Abstract
One undisputed milestone of traditional oncology is neoplastic progression, which consists of a progressive selection of dedifferentiated cells driven by a chance sequence of genetic mutations. Recently it has been demonstrated that the overexpression of well-defined transcription factors reprograms somatic cells to the pluripotent stem status. The demonstration raises crucial questions as to whether and to what extent this reprogramming contributes to tumorigenesis, and whether the epigenetic changes involved in it are reversible. Here, we show for the first time that a tumor produced in vivo by a chemical carcinogen is the product of the interaction between neoplastic progression and reprogramming. The experimental model employed the prototype of ascites tumors, the Yoshida AH130 hepatoma and other neoplasias, including human melanoma. AH130 hepatoma was started in the liver by the carcinogen o-aminoazotoluene. This compound binds to and abolishes the p53 protein, producing a genomic instability that promotes both the neoplastic progression and the hepatoma reprogramming. Eventually this tumor contained 100% CD133+ elements and pO2-dependent percentages of the three embryonic transcription factors Nanog, Klf4 and c-Myc. Once transferred into aerobic cultures, the minor cellular fraction expressing this triad generates various types of adherent cells, which are progressively substituted by non-tumorigenic elements committed to fibromuscular, neuronal and glial differentiation. This reprogramming appears to be accomplished stepwise, with the assembly of the triad into a sophisticated transcriptional, oxygen-dependent circuitry, in which Nanog and Klf4 antagonistically regulate c-Myc, and hence, cell hypoxia survival and cell cycle activation.
Introduction
Cancer phenotype is subject to a multistep drift that transforms differentiated tissues into an immature cell population, a process traditionally referred to as “neoplastic progression.”Citation1-Citation3 This definition implied an irreversible combination of genetic and epigenetic changes, brought about by DNA mutations and clonal selection and driven by clonal selection.Citation4,Citation5 At the final stage of this stochastic sequence, it is often not possible to trace tumor hystogenesis, due to the morphological and biochemical confluence of various neoplasias into a “convergent phenotype.”Citation2,Citation6 Such phenotype does not reveal any signs of differentiation, while it is characterized by an indefinite self-renewal and a glycolytic-oriented profile, which compensates for a poor mitochondrial apparatus.Citation7-Citation15
Nowadays, certain features suggest that the neoplastic progression might be intertwined to reprogramming, which is the ordered epigenetic process of inducing highly differentiated cells to reverse their developmental potential to attain the undifferentiated, pluripotent state.Citation16-Citation20 Actually, the introduction of genes of embryonic transcription factors (ETFs) (Oct4, Sox2, c-Myc and Klf4) into gastrointestinal cancer cells resulted in reprogramming of cells to a pluripotent state and sensitized them to differentiation induction.Citation16-Citation20 This new perspective might help answering two related questions which are of the greatest importance in modern oncology: to what extent does reprogramming contribute to tumorigenesis and whether the epigenetic changes involved in it are reversible.Citation17,Citation18 Significant answers should be obtained by unraveling an ETF’s signature of anaplastic tumors and by the integration of these factors in the biochemical pathways underlying the embryonic hallmarks of cancer stem cells, foremost adaptation to hypoxia.
We approached this topic by using the AH130 hepatoma generated by Yoshida by treating Wistar rats with the carcinogen o-aminoazotoluene and widely used as in vivo model of experimental cancer.Citation21,Citation22 After serial transplantations in rat peritoneal cavity, this tumor became composed of isolated spheroidal cells, fed by the ascites fluid exuded from the peritoneal vessels. These cells displayed a totally undifferentiated phenotype, with a high nucleus/cytoplasm ratio and few mitochondria. This nude phenotype implies a silencing of most differentiation genes, like that nowadays reproducible by transfecting definite ETFs.Citation23 In addition, they lost the p53 protein, a condition facilitating a stem cell-like cellular plasticity.Citation24
At advanced stages after transplantation, when the intraperitoneal pO2 is near zero, the tumor cell bulk survives as a G0/G1 naturally synchronized population. We have established that, in order to alternate their hypoxic survival and their oxygen-dependent growth, this population exploits a complex metabolic checkpoint (MCP) at the G1/S transition, consisting of a tight complementation of aerobic glycolysis, redox state and purine metabolism.Citation6 The existence of such a sophisticated and self-perpetuated MCP reflects a primordial transcription signature of stem cells destined to survive in hypoxia. We hypothesized that AH130 cells represent the outcome of a spontaneous intertwined neoplastic progression and reprogramming processes, an assumption sustained by the finding of the expression of EFTs responsible for differentiated cell reprogramming to the induced pluripotent stem cells (iPSCs) state.Citation25,Citation26
Here we present evidence that within the AH130 population, a significant proportion of the cells express an embryonic phenotype, modulated by pO2 through a transcriptional loop of three fundamental ETFs, Nanog, Klf4 and c-Myc, which regulates the cell cycle, working in concert with the MCP. Upon transfer in vitro, this subpopulation manifested differentiated phenotypes deprived of tumorigenic potency.
Results
The embryonic-like morphology and lack of p53 protein in AH130 cells: the flow-chart of experimental procedures
We first characterized AH130 cells by analyzing their cellular morphology and cell cycle distribution at different pO2 concentrations. shows the typical morphology of AH130 cells during tumor development in vivo and in vitro. Cells are round-shaped, randomly heaped, with a high nucleus/cytoplasm ratio () and exhibit a reduced mitochondrial apparatus (), features profoundly different from those of normal liver (), in keeping with immunocytochemical negativity to albumin and α-fetoprotein ().
Figure 1. The AH130 experimental model. (A and B) Phase-contrast (A) and electron microscopy (B) images of AH130 cell population as soon as recovered from the host animal at day 11 after transplantation. Bar = 100 µm and 10 µm, respectively. (C) An electron microscopy image of a normal rat adult liver. Bar = 10 µm. (D and E). The immunocytochemical technique revealed the negativity to Albumin (D) and α fetoprotein (E) of AH130 hepatoma cells. Bars = 50 µm. (F) The loss of p53 in ascites cells. Lysates of AH130 cells were obtained and processed in parallel with those of a positive and a negative control (Kasumi acute myeloid leukemia and K562 chronic myeloid leukemia cells, respectively). Vinculine expression was tested as immunoblot control. (G) Flow chart of experimental procedures. Six days after transplantation (mild hypoxia), aliquots of the tumor were transplantated intraperitoneally in new hosts or utilized for immunophenotypical characterization. Eleven days after transplantation (severe hypoxia), the tumor was harvested and utilized for the in vitro experiments.
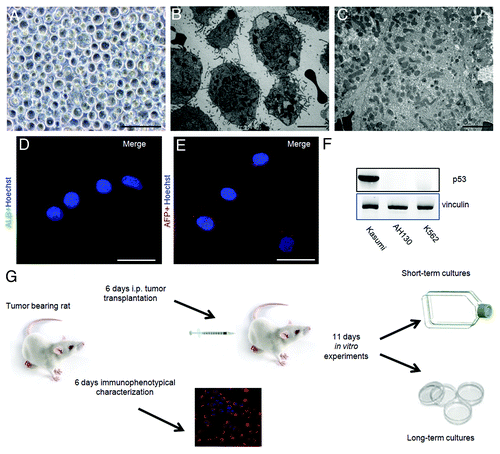
An immunoblot analysis of p53 expression in lysates of AH130 cells demonstrates the total lack of this fundamental tumor suppressor gene in these hepatoma cells ().
Our experiments were performed both in vivo and in vitro, and their time course is summarized in the flow chart in .
Cytokinetics and pO2 changes at various stages of tumor development in vivo and in vitro
As typical of ascites tumors, cells grow suspended in the fluid (ascites) exuded from peritoneal vessels into the peritoneal cavity. The standard growth kinetics in vivo () were initially exponential, slowing down thereafter until growth arrest was reached, at day 11, when all cells were blocked in G0/G1 (). As shown in , these growth kinetics were strictly conditioned by the intraperitoneal pO2, which reached 5 mmHg around day 6 (mild hypoxia) and approached zero around day 11 (severe hypoxia). This parameter was measured by polarography.Citation27
Figure 2. The role of the mitochondrial respiration on the G1/S transition. (A) Time-course of pO2 (open circles) and AH130 tumor development in vivo (closed circles) at various time after transplantation. The pO2 measurements were obtained by a Clark-type microelectrode (Beckman, 315780) mounted on the Beckman Physiological Gas Analyzer (Mod.160) connected to a recorder, according to Del Monte (1967). (B) Cell cycle distribution of AH130 cells at day 11 after transplantation as measured by cytofluorimetric technique. (C). Linear correlation between the number of cells recruited into S as measured by the R = 14C-Thymidine pulse (DPM/90 min/106 viable cells) and by cytofluorimetric technique at the corresponding times. The latter parameter was estimated by multiplying the cytofluorimetric percentage of cells in S for the total number of cells present in the sample at the corresponding time. Note the synchronization of the cells in the S phase at t = 18 as compared with t = 0.
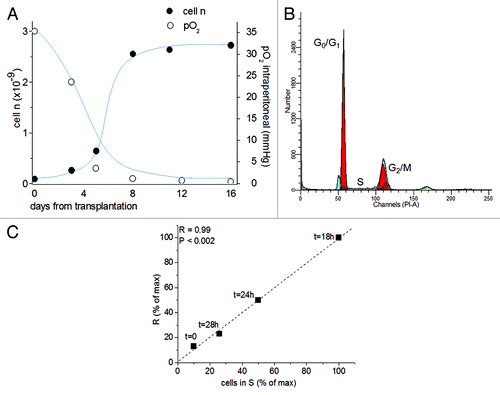
Upon transfer from the anaerobic peritoneal cavity at day 11 into an aerobic culture in vitro (pO2 = 160 mmHg), the AH130 cells undergo a synchronized recruitment from G0/G1 into the S phase, as determined by the strict relationship of flow cytometry data to those obtained by pulse labeling with 14C-thymidine ().Citation6 The maximum value of this parameter was reached after 18 h of incubation in air (R, t = 18 h), when 100% of the cells accumulated in S phase.
The metabolic check point (MCP) at the G1/S transition
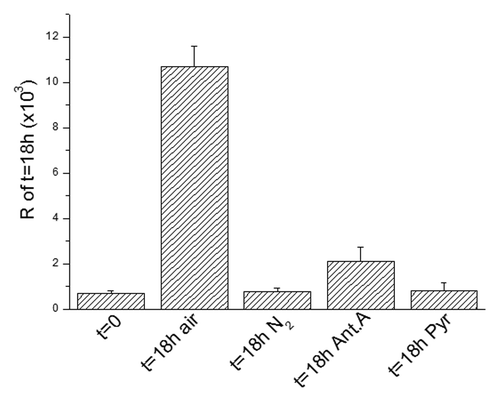
We previously demonstrated that the recruitment of G1-blocked cells into the S phase is subjected to an MCP based on a respiration-linked limiting step.Citation6 This MCP implies a tight complementation of aerobic glycolysis, cellular redox state and folate metabolism and controls the purine pool necessary to promote the neo-synthesis of DNA.Citation28 This promotion requires a cytosolic NADP-dependent step of folate utilization in the synthesis of purine ring, where NADPH produced must be reoxidated through the transport of reducing equivalents (electrons) to the mitochondrial respiratory chain.Citation28 Thus, this step can be limited in two ways: (1) hindering respiration, either by hypoxia or by impairing the electron transport to oxygen with specific inhibitors of the respiratory chain (antimycin A), or (2) saturating the respiratory chain with an excess of oxidizable substrates, foremost pyruvate. In the latter case, the differentiation state of cells is critical. Indeed, saturation is easily prevented in differentiated cells endowed with large mitochondrial equipment but easily produced in cells with few mitochondria, such as AH130 or cancer stem cells. According to all above, AH130 cell recruitment into S phase could be inhibited in anaerobiosis in the presence of antimycin A or in normoxic cultures supplemented with an excess of pyruvate ().
Figure 3. The metabolic check point (MCP) at the G1/S transition. The recruitment into S of ascites cells as measured by R at 18 h as compared with t = 0 and the effects of nitrogen incubation or antimycin A (6 × 10−6M) or pyruvate (10 mM) on this recruitment. Values are expressed as mean ± SEM of three experiments.
ETF and immunophenotypical markers at various stages in vivo and in vitro
To study the modulation of ETF at different pO2 concentrations in vivo, AH130 cells were recovered from animals at day 6, when the intraperitoneal pO2 was about 5 mmHg (mild hypoxia), and at day 11, when peritoneal hypoxia was severe (pO2~0). Immunocytochemical analyses showed that at day 6, AH130 cells expressed (1) CD133 (in 44% of the cells), a marker localized on the plasma membrane of several types of somatic stem cells, including hematopoietic and neural stem cells; (2) p75 (28%), a marker of neural crest stem cells; (3) nestin (36%), a common marker of immature and malignant neuronal cells; (4) (100%) vimentin, a common component of cytoskeleton.Citation29 The ETF “signature” was: Nanog 21%, Klf4 16.5%, c-Myc 12.3% ().
Figure 4. Immunocytochemical profile of AH130 cells at 6 and 11 d after transplantation or after 18 h of incubation in air. (A‒F) The marker signature of the cell population at day 6. Note the co-expression of Nanog and Klf4 revealed by the simultaneous exposure to the corresponding antibodies (E). Bars = 50 µm. (G‒H) The marker signature at day 11. Note the expression of Nanog alone, despite simultaneous exposure to anti-Nanog and anti-Klf4 antibodies (panel (G). Bars = 50 µm. (I‒K) The marker signature after 18 h of incubation in air. Note the expression of Klf4 alone, despite simultaneous exposure to anti-Nanog and anti-Klf4 antibodies (I) Bars = 50 µm. Cell nuclei were stained with Hoechst 33258. (L) Summary of AH130 cells immunocytochemical profiles under the various above conditions. Note the 100% expression of Nanog in N2 atmosphere (black column). Values are expressed as mean ± SEM of three experiments.
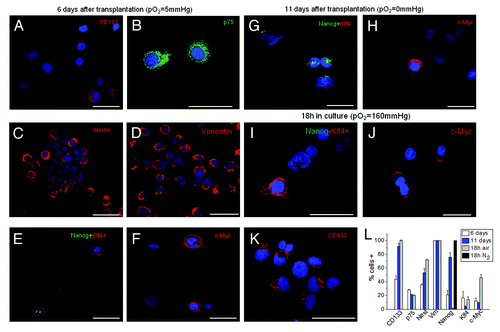
At day 11 in vivo, when peritoneal hypoxia was severe and all cells were growth-arrested (cell concentration in ascites fluid reaching 80‒100 × 106/ml), the ETF signature was characterized by a strong increase in the percentage of Nanog-positive cells with respect to day 6, a strong decrease of Klf4 positivity and a substantial stability of c-Myc positivity (). CD133 also markedly increased with respect to day 6, resulting in expression in more than 90% of cells. Nestin changed from 36 to 53%, whereas p75 and vimentin remained unchanged. The most important result of these experiments is that at day 11 in vivo, the vast majority of cells exhibited a marker of immaturity (CD133), and that three-fourths of cells attained an immaturity level sufficiently high to express at least one ETF (Nanog). Actually, this is an under-estimation of the effects of hypoxia/anoxia on Nanog expression, as 100% of cells expressed Nanog incubated for 18 h in nitrogen atmosphere ().
The transfer in air (pO2 = 160 mmHg) of AH130 cells harvested ex vivo at day 11 produced a synchronized cell recruitment into S phase () and was accompanied by the following changes in the ETF signature: drastic reduction of Nanog+ cells (from 76 to 0.8%) and increase in Klf4+ and c-Myc+ cells (from 4 to 14% and from 9 to 46%, respectively). Nestin slightly increased (from 53 to 72%), whereas CD133, vimentin and p75 remained unchanged ().
Taken together, the results of indicate that (1) the entire population has attained, in hypoxia in vivo and after 18 h incubation in normoxia, an immaturity level sufficiently high to express at least one ETF; (2) 75% of the AH130 population is enabled to express Nanog 11 d after transplantation (100% in nitrogen atmosphere); (3) 17% Klf4 and 46% c-Myc in air: this leads to calculate that about 7% have the probability to assemble the triad in the transcriptional loop that controls the cell cycle in these cells. Thus, the AH130 tumor emerges as a heterogeneous cell population containing a variety of immature cells, whose signatures characterize pluripotent stem cells (Nanog, Klf4, c-Myc) and multipotent stem cells (CD133).
Short-term incubation in air modulates ETF mRNA expression
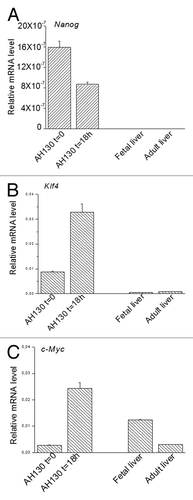
The ETF modulation by pO2 in AH130 cell was quantified by the real-time PCR (qPCR) as follows. Cells were recovered from animals at the hypoxic stage of tumor development in vivo (day 11) and ETF expression was tested immediately after recovery (t = 0) and after 18 h incubation in air. As shown in , Nanog expression was reduced by half at 18 h in air as compared with t = 0. On the other hand, the expression of Klf4 () and c-Myc () was 3-fold and 9-fold increased, respectively. These results confirm that in AH130 cells pO2 modulates the level of expression of Nanog negatively and that of Klf4 and c-Myc positively. The pattern of Nanog, Klf4 and c-Myc expression was also determined in normal liver as well as in hepatocytes characterized by intense proliferation, such as those of fetal liver. The results of showed that fetal and adult liver do not express Nanog or Klf4 at all but expresses c-Myc. Taken together, the data of indicate that the expression of Nanog and Klf4 is limited to dedifferentiated cells of Yoshida hepatoma, while the expression of c-Myc is rather related to the growth rate of the cell population.
Figure 5. The intracellular modulation of ETFs quantified by RT-PCR experiments. (A‒C). Relative mRNA levels of Nanog, Klf4 and c-Myc in AH130 ascites cells at t = 0 and t = 18 h in fetal and adult liver. Values are expressed as mean ± SEM of three experiments.
AH130 cells maintained in long-term aerobic conditions
Pathways of differentiation
We tested the long-term effects of air in AH130 cells transferred in vitro in MEM/F12 medium containing 10% fetal calf serum. Under these conditions, and throughout the experimental time, the bulk of cell population did not adhere to the plastic surface and progressively die as revealed by the PI incorporation ( and ).
Figure 6. Time course of viability and immunophenotipycal analysis of non-adherent population. (A‒C) Cellular incorporation of propidium iodide at 24 h, 72 h and 5 d after seeding. The percentage of necrotic cells increased up to 95% throughout 5 d in culture. (D‒F1) The immunocytochemical analysis performed in the first 5 d of aerobic culture demonstrated the AH130 population is negative to CD34, CD68, calretinin, platelet/endothelial cell adhesion molecule-1, fibroblast activation protein 1. The overlay of anti-mouse Cy3-, anti-rabbit fluorescein- or anti-goat fluorescein-conjugated antibody staining and Hoechst 33258 signals is shown in the panels labeled “Merge.”
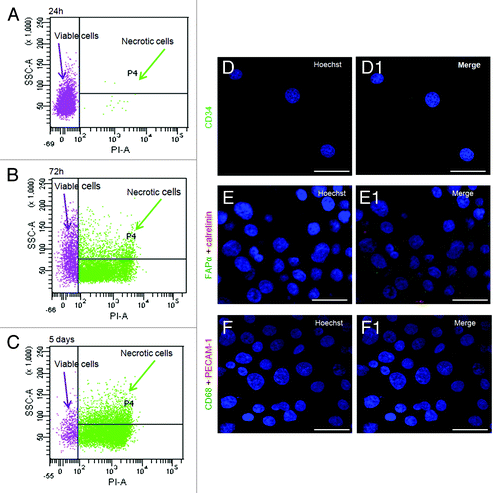
Table 1. Propidium iodide (PI) incorporation in AH130 cells
The contamination of the adherent population by the cells, derived from the host, was excluded principally by the fact that all these cells express the ETFs before adhesion, characterizing the AH130 ascites cells: in addition to that, they resulted negative to CD68, calretinin, platelet/endothelial cell adhesion molecule-1, CD34 and fibroblast activation protein 1, ruling out their derivation from macrophages, mesothelial (peritoneal), endothelial, hematopoietic cells and fibroblasts, respectively ( and ).
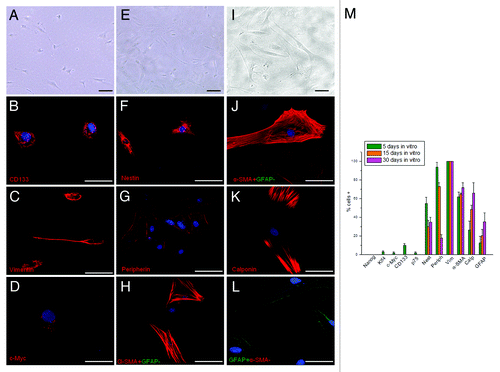
About 5–7% of cells adhered and survived (), undergoing a very slow growth, which arrests completely after 1 mo in culture. This percentage corresponds to cells expressing the ETF triad (Nanog, Klf4 and c-Myc). At day 5 in culture, which is the time required for cell adhesion, Klf4 and c-Myc were both expressed in about 5% of the entire adherent population (), while Nanog was not expressed at all as expected from the aerobic conditions. The rest of this population manifested signs of commitment to various differentiation lineages, as indicated by the expression of peripherin, α-smooth muscle actin (α-SMA), calponin and glial fibrillary acid protein (GFAP). At day 15, the three ETFs, CD133 and p75, were no longer expressed, while nestin persisted as the only sign of immaturity (). At day 30, two cell subpopulations were observed. The major one included cells strongly positive for α-SMA, calponin and vimentin (), suggesting cell differentiation along the fibromuscular lineage; the second one was positive for nestin, peripherin and GFAP, suggesting cell differentiation along neuronal and glial lineages.Citation29
Figure 7. Morphological and immunocytochemical profile of the tumor during a long-term aerobic incubation. (A) Phase-contrast image of AH130 cell population after 5 d in aerobic culture. Bar = 50 µm. (B‒D) Representative signature of ascites cells after 5 d in aerobic culture. Bar = 50 µm. (E) Phase-contrast image of AH130 cell population after 15 d in aerobic culture. Bar = 50 µm. (F‒H) Representative signature of ascites cells after 15 d in aerobic culture. Bar = 50 µm. (I) Phase-contrast image of AH130 cell population after 30 d in aerobic culture. Bar = 50 µm. (J‒L) Representative signature of ascites cells after 30 d in aerobic culture. Bar = 50 µm. (M) Summary of AH130 cells’ immunocytochemical profiles during a long-term aerobic incubation. Values are expressed as mean ± SEM of three experiments.
Loss of tumorigenicity
The tumorigenicity of the adherent population rescued ex vitro was tested in syngenic animals, comparing the effect of cells normally utilized for serial transplantation (control). 106 cells of both these populations were inoculated intraperitoneally in normal Wistar rats. Two months after transplantation, all control rats had died due to the development of a large neoplastic population containing about 3 × 109 cells in 80 ml of ascites fluid. On the contrary, rats transplanted with adherent cells did not develop any neoplasia and survived normally. These results suggest that upon long-term adhesion in aerobic conditions, AH130 cells undergo a progressive loss of their stem phenotype and tumorigenic potential.
The ETFs transcription loop and its role in the cell cycle regulation
The effects of pO2
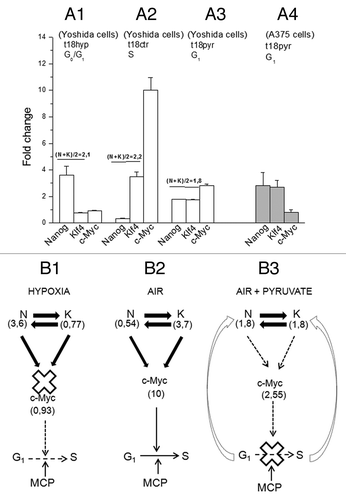
In we reported the ETFs signatures expressed in fold change with respect to t = 0. These are: after incubation in hypoxia (0.1% O2) (Fig. 8A1) (t = 18 h hyp) Nanog = 3.6, Klf4 = 0.77, c-Myc = 0.93; in air (Fig. 8A2) (t = 18 h ctr) Nanog = 0.54, Klf4 = 3.7, c-Myc = 10. Thus, the hypoxia/air transition produced changes of Nanog (N) opposite and quantitatively equivalent to those of Klf4 (K), so that (N + K)/2 remains constant (~2). The corresponding changes of c-Myc varied from 0.90- to 10-fold change. All these findings indicate that Nanog and Klf4 compete with each other to move c-Myc in opposing directions according to the functions: c-Myc = f(N) and c-Myc = g(K).
Figure 8. The role of the ETF triad operating in the AH130 reprogrammed cells to regulate cell cycle activation. (A) The pO2 modulation of the Nanog, Klf4, c-Myc transcriptional circuitry. (A1) Hypoxia (t18hyp) enhances Nanog, while it represses Klf4 and c-Myc. (A2) After transfer to aerobic culture (t18ctr), the increase of pO2 represses Nanog and enhances Klf4 and c-Myc. The fold change of Nanog and Klf4 (A1 vs A2) are equivalent in quantity but opposite in sign, so that N + K = C. (B). Diagrammatic representation of the check-points regulating the G1/S transition: the role of ETFs. (B1) The hypoxic ratio N/K = 3, 6/0, 77 is inadequate to generate the mutual interaction necessary to activate c-Myc. This produces a cell cycle check point that restricts the G1/S transition upstream to the pyruvate-sensitive MCP. (B2) The aerobic ratio N/K = 0, 54/3, 7 is adequate to activate c-Myc providing the consensus to the G1/S transition upstream to MCP. (A3) The aerobic incubation in the presence of pyruvate produces the equivalence N = K (1, 8/1, 8), again inadequate to generate the c-Myc activation. (A4) The equivalence N = K and the consequent inhibition of c-Myc activation was also produced by pyruvate in A375 human melanoma cells. (B3) In the presence of pyruvate, the inhibition of c-Myc activation brought about by the N/K equivalence reinforces upstream the block of the G1/S transition, brought about by pyruvate saturation of the respiratory chain (MCP).
The effects of pyruvate
The results reported above did not answer the question as to whether the ETF changes produced by the hypoxia to normoxia transition were brought about by the increase of pO2 directly or by the consequent cell recruitment into S. To address this question, cells rescued from animals at day 11 after transplantation were transferred in vitro and incubated for 18 h under the following conditions: severe hypoxia, air, air plus 10 mM pyruvate ().
The rationale of these experiments lies in the fact that hypoxia blocks cell cycle, but also abolishes any direct effect of oxygen on ETFs. On the other hand, the addition of pyruvate inhibits cell cycling even when cells are maintained in aerobiosis. Thus, changes of ETFs produced by pyruvate are to be related exclusively to the impairment of cell recruitment into S phase. As shown above, hypoxia to air transition produced opposite changes of Nanog on one side and of Klf4 and c-Myc on the other. Pyruvate modified these effects (Fig. 8A3), leading Nanog from 3.6 in hypoxia to 1.8 (instead of 0.54 in air), Klf4 from 0.77 in hypoxia to 1.8 (instead of 3.7 in air) and c-Myc from 0.93‒2.55 (instead of 10 in air). Altogether, pyruvate treatment revealed that 50% of the changes in ETFs expression induced by oxygen are direct (pyruvate insensitive), and 50% are mediated through the activation of the cell cycle (pyruvate sensitive).
This above peculiar effect of pyruvate was confirmed in human melanoma stem cells (A375) (Fig. 8A4), which exhibit a stem cell phenotype characterized by the expression of Nanog, Klf4 and c-Myc.Citation24 It is to note that changes of Nanog and Klf4 appeared inversely proportional, whereas their sum is constant across the different experimental variants (hypoxia, 4.2; air, 4.4; air plus pyruvate, 3.6). This implies that the level of c-Myc expression is regulated of the Nanog/Klf4 ratio (). In air (Fig. 8B2), this balance is settled to determine a maximal expression of c-Myc and thereby G1-S transition, whereas in hypoxia (Fig. 8B1), it becomes such as to block c-Myc activation and, hence, cycling. In the presence of pyruvate (Fig. 8B3), the initial event is the block of G1-S transition, due to impairment of the redox steps of folate utilization in purine synthesis.Citation6 This impairment, in turn, induces a negative feedback on the Nanag/ Klf4 ratio and, hence, the deficit of c-Myc.
Discussion
The AH130 hepatoma emerges from this study as the product of a complex intertwining between neoplastic progressions and reprogramming. Both these processes contributed to a progressive cell dedifferentiation that began in the liver by the cancerogenetic treatment and ended with a composite stem cell population containing up to 100% CD133+ elements and pO2-dependent percentages of Nanog-, Klf4- and c-Myc-positive cells. Such signature implies a dedifferentiation proceeded far beyond the liver organogenesis, in some cases right down to pluripotent developmental stages.
At a first glance, this population may appear generated at random, alternating variously dedifferentiated cells in an aberrant evolution driven exclusively by the neoplastic progression. Indeed, this view is contradicted by the following analysis of the expression of ETFs under various experimental conditions. We calculated that about 7% of cells are enabled to assemble the triad in the transcriptional loop that controls the cell cycle in these cells (). This pO2-dependent control consists of an enhancement of Nanog at the expense of Klf4 in hypoxia, and vice versa in air, so that the Nanog/Klf4 ratio gears the c-Myc expression, which acts as the final activator of the G1/S transition.
The interplay between ETFs and cell cycle defined in this work is in keeping with the following references: (1) the expression of endogenous Nanog is strongly increased in hypoxia;Citation30 (2) Nanog and Klf4 work in concert to provide circuitries that regulate the fate of embryonic stem cells, co-occupying various common binding sites of c-Myc.Citation31,Citation32 Assuming that the number of these sites are limited, their engagement by Nanog will happen at the expense of Klf4 and vice versa, so that the increase of the one is associated with an equivalent decrease of the other. The ETFs- dependent control of the cell cycle works in concert with the MCP, the check-point we discovered at the guard of the G1/S transition of all the AH130 cells and is shared by most anaplastic tumors and hemopoietic and lymphopoietic progenitors.Citation33,Citation34 The MCP consensus is inhibited either in hypoxia by the block of the respiratory chain, or in air through the saturation of this chain. It is noteworthy that the saturating effect of pyruvate is associated with the settlement of the ratio Nanog/Klf4 at the equivalence (N = K), implying an influence of this physiological substrate on the EFTs triad, either direct or mediated through the block of the G1/S transition. c-Myc promotes the G1/S transition by regulating cyclins and cyclin kinase inhibitors.Citation35-Citation37 Some of these factors might play a role in the putative feedback between cell cycle activation and Nanog and Klf4 modulation registered in this paper.
To sum up, the hepatoma contains two distinct populations as far as their control of the G1/S transition is concerned: the component governed by the sole MCP and that governed by the ETFs triad in concert with MCP.
A strict control of the G1/S transition is essential to prevent a premature cell engagement in the S phase and the consequent death.Citation38 Evidently, the MCP control provides a sufficient protection against the hypoxic death of the entire population, which, in fact, survive in hypoxia, synchronized in G1. The reinforcement of this control in cells that reprogram the ETFs triad indicates that such a sophisticated device is required to make practically impossible for embryonic stem cells to enter the S phase prematurely in their hypoxic niche. In other words, we interpret the ETFs triad as a crucial step of cell reprogramming to the embryonic stage, which implies the highest level of defense from premature oxygen stimulation. In this perspective, the contemporary expression of Nanog, Klf4, c-Myc represents a very late and, quite possibly, the final step in the process, which began in the liver cells and continued stepwise up to the triad assembly. Any incomplete or defective accomplishment of this program would account for the erratic expression of one single member of the triad within the cell population.
This interpretation is confirmed by the results of the long-term incubation of the tumor. In fact, this procedure leads to death of 95% of the entire population, which does not adhere to the cultures dishes, while the 5% adherent fraction undergoes the multiform differentiation reported in . This small subpopulation corresponds to the ca. 7% of cells enabled to assemble the triad in the original tumor. Notably, the adherent cells maintain this triad in culture only up to the appearance of the differentiated cells, so that at day 5, only 3–4% of the adherent population maintains the ETFs expression, and at day 15, all cells are differentiated and no traces of ETFs signature persists. Altogether, the cell reprogramming brought about spontaneously in the tumor in vivo up to the assembly of the ETFs triad can be furthered to higher level of differentiation. This is simply by cell adhesion and enriching the culture medium with FCS. A relevant corollary of these data are that the lack of p53 is associated in AH130 tumor with a reprogramming efficiency ranging about 5%, a figure of the same order of that (10%) obtained by the Yamanaka group, producing iPSCs cells from mouse embryonic fibroblasts (MEF) deprived of p53.Citation39
All in all, although the AH130 hepatoma reveals a significant tribute to reprogramming, it is still prevalently contributed by the neoplastic progression. The latter is testified by the predominance of the cells whose extreme dedifferentiation cannot be tracked to any plausible normal embryonic phenotype. This large contribution is well in keeping with the p53 total deletion that characterizes this tumor.
Evidently, the multitude of genetic mutations driven by the p53 deletion does not impair a significant reprogramming. Actually, a vast literature is nowadays providing evidence that even a vast amount of genetic mutations, such as that produced by the p53 deletion, not only is compatible with reprogramming, but favors it highly.Citation40-Citation45
Reprogramming and neoplastic progression essentially differ with regard to the nature of the steps involved, which are prevalently epigenetic events or genetic mutations, respectively. Moreover, neoplastic progression is started by oncogenic stimuli (physical, chemical, viral), while the initiating events of reprogramming are unknown, except the experimental transfection of definite ETFs.Citation1,Citation24 However, the two processes share one fundamental feature, which consists in driving a progressive selection of extremely simplified cells, endowed with few mitochondria, a powerful glycolytic apparatus, an indefinite self-renewal and hypoxia adaptation. In this light, it is conceivable that the neoplastic progression, in so far as it selects anaplastic elements, progressively pushes the cell population toward a status that in some cases, facilitates the reprogramming. This status has been identified in the genetic instability displayed by the cell populations with null or mutated expression of p53.Citation42 Mutations of this key regulator protein appear to be especially concentrated among tumors showing plasticity and loss of differentiation. Consistently, the p53 deletion eliminates one of the barriers to the acquisition of the epigenetic changes that underlie the dedifferentiation down to the iPSCs.Citation23,Citation40-Citation45 In this context, the fact that o-aminoazotoluene binds to proteins and not to DNA, besides deleting p53, may involve epigenetic changes of chromatin at the same time it sustains the neoplastic progression.Citation7,Citation41
All these hints suggest that a strict intertwining between reprogramming and neoplastic progression might proceed in parallel so far as genetic and epigenetic changes silence or mutate the differentiation genes, associating immaturity with self-renewal and hypoxia adaptation. Cancer might intervene with the loss of cell cycle control as a stochastic irreversible deviation consequent to the generation of appropriate oncogenic lesions.Citation26
Materials and Methods
Cells
AH130 ascites hepatoma was obtained by T. Yoshida (1932) by treating Wistar rats with the carcinogen o-aminoazotoluene. After serial transplantations in rat peritoneal cavity, the tumor became composed of isolated spheroidal cells fed by the ascites fluid exuded from the peritoneal vessels. These cells displayed a totally undifferentiated phenotype, with a high nucleus/cytoplasm ratio and few mitochondria. Tumor was maintained by weekly intraperitoneal inoculation of 3 × 107 cells in tumor-adapted male Wistar rats weighing 150–200 g, given water and food ad libitum. At the time of the experiment (11 d after transplantation), one tumor-bearing animal was sacrificed and the tumor withdrawn under sterile conditions. Seventy to 80 ml, was centrifuged at 1,000 g for 20 min in order to separate the cells from the ascitic plasma, which was collected and used for the preparation of the incubation medium. Portions of the cell suspension were incubated at 38°C in a Warburg apparatus. At the end of incubation, the contents of the flasks were spun at 1,000 g for 10 min and the sedimented cells were processed for radioactivity measurements. The standard procedure for labeling DNA and proteins was done essentially as previously described.Citation21
Contrast-phase images of cell populations were acquired using a Nikon TMS microscope with 20 or 40 objective using a digital camera.
Electron microscopy
Cytocentrifugates were fixed in Karnovsky fixative (2% formaldehyde and 2.5% glutaraldehyde in 0.1M cacodylate buffer, pH 7.4), osmicated and embedded in epoxy resin. Sections approximately 70 nm thick were stained with lead acetate and uranyl acetate and observed in a JEM 1010 electron microscope (Jeol) at 80 kV.
A375 human melanoma cells: Spheres and in vitro clonal assay
Human melanoma cells A375 (ATCC) were grown in DMEM with fetal bovine serum (FBS) for adherent cultures or seeded at a concentration of 5 cells/µl in DMEM-F12 serum-free medium with N2 supplement, Insulin (20 µg/ml), EGF (10 ng/ml), FGF-2 (10 ng/ml) (all from Invitrogen) and 0.6% (33 mM) glucose in low adhesion plates for melanoma sphere cultures.Citation46 Melanoma spheres were dissociated, plated at 5 cells/µl and treated with PBS or 10 mM and 20 mM pyruvate for 7 d lowering EGF and FGF-2 concentration to 2.5 ng/ml. Melanoma spheres were dissociated and living cells were counted.
Flow cytometry
To determine cell cycle distribution, 5 × 105 cells were centrifugated for 6 min at 1,200 rpm. Once the pellet was discarded, cells were resuspended in 500 µl of propidium iodide (PI) solution (trisodium citrate 0.1% w/v, NP40 0.1% w/v, PI 50 µg/ml) (Merck 4 Biosciences, Calbiochem, # 537059), incubated for 30 min at 4°C in darkness and subjected to flow cytometry (BD FACS Canto). Analysis for cycle distribution was performed by Mod fit LT software.
To determine the cell viability 1 × 105 cells were centrifuged for 5 min at 1,000 rpm; once the pellet discarded, the cells were incubated in 1:100 PI solution (10 mg/ml in PBS) at r.t. for 5 min. Analysis for cell viability was performed by BD FACS Diva 6.1.3 software.
Additions
All the substances tested were purchased from Sigma Aldrich and added at time 0 at the concentration indicated in the figure legends.
Culture conditions
Incubation was performed at 38°C in a 95% air, 5% CO2 atmosphere in a humidified incubator, unless otherwise specified.
To obtain complete anaerobiosis, the incubation mixture was thoroughly flushed with nitrogen, and a stick of yellow phosphorus was put in the wells of the Warburg flasks. Complete air replacement was obtained by maintaining nitrogen flushing for 5 min before time 0. Alternatively, an hypoxic apparatus was used. Hypoxic conditions (0.1% O2) were achieved by culturing the cells in an anaerobic workstation incubator (Concept 400, Jouan) flushed with a gas mixture containing 0.1% O2, 5% CO2 and balanced N2 at 37°C in a humidified atmosphere. Oxygen tension in the medium was measured with a portable trace oxygen analyzer (Drager PACIII).
Immunofluorescence
Immunofluorescence for adherent cells and cells in suspension (cytocentrifuged at 800 rpm for 15 min) was done essentially as previously described in Biagiotti et al.Citation47 Ascites stem cells were cultured in a MEM+F12 medium containing 10% FCS. Each cell population was tested for the following list of antibodies: mouse anti-α-smooth muscle actin, 1:800; mouse anti-calponin, 1:3,000; mouse anti-vimentin, 1:3,000; mouse anti-microtubule-associated protein 2, 1:2,500; mouse anti-glial fibrillary acidic protein, 1:2,000; mouse anti-peripherin, 1:600 (Sigma Aldrich); mouse anti-nestin,1:800 (BD Transduction Laboratories); rabbit anti-p75, 1:200 (Alomone Labs); mouse anti-CD133, 1:11 (Miltenyi Biotec); goat anti-neural cell adhesion molecule-L1, 1:1,000; goat anti-S-100, 1:1,000; goat anti-Tyr-related protein 1, 1:250; rabbit anti-CD34, 1:1,000; rabbit anti-calretinin, 1:500; mouse anti-platelet/endothelial cell adhesion molecule-1, 1:500; goat anti-fibroblast activation protein α, 1:500; rabbit anti-CD68, 1:500; mouse anti-c-Myc, 1:1,000; rabbit anti-α-fetoprotein, 1:500; goat anti-albumin, 1:500 (Santa Cruz Biotechnology Inc.); rabbit anti-Nanog, 1:800; mouse anti-Klf4, 1:800 (Stemgent). After washing with PBST, the cell cultures were incubated with anti-mouse Cy3-conjugated (Chemicon), anti-rabbit fluorescein-conjugated (Chemicon) or anti-goat fluorescein-conjugated (Calbiochem) secondary antibody (1:800 dilution in PBST/3% BSA) for 1 h at room temperature.
Quantitative real-time PCR
Quantitative real-time PCR (qPCR) was performed at 60°C on a 7500 fast real-time PCR machine (Applied Biosystem) using Power SYBR green mix (Applied Biosystem) and values calculated by delta-Ct method. The quantitative real-time PCR amplifications were performed at 60°C using Power SYBR green mix (Applied Biosystem).
Primer sequences for human genes were the following (5′ to 3′): NANOG-F, ACCTTGGCTGCCGTCTCTGG; NANOG-R, AGCAAAGCCTCCCAATCCCAAACA; KLF4-F, GCAGCCACCTGGCGAGTCTG; KLF4-R, CCGCCAGCGGTTATTCGGGG; cMYC-F, TTCGGGTAGTGGAAAACCAG; cMYC-R, CAGCAGCTCGAATTTCTTCC.
Primers for human reference genes were (5′ to 3′): GAPDH-F, GACGCTGGGGCTGGCATTG; GAPDH-R, GCTGGTGGTCCAGGGGTC; α-ACTIN-F, GAAAATCTGGCACCACACCT; α-ACTIN-R, TAGCACAGCCTGGATAGCAA.
Primer sequences for rat genes were the following (5′ to 3′) Nanog-F, AACGGCCTGACTCAGAAGGGCT; Nanog-R, GGAAGGTTTCCAGACGCGTTCATCA; Klf4-F,TTCTCCACGTTCGCGTCCGG; Klf4-R,TCTCGCCAACGGTTAGTCGGGG; cMyc-F,TCTGCGAGCCAGGACAGGACT; cMyc-R, GTCGTGACTGTCGGGTTTTCCACC.
Primers for rat reference genes were (5′ to 3′): Gapdh-F, GGGCTCTCTGCTCCTCCCTGTT; Gapdh-R, TACGGCCAAATCCGTTCACACCG; α-Actin-F, CGTCCACCCGCGAGTACAACC; α-Actin-R, ATCCATGGCGAACTGGTGGCG.
Statistical analysis
Significance levels were calculated with a Student’s t-test: * = p > 0.05; ** = p > 0. 02; *** = p < 0.005.
Male adult Wistar rats were used (Harlan). All animal manipulations were performed according to the European Community guidelines for animal care (DL 116/92, application of the European Communities Council Directive 86/609/EEC). All efforts were made to minimize animal sufferings and to use only the number of animals necessary to produce reliable scientific data.
Acknowledgments
The authors are indebted to Professor Paolo Romagnoli, Department of Anatomy and Histology of the University of Florence, for the electron microscopic figures. We are grateful Prof. P. Boyde, University of Cambridge, UK, for his help in the revision of the manuscript. This paper is dedicated to the memory of Prof. E. Ciaranfi and Prof. A. Fonnesu, who directly from Warburg’s and Krebs’s laboratories transmitted to us the intellectual commitment and the rigorous experimental skill required for studying cancer development. This work was supported by grants from Ente Cassa di Risparmio di Firenze, Istituto Toscano Tumori (ITT), Associazione Italiana per la Ricerca sul Cancro (AIRC), Regione Toscana and Ministero della Salute.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
References
- Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science 2009; 324:1670 - 3; http://dx.doi.org/10.1126/science.1171837; PMID: 19556499
- Klein CA. The systemic progression of human cancer: a focus on the individual disseminated cancer cell--the unit of selection. Adv Cancer Res 2003; 89:35 - 67; http://dx.doi.org/10.1016/S0065-230X(03)01002-9; PMID: 14587870
- Foulds L. The natural history of cancer. J Chronic Dis 1958; 8:2 - 37; http://dx.doi.org/10.1016/0021-9681(58)90039-0; PMID: 13563591
- Klein G. Fould’s dangerous idea revisited: the multistep development of tumors 40 years later. Adv Cancer Res 1998; 72:25 - 56; PMID: 9338073
- Loeb LA. Cancer cells exhibit a mutator phenotype. Adv Cancer Res 1998; 72:25 - 56; http://dx.doi.org/10.1016/S0065-230X(08)60699-5; PMID: 9338073
- Olivotto M, Dello Sbarba P. Environmental restrictions within tumor ecosystems select for a convergent, hypoxia-resistant phenotype of cancer stem cells. Cell Cycle 2008; 7:176 - 87; http://dx.doi.org/10.4161/cc.7.2.5315; PMID: 18256528
- Warburg O. On respiratory impairment in cancer cells. Science 1956; 124:269 - 70; PMID: 13351639
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008; 452:230 - 3; http://dx.doi.org/10.1038/nature06734; PMID: 18337823
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029 - 33; http://dx.doi.org/10.1126/science.1160809; PMID: 19460998
- St John JC, Ramalho-Santos J, Gray HL, Petrosko P, Rawe VY, Navara CS, et al. The expression of mitochondrial DNA transcription factors during early cardiomyocyte in vitro differentiation from human embryonic stem cells. Cloning Stem Cells 2005; 7:141 - 53; http://dx.doi.org/10.1089/clo.2005.7.141; PMID: 16176124
- Varum S, Momcilović O, Castro C, Ben-Yehudah A, Ramalho-Santos J, Navara CS. Enhancement of human embryonic stem cell pluripotency through inhibition of the mitochondrial respiratory chain. Stem Cell Res 2009; 3:142 - 56; http://dx.doi.org/10.1016/j.scr.2009.07.002; PMID: 19716358
- Ježek P, Plecitá-Hlavatá L, Smolková K, Rossignol R. Distinctions and similarities of cell bioenergetics and the role of mitochondria in hypoxia, cancer, and embryonic development. Int J Biochem Cell Biol 2010; 42:604 - 22; http://dx.doi.org/10.1016/j.biocel.2009.11.008; PMID: 19931409
- Rehman J. Empowering self-renewal and differentiation: the role of mitochondria in stem cells. J Mol Med (Berl) 2010; 88:981 - 6; http://dx.doi.org/10.1007/s00109-010-0678-2; PMID: 20809088
- Guido C, Whitaker-Menezes D, Lin Z, Pestell RG, Howell A, Zimmers TA, et al. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget 2012; 3:798 - 810; PMID: 22878233
- Sheffer M, Simon AJ, Jacob-Hirsch J, Rechavi G, Domany E, Givol D, et al. Genome-wide analysis discloses reversal of the hypoxia-induced changes of gene expression in colon cancer cells by zinc supplementation. Oncotarget 2011; 2:1191 - 202; PMID: 22202117
- Miyoshi N, Ishii H, Nagai K, Hoshino H, Mimori K, Tanaka F, et al. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc Natl Acad Sci USA 2010; 107:40 - 5; http://dx.doi.org/10.1073/pnas.0912407107; PMID: 20018687
- Hochedlinger K, Jaenisch R. Nuclear reprogramming and pluripotency. Nature 2006; 441:1061 - 7; http://dx.doi.org/10.1038/nature04955; PMID: 16810240
- Hochedlinger K, Plath K. Epigenetic reprogramming and induced pluripotency. Development 2009; 136:509 - 23; http://dx.doi.org/10.1242/dev.020867; PMID: 19168672
- Welstead GG, Schorderet P, Boyer LA. The reprogramming language of pluripotency. Curr Opin Genet Dev 2008; 18:123 - 9; http://dx.doi.org/10.1016/j.gde.2008.01.013; PMID: 18356040
- Lipchina I, Studer L, Betel D. The expanding role of miR-302-367 in pluripotency and reprogramming. Cell Cycle 2012; 11:1517 - 23; http://dx.doi.org/10.4161/cc.19846; PMID: 22436490
- Olivotto M, Caldini R, Chevanne M, Cipolleschi MG. The respiration-linked limiting step of tumor cell transition from the non-cycling to the cycling state: its inhibition by oxidizable substrates and its relationships to purine metabolism. J Cell Physiol 1983; 116:149 - 58; http://dx.doi.org/10.1002/jcp.1041160205; PMID: 6863398
- Sani BP, Mott DM, Jasty V, Sorof S. Properties of the principal liver target protein of a hepatic carcinogen. Cancer Res 1974; 34:2476 - 81; PMID: 4213321
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131:861 - 72; http://dx.doi.org/10.1016/j.cell.2007.11.019; PMID: 18035408
- Spike BT, Wahl GM. p53, stem cells and reprogramming: tumor suppression beyond guarding the genome. Genes Cancer 2011; 2:404 - 19; http://dx.doi.org/10.1177/1947601911410224; PMID: 21779509
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126:663 - 76; http://dx.doi.org/10.1016/j.cell.2006.07.024; PMID: 16904174
- Ramos-Mejia V, Fraga MF, Menendez P. iPSCs from cancer cells: challenges and opportunities. Trends Mol Med 2012; 18:245 - 7; http://dx.doi.org/10.1016/j.molmed.2012.04.001; PMID: 22521522
- Del Monte U. Changes in oxygen tension in Yoshida ascites hepatoma during growth. Proc Soc Exp Biol Med 1967; 125:853 - 6; PMID: 15938283
- Maden BEH. Tetrahydrofolate and tetrahydromethanopterin compared: functionally distinct carriers in C1 metabolism. Biochem J 2000; 350:609 - 29; http://dx.doi.org/10.1042/0264-6021:3500609; PMID: 10970772
- Marzi I, D’Amico M, Biagiotti T, Giunti S, Carbone MV, Fredducci D, et al. Purging of the neuroblastoma stem cell compartment and tumor regression on exposure to hypoxia or cytotoxic treatment. Cancer Res 2007; 67:2402 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-06-3208; PMID: 17363556
- Yoshida Y, Takahashi K, Okita K, Ichisaka T, Yamanaka S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 2009; 5:237 - 41; http://dx.doi.org/10.1016/j.stem.2009.08.001; PMID: 19716359
- Jiang J, Chan YS, Loh YH, Cai J, Tong GQ, Lim CA, et al. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol 2008; 10:353 - 60; http://dx.doi.org/10.1038/ncb1698; PMID: 18264089
- McConnell BB, Yang VW. Mammalian Krüppel-like factors in health and diseases. Physiol Rev 2010; 90:1337 - 81; http://dx.doi.org/10.1152/physrev.00058.2009; PMID: 20959618
- Cipolleschi MG, Dello Sbarba P, Olivotto M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood 1993; 82:2031 - 7; PMID: 8104535
- Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010; 7:380 - 90; http://dx.doi.org/10.1016/j.stem.2010.07.011; PMID: 20804973
- Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol 2006; 16:253 - 64; http://dx.doi.org/10.1016/j.semcancer.2006.07.014; PMID: 16904903
- Johnson DG, Walker CL. Cyclins and cell cycle checkpoints. Annu Rev Pharmacol Toxicol 1999; 39:295 - 312; http://dx.doi.org/10.1146/annurev.pharmtox.39.1.295; PMID: 10331086
- Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol 2005; 6:635 - 45; http://dx.doi.org/10.1038/nrm1703; PMID: 16064138
- Hastak K, Paul RK, Agarwal MK, Thakur VS, Amin AR, Agrawal S, et al. DNA synthesis from unbalanced nucleotide pools causes limited DNA damage that triggers ATR-CHK1-dependent p53 activation. Proc Natl Acad Sci USA 2008; 105:6314 - 9; http://dx.doi.org/10.1073/pnas.0802080105; PMID: 18434539
- Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009; 460:1132 - 5; http://dx.doi.org/10.1038/nature08235; PMID: 19668191
- Lin C-P, Choi YJ, Hicks GG, He L. The emerging functions of the p53-miRNA network in stem cell biology. Cell Cycle 2012; 11:2063 - 72; http://dx.doi.org/10.4161/cc.20207; PMID: 22580472
- Vicente-Dueñas C, González-Herrero I, García Cenador MB, García Criado FJ, Sánchez-García I. Loss of p53 exacerbates multiple myeloma phenotype by facilitating the reprogramming of hematopoietic stem/progenitor cells to malignant plasma cells by MafB. Cell Cycle 2012; 11:3896 - 900; http://dx.doi.org/10.4161/cc.22186; PMID: 22983007
- Vazquez-Martin A, Vellon L, Quirós PM, Cufí S, Ruiz de Galarreta E, Oliveras-Ferraros C, et al. Activation of AMP-activated protein kinase (AMPK) provides a metabolic barrier to reprogramming somatic cells into stem cells. Cell Cycle 2012; 11:974 - 89; http://dx.doi.org/10.4161/cc.11.5.19450; PMID: 22333578
- Ombrato L, Lluis F, Cosma MP. Regulation of self-renewal and reprogramming by TCF factors. Cell Cycle 2012; 11:39 - 47; http://dx.doi.org/10.4161/cc.11.1.18759; PMID: 22186782
- Loeb LA, Cheng KC. Errors in DNA synthesis: a source of spontaneous mutations. Mutat Res 1990; 238:297 - 304; http://dx.doi.org/10.1016/0165-1110(90)90021-3; PMID: 2188126
- Wang X-M, Yang L-Y, Guo L, Fan C, Wu F. p53-induced RING-H2 protein, a novel marker for poor survival in hepatocellular carcinoma after hepatic resection. Cancer 2009; 115:4554 - 63; http://dx.doi.org/10.1002/cncr.24494; PMID: 19551892
- Santini R, Vinci MC, Pandolfi S, Penachioni JY, Montagnani V, Olivito B, et al. Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells 2012; 30:1808 - 18; http://dx.doi.org/10.1002/stem.1160; PMID: 22730244
- Biagiotti T, D’Amico M, Marzi I, Di Gennaro P, Arcangeli A, Wanke E, et al. Cell renewing in neuroblastoma: electrophysiological and immunocytochemical characterization of stem cells and derivatives. Stem Cells 2006; 24:443 - 53; http://dx.doi.org/10.1634/stemcells.2004-0264; PMID: 16100002