harvested by trypsinization, washed with 4°C PBS and lysed in Tris-Cl buffer (50 mM Tris-Cl pH 8.0, 150 mM NaCl, 5 mM EDTA) containing 0.1% NP-40, 1% Sigma Protease Inhibitor Cocktail, 1 mM sodium orthovanadate, 10 mM β-glycerolphosphate, 1 mM sodium fluoride and 2 mM DTT. Protein concentration was determined using the BioRad Dc Assay. Equivalent amounts of protein per well were loaded onto BioRad Criterion-TGX 4–15% gradient gels. Size-separated proteins were transferred onto nitrocellulose using a BioRad Criterion Blotter.
The following antibodies were used for immunoblotting: goat anti-ATR, rabbit anti-CLASPIN, mouse anti-CHK1 (Santa Cruz); rabbit anti- TIMELESS, rabbit anti-TIPIN, rabbit anti-ATM (Bethyl); rabbit anti-P-CHK1 S345, rabbit anti-P-CHK2 T68, rabbit anti-P-p53 S15, rabbit anti-α-tubulin (Cell signaling); mouse anti-p53, mouse anti-p21Waf1 (NeoMarkers); mouse anti-CHK2 (BD Transduction Laboratories); rabbit anti-P-ATM S1981 (Epitomics).
The degree of protein depletion was determined by using ImageJ software (ImageJ US, National Institute of Health, www.rsb.info.nih.gov/ij/, 1997–2009) to obtain the pixel density of protein bands from scanned images of exposed Amersham Hyperfilm (GE Healthcare). Protein levels were first normalized against the anti-α-tubulin loading control and then expressed as the percent of the NTC protein level. Phospho-protein signals were normalized to their total protein levels.
Metaphase preparations
Giemsa-stained metaphases were prepared as previously described.Citation14 Twenty-five to 50 metaphases were evaluated per treatment. The experimenter was blind to treatment during collection of metaphase data and analysis of chromosomal aberrations. Metaphases with bifilar sister chromatids were evaluated for chromatid breaks, gaps and exchanges.
Clonogenic survival assay
One million NHF1-hTERT that were electroporated with siRNA were serially diluted and counted using a Coulter counter in order to seed cells at a density that would result in ~150 colonies per 10 cm dish for the NTC siRNA control. Each independent experiment was seeded in triplicate and the experiment was repeated three or more times per siRNA. Medium was changed every third day. Cells were fixed and stained in a solution of 0.05% Crystal Violet in 40% methanol on day 14 after seeding. Colonies of 50 cells or more were counted. NHF1-hTERT that were not plated for colony formation were used to verify protein depletion at 24 h after electroporation.
Heatmap comparison of depletions
To visually represent the magnitude of the phenotypic outcomes resulting from depletion of checkpoint proteins and to sort depletions and phenotypes into clades, the Partek Genomic Suite 6.6 program was used to generate a heat map depicting unsupervised hierarchical clustering of depletions and endpoints.
Statistical analysis
Statistical comparisons were performed in order to determine whether change varied significantly across different treatments for fold change compared with the control for cell cycle analysis and the frequency and incidence of chromosomal aberrations in knockdowns compared with the control. The generalized linear model framework was used to handle standard data analysis for estimating various parameters of interest with appropriate 95% confidence intervals, and hypothesis testing. Specifically, a Linear Mixed model with random effect was used to model Log-transformed fold change, and the Zero-inflated Poisson model in NLMIXED as well as the generalized Poisson model in GENMOD was used to model the incidences of chromosomal aberrations. The likelihood ratio statistic was used to determine the statistical significance of the difference across treatments. All statistical analyses were performed using SAS 9.2 (SAS Institute Inc.).
| Abbreviations: | ||
| ATM | = | Ataxia-telangiectasia mutated |
| ATR | = | Ataxia-telangiectasia mutated and Rad3-related |
| CMG | = | CDC45/MCM2-7/GINS |
| CHK1 | = | checkpoint kinase 1 |
| CHK2 | = | checkpoint kinase 2 |
| DDR | = | DNA damage response |
| dsDNA | = | double-stranded DNA |
| NHF | = | normal human fibroblasts |
| PCNA | = | proliferating cell nuclear antigen |
| RPA | = | replication protein A |
| SCC | = | sister chromatid cohesion |
| siRNA | = | small interfering RNA |
| ssDNA | = | single-stranded DNA |
| TIM-TIPIN | = | TIMELESS-TIPIN |
| TIPIN | = | TIMELESS-interacting protein |
Additional material
Download Zip (1.7 MB)Acknowledgments
We wish to express our gratitude to Drs. Cyrus Vaziri, Michael G. Kemp and Shobhan Gaddameedhi for critical review of the manuscript and Dr. Shay V. Covo for helpful discussion. We thank Dr. Jacquelyn J. Bower for sharing unpublished mitotic entry data for ATR and CHK1 knockdowns in NHF1-hTERT. The manuscript was seen and approved by all authors. The authors have no conflicts of interests or competing financial interests to declare. This work was supported by Public Health Science grants P30ES10126, P30CA16086, ES015856, ES014635 and Environmental Pathology Training Grant ES007017 to SLS-R. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. The UNC Flow Cytometry Core Facility is supported in part by an NCI Center Core Support Grant (P30CA06086) to the UNC Lineberger Comprehensive Cancer Center.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Paulsen RD, Cimprich KA. The ATR pathway: fine-tuning the fork. DNA Repair (Amst) 2007; 6:953 - 66; http://dx.doi.org/10.1016/j.dnarep.2007.02.015; PMID: 17531546
- Kaufmann WK. The human intra-S checkpoint response to UVC-induced DNA damage. Carcinogenesis 2010; 31:751 - 65; http://dx.doi.org/10.1093/carcin/bgp230; PMID: 19793801
- Nam EA, Cortez D. ATR signalling: more than meeting at the fork. Biochem J 2011; 436:527 - 36; http://dx.doi.org/10.1042/BJ20102162; PMID: 21615334
- MacDougall CA, Byun TS, Van C, Yee MC, Cimprich KA. The structural determinants of checkpoint activation. Genes Dev 2007; 21:898 - 903; http://dx.doi.org/10.1101/gad.1522607; PMID: 17437996
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003; 300:1542 - 8; http://dx.doi.org/10.1126/science.1083430; PMID: 12791985
- Zou L, Cortez D, Elledge SJ. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev 2002; 16:198 - 208; http://dx.doi.org/10.1101/gad.950302; PMID: 11799063
- Bermudez VP, Lindsey-Boltz LA, Cesare AJ, Maniwa Y, Griffith JD, Hurwitz J, et al. Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc Natl Acad Sci USA 2003; 100:1633 - 8; http://dx.doi.org/10.1073/pnas.0437927100; PMID: 12578958
- Ellison V, Stillman B. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol 2003; 1:E33; http://dx.doi.org/10.1371/journal.pbio.0000033; PMID: 14624239
- Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev 2007; 21:1472 - 7; http://dx.doi.org/10.1101/gad.1547007; PMID: 17575048
- Chini CC, Chen J. Human claspin is required for replication checkpoint control. J Biol Chem 2003; 278:30057 - 62; http://dx.doi.org/10.1074/jbc.M301136200; PMID: 12766152
- Unsal-Kaçmaz K, Mullen TE, Kaufmann WK, Sancar A. Coupling of human circadian and cell cycles by the timeless protein. Mol Cell Biol 2005; 25:3109 - 16; http://dx.doi.org/10.1128/MCB.25.8.3109-3116.2005; PMID: 15798197
- Unsal-Kaçmaz K, Chastain PD, Qu PP, Minoo P, Cordeiro-Stone M, Sancar A, et al. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol Cell Biol 2007; 27:3131 - 42; http://dx.doi.org/10.1128/MCB.02190-06; PMID: 17296725
- Yoshizawa-Sugata N, Masai H. Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J Biol Chem 2007; 282:2729 - 40; http://dx.doi.org/10.1074/jbc.M605596200; PMID: 17102137
- Smith-Roe SL, Patel SS, Simpson DA, Zhou YC, Rao S, Ibrahim JG, et al. Timeless functions independently of the Tim-Tipin complex to promote sister chromatid cohesion in normal human fibroblasts. Cell Cycle 2011; 10:1618 - 24; http://dx.doi.org/10.4161/cc.10.10.15613; PMID: 21508667
- Gotter AL, Suppa C, Emanuel BS. Mammalian TIMELESS and Tipin are evolutionarily conserved replication fork-associated factors. J Mol Biol 2007; 366:36 - 52; http://dx.doi.org/10.1016/j.jmb.2006.10.097; PMID: 17141802
- Kemp MG, Akan Z, Yilmaz S, Grillo M, Smith-Roe SL, Kang TH, et al. Tipin-replication protein A interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress. J Biol Chem 2010; 285:16562 - 71; http://dx.doi.org/10.1074/jbc.M110.110304; PMID: 20233725
- Serçin O, Kemp MG. Characterization of functional domains in human Claspin. Cell Cycle 2011; 10:1599 - 606; http://dx.doi.org/10.4161/cc.10.10.15562; PMID: 21478680
- De Piccoli G, Katou Y, Itoh T, Nakato R, Shirahige K, Labib K. Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Mol Cell 2012; 45:696 - 704; http://dx.doi.org/10.1016/j.molcel.2012.01.007; PMID: 22325992
- Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 2000; 14:397 - 402; PMID: 10691732
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14:1448 - 59; PMID: 10859164
- Gotter AL, Manganaro T, Weaver DR, Kolakowski LF Jr., Possidente B, Sriram S, et al. A time-less function for mouse timeless. Nat Neurosci 2000; 3:755 - 6; http://dx.doi.org/10.1038/77653; PMID: 10903565
- Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell 2002; 111:779 - 89; http://dx.doi.org/10.1016/S0092-8674(02)01113-3; PMID: 12526805
- Durkin SG, Arlt MF, Howlett NG, Glover TW. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene 2006; 25:4381 - 8; http://dx.doi.org/10.1038/sj.onc.1209466; PMID: 16732333
- Lam MH, Liu Q, Elledge SJ, Rosen JM. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell 2004; 6:45 - 59; http://dx.doi.org/10.1016/j.ccr.2004.06.015; PMID: 15261141
- Focarelli ML, Soza S, Mannini L, Paulis M, Montecucco A, Musio A. Claspin inhibition leads to fragile site expression. Genes Chromosomes Cancer 2009; 48:1083 - 90; http://dx.doi.org/10.1002/gcc.20710; PMID: 19760606
- Leman AR, Noguchi C, Lee CY, Noguchi E. Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J Cell Sci 2010; 123:660 - 70; http://dx.doi.org/10.1242/jcs.057984; PMID: 20124417
- Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 1998; 2:329 - 40; http://dx.doi.org/10.1016/S1097-2765(00)80277-4; PMID: 9774971
- Myung K, Datta A, Kolodner RD. Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell 2001; 104:397 - 408; http://dx.doi.org/10.1016/S0092-8674(01)00227-6; PMID: 11239397
- Sørensen CS, Syljuåsen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res 2012; 40:477 - 86; http://dx.doi.org/10.1093/nar/gkr697; PMID: 21937510
- Jones RM, Petermann E. Replication fork dynamics and the DNA damage response. Biochem J 2012; 443:13 - 26; http://dx.doi.org/10.1042/BJ20112100; PMID: 22417748
- Voineagu I, Freudenreich CH, Mirkin SM. Checkpoint responses to unusual structures formed by DNA repeats. Mol Carcinog 2009; 48:309 - 18; http://dx.doi.org/10.1002/mc.20512; PMID: 19306277
- McFarlane RJ, Mian S, Dalgaard JZ. The many facets of the Tim-Tipin protein families’ roles in chromosome biology. Cell Cycle 2010; 9:700 - 5; http://dx.doi.org/10.4161/cc.9.4.10676; PMID: 20139726
- Dheekollu J, Wiedmer A, Hayden J, Speicher D, Gotter AL, Yen T, et al. Timeless links replication termination to mitotic kinase activation. PLoS One 2011; 6:e19596; http://dx.doi.org/10.1371/journal.pone.0019596; PMID: 21573113
- Dheekollu J, Lieberman PM. The replisome pausing factor Timeless is required for episomal maintenance of latent Epstein-Barr virus. J Virol 2011; 85:5853 - 63; http://dx.doi.org/10.1128/JVI.02425-10; PMID: 21490103
- Liu G, Chen X, Gao Y, Lewis T, Barthelemy J, Leffak M. Altered replication in human cells promotes DMPK (CTG)(n) · (CAG)(n) repeat instability. Mol Cell Biol 2012; 32:1618 - 32; http://dx.doi.org/10.1128/MCB.06727-11; PMID: 22354993
- Leman AR, Dheekollu J, Deng Z, Lee SW, Das MM, Lieberman PM, et al. Timeless preserves telomere length by promoting efficient DNA replication through human telomeres. Cell Cycle 2012; 11:2337 - 47; http://dx.doi.org/10.4161/cc.20810; PMID: 22672906
- Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, et al. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 2003; 424:1078 - 83; http://dx.doi.org/10.1038/nature01900; PMID: 12944972
- Bando M, Katou YM, Komata M, Tanaka H, Itoh T, Sutani T, et al. Csm3, Tof1, and Mrc1 form a heterotrimeric mediator complex that associates with DNA replication forks. J Biol Chem 2009; 284:34355 - 65; http://dx.doi.org/10.1074/jbc.M109.065730; PMID: 19819872
- Chou DM, Elledge SJ. Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc Natl Acad Sci USA 2006; 103:18143 - 7; http://dx.doi.org/10.1073/pnas.0609251103; PMID: 17116885
- Yang XH, Shiotani B, Classon M, Zou L. Chk1 and Claspin potentiate PCNA ubiquitination. Genes Dev 2008; 22:1147 - 52; http://dx.doi.org/10.1101/gad.1632808; PMID: 18451105
- Smith KD, Fu MA, Brown EJ. Tim-Tipin dysfunction creates an indispensible reliance on the ATR-Chk1 pathway for continued DNA synthesis. J Cell Biol 2009; 187:15 - 23; http://dx.doi.org/10.1083/jcb.200905006; PMID: 19805627
- Tanaka K. Multiple functions of the S-phase checkpoint mediator. Biosci Biotechnol Biochem 2010; 74:2367 - 73; http://dx.doi.org/10.1271/bbb.100583; PMID: 21150122
- Noguchi E, Noguchi C, McDonald WH, Yates JR 3rd, Russell P. Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol Cell Biol 2004; 24:8342 - 55; http://dx.doi.org/10.1128/MCB.24.19.8342-8355.2004; PMID: 15367656
- Nedelcheva MN, Roguev A, Dolapchiev LB, Shevchenko A, Taskov HB, Shevchenko A, et al. Uncoupling of unwinding from DNA synthesis implies regulation of MCM helicase by Tof1/Mrc1/Csm3 checkpoint complex. J Mol Biol 2005; 347:509 - 21; http://dx.doi.org/10.1016/j.jmb.2005.01.041; PMID: 15755447
- Errico A, Costanzo V. Mechanisms of replication fork protection: a safeguard for genome stability. Crit Rev Biochem Mol Biol 2012; 47:222 - 35; http://dx.doi.org/10.3109/10409238.2012.655374; PMID: 22324461
- Chini CC, Wood J, Chen J. Chk1 is required to maintain claspin stability. Oncogene 2006; 25:4165 - 71; http://dx.doi.org/10.1038/sj.onc.1209447; PMID: 16501606
- Petermann E, Maya-Mendoza A, Zachos G, Gillespie DA, Jackson DA, Caldecott KW. Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol Cell Biol 2006; 26:3319 - 26; http://dx.doi.org/10.1128/MCB.26.8.3319-3326.2006; PMID: 16581803
- Chastain PD 2nd, Heffernan TP, Nevis KR, Lin L, Kaufmann WK, Kaufman DG, et al. Checkpoint regulation of replication dynamics in UV-irradiated human cells. Cell Cycle 2006; 5:2160 - 7; http://dx.doi.org/10.4161/cc.5.18.3236; PMID: 16969085
- Petermann E, Helleday T, Caldecott KW. Claspin promotes normal replication fork rates in human cells. Mol Biol Cell 2008; 19:2373 - 8; http://dx.doi.org/10.1091/mbc.E07-10-1035; PMID: 18353973
- Scorah J, McGowan CH. Claspin and Chk1 regulate replication fork stability by different mechanisms. Cell Cycle 2009; 8:1036 - 43; http://dx.doi.org/10.4161/cc.8.7.8040; PMID: 19270516
- Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet 2007; 41:169 - 92; http://dx.doi.org/10.1146/annurev.genet.41.042007.165900; PMID: 17608616
- Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol 2010; 11:683 - 7; http://dx.doi.org/10.1038/nrm2974; PMID: 20842177
- Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 2003; 17:615 - 28; http://dx.doi.org/10.1101/gad.1067403; PMID: 12629044
- Jiang K, Pereira E, Maxfield M, Russell B, Goudelock DM, Sanchez Y. Regulation of Chk1 includes chromatin association and 14-3-3 binding following phosphorylation on Ser-345. J Biol Chem 2003; 278:25207 - 17; http://dx.doi.org/10.1074/jbc.M300070200; PMID: 12676962
- Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, et al. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell 2005; 19:607 - 18; http://dx.doi.org/10.1016/j.molcel.2005.07.019; PMID: 16137618
- Smits VA, Reaper PM, Jackson SP. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr Biol 2006; 16:150 - 9; http://dx.doi.org/10.1016/j.cub.2005.11.066; PMID: 16360315
- Speroni J, Federico MB, Mansilla SF, Soria G, Gottifredi V. Kinase-independent function of checkpoint kinase 1 (Chk1) in the replication of damaged DNA. Proc Natl Acad Sci USA 2012; 109:7344 - 9; http://dx.doi.org/10.1073/pnas.1116345109; PMID: 22529391
- Arlt MF, Glover TW. Inhibition of topoisomerase I prevents chromosome breakage at common fragile sites. DNA Repair (Amst) 2010; 9:678 - 89; http://dx.doi.org/10.1016/j.dnarep.2010.03.005; PMID: 20413351
- Tanaka H, Kubota Y, Tsujimura T, Kumano M, Masai H, Takisawa H. Replisome progression complex links DNA replication to sister chromatid cohesion in Xenopus egg extracts. Genes Cells 2009; 14:949 - 63; http://dx.doi.org/10.1111/j.1365-2443.2009.01322.x; PMID: 19622120
- Sjögren C, Nasmyth K. Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr Biol 2001; 11:991 - 5; http://dx.doi.org/10.1016/S0960-9822(01)00271-8; PMID: 11448778
- Urtishak KA, Smith KD, Chanoux RA, Greenberg RA, Johnson FB, Brown EJ. Timeless Maintains Genomic Stability and Suppresses Sister Chromatid Exchange during Unperturbed DNA Replication. J Biol Chem 2009; 284:8777 - 85; http://dx.doi.org/10.1074/jbc.M806103200; PMID: 19112184
- Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, et al. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell 2004; 16:991 - 1002; http://dx.doi.org/10.1016/j.molcel.2004.11.027; PMID: 15610741
- Sabouri N, McDonald KR, Webb CJ, Cristea IM, Zakian VA. DNA replication through hard-to-replicate sites, including both highly transcribed RNA Pol II and Pol III genes, requires the S. pombe Pfh1 helicase. Genes Dev 2012; 26:581 - 93; http://dx.doi.org/10.1101/gad.184697.111; PMID: 22426534
- Petermann E, Woodcock M, Helleday T. Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci USA 2010; 107:16090 - 5; http://dx.doi.org/10.1073/pnas.1005031107; PMID: 20805465
- Shechter D, Costanzo V, Gautier J. ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol 2004; 6:648 - 55; http://dx.doi.org/10.1038/ncb1145; PMID: 15220931
- Maya-Mendoza A, Petermann E, Gillespie DA, Caldecott KW, Jackson DA. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J 2007; 26:2719 - 31; http://dx.doi.org/10.1038/sj.emboj.7601714; PMID: 17491592
- Boyer JC, Kaufmann WK, Brylawski BP, Cordeiro-Stone M. Defective postreplication repair in xeroderma pigmentosum variant fibroblasts. Cancer Res 1990; 50:2593 - 8; PMID: 2109654
- Heffernan TP, Simpson DA, Frank AR, Heinloth AN, Paules RS, Cordeiro-Stone M, et al. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol Cell Biol 2002; 22:8552 - 61; http://dx.doi.org/10.1128/MCB.22.24.8552-8561.2002; PMID: 12446774
- Simpson DA, Livanos E, Heffernan TP, Kaufmann WK. Telomerase expression is sufficient for chromosomal integrity in cells lacking p53 dependent G1 checkpoint function. J Carcinog 2005; 4:18; http://dx.doi.org/10.1186/1477-3163-4-18; PMID: 16209708
- Zhou T, Chou JW, Simpson DA, Zhou Y, Mullen TE, Medeiros M, et al. Profiles of global gene expression in ionizing-radiation-damaged human diploid fibroblasts reveal synchronization behind the G1 checkpoint in a G0-like state of quiescence. Environ Health Perspect 2006; 114:553 - 9; http://dx.doi.org/10.1289/ehp.8026; PMID: 16581545
- Anderson EM, Birmingham A, Baskerville S, Reynolds A, Maksimova E, Leake D, et al. Experimental validation of the importance of seed complement frequency to siRNA specificity. RNA 2008; 14:853 - 61; http://dx.doi.org/10.1261/rna.704708; PMID: 18367722