Abstract
High rates of inherent primary resistance to the humanized monoclonal antibody trastuzumab (Herceptin) are frequent among HER2 gene-amplified breast carcinomas in both metastatic and adjuvant settings. The clinical efficacy of trastuzumab is highly correlated with its ability to specifically and efficiently target HER2-driven populations of breast cancer stem cells (CSCs). Intriguingly, many of the possible mechanisms by which cancer cells escape trastuzumab involve many of the same biomarkers that have been implicated in the biology of CS-like tumor-initiating cells. In the traditional, one-way hierarchy of CSCs in which all cancer cells descend from special self-renewing CSCs, HER2-positive CSCs can occur solely by self-renewal. Therefore, by targeting CSC self-renewal and resistance, trastuzumab is expected to induce tumor shrinkage and further reduce breast cancer recurrence rates when used alongside traditional therapies. In a new, alternate model, more differentiated non-stem cancer cells can revert to trastuzumab-refractory, CS-like cells via the activation of intrinsic or microenvironmental paths-to-stemness, such as the epithelial-to-mesenchymal transition (EMT). Alternatively, stochastic transitions of trastuzumab-responsive CSCs might also give rise to non-CSC cellular states that lack major attributes of CSCs and, therefore, can remain “hidden” from trastuzumab activity. Here, we hypothesize that a better understanding of the CSC/non-CSC social structure within HER2-overexpressing breast carcinomas is critical for trastuzumab-based treatment decisions in the clinic. First, we decipher the biological significance of CSC features and the EMT on the molecular effects and efficacy of trastuzumab in HER2-positive breast cancer cells. Second, we reinterpret the genetic heterogeneity that differentiates trastuzumab-responders from non-responders in terms of CSC cellular states. Finally, we propose that novel predictive approaches aimed at better forecasting early tumor responses to trastuzumab should identify biological determinants that causally underlie the intrinsic flexibility of HER2-positive CSCs to “enter” into or “exit” from trastuzumab-sensitive states. An accurate integration of CSC cellular states and EMT-related biomarkers with the currently available breast cancer molecular taxonomy may significantly improve our ability to make a priori decisions about whether patients belonging to HER2 subtypes differentially enriched with a “mesenchymal transition signature” (e.g., luminal/HER2 vs. basal/HER2) would distinctly benefit from trastuzumab-based therapy ab initio.
Because they cannot benefit from hormonal therapy with HER2-targeted drugs,Citation1 there is no established systemic therapy other than chemotherapy for patients affected by the basal-like subtype of breast cancer, which is the most impacted breast cancer disease among the intrinsic spectrum of tumor entities originally identified by applying breast cancer molecular taxonomy in 2000 (i.e., luminal A, luminal B, HER2-enriched, basal-like and a normal breast-like group).Citation2 Conversely, the HER2-enriched breast cancer subtype, which originally carried a poor prognosis in landmark studies of breast cancer genomics,Citation3,Citation4 has largely benefited from the era of HER2-targeting therapies. In 2012, an institutional-based review confirmed that women with HER2-positive breast cancer disease who received the anti-HER2 monoclonal antibody trastuzumab (Herceptin™, Roche Biomedical Laboratories) had an improved prognosis compared with women with HER2-negative breast cancer disease.Citation5 Indeed, there is no doubt that pivotal trials showing the clinical benefit of trastuzumab in combination with chemotherapy have led to a new standard of care for women with metastatic and early-stage HER2-positive breast cancer.Citation6-Citation9 Based upon the high-risk assessment of recurrence among HER2-positive patients with small and node-negative breast carcinomas (pT1a,b,N0,M0), it has been suggested that prospective studies and randomized clinical trials should clarify whether women with node-negative, sub-centimeter, HER2-positive tumors could also benefit from trastuzumab-based adjuvant systemic therapy.Citation10,Citation11 However, it should be noted that while the addition of trastuzumab to adjuvant chemotherapy can impressively reduce the recurrence rate by almost 50% and improve survival in patients with early-stage HER2-positive breast cancer, approximately 15% of women diagnosed with early HER2-positive disease show de novo (i.e., primary or intrinsic) resistance to trastuzumab and relapse in spite of treatment with trastuzumab-based therapies.Citation12-Citation18 In the same regard, the overall response rate of women diagnosed with HER2-positive metastatic breast cancer and treated with trastuzumab as a single, first-line agent is only 26%. Thus, > 70% of HER2-overexpressing metastatic breast carcinomas show resistance to trastuzumab ab initio.
Breast cancer heterogeneity, in which biologically aggressive sub-clonal cell populations occur spontaneously within an individual tumor, has been suggested to underlie both the metastatic recurrence capacity and the variable response of HER2-positive tumors to selective HER2-targeting inhibitors, including trastuzumab.Citation19,Citation20 Unfortunately, the precise genomic and biological features of HER2-positive breast cancer with intrinsic aggressive behavior and de novo non-sensitivity to trastuzumab remain largely obscure. Estrogen receptor (ER)-negative breast carcinomas are of either the basal-like or HER2-enriched subtype. ER-positive, clinically defined HER2-positive tumors are generally enriched with the luminal gene cluster, and rather than belonging to the HER2-enriched subtype, these tumors fall into the luminal (i.e., luminal B) subtype of breast carcinomas. Therefore, at least two types of HER2 gene-amplified breast carcinomas exist (ER-positive and ER-negative). Because several risk factors for developing basal-like breast cancer have been identified (e.g., BRCA1 mutations carriers and young African-American women), and there are no known specific risk factors (including no apparent interaction with race or age) for the HER2-enriched subtype,Citation1,Citation21,Citation22 it appears reasonable to etiologically consider basal-like and HER2-enriched breast carcinomas as two different breast cancer diseases.Citation23 Moreover, the basal-like breast cancer subtype displays an obvious and strong common biology, whereas the HER2-enriched subtype solely describes the occurrence of a high but imperfect correlation with HER2 gene-amplification status. That is, not all tumors of this molecular subtype exhibit HER2 gene amplification/HER2 protein overexpression, and some, but not all, clinically defined HER2-positive breast tumors fall into the HER2-enriched subtype. Although we should acknowledge that ~25% of basal-like breast carcinomas are not equivalent in clinical terms to immunohistochemically defined (ER, PR and HER2) triple-negative breast cancer,Citation1 it should be noted that up to 45% of basal-like breast carcinomas, which by rigorous definition depends on the assessment of the mRNA expression status of approximately 500 genes, are not triple-negative,Citation24-Citation26 because they do express ER, PR and/or HER2. In this complex scenario, we hypothesize that the occurrence of HER2 gene amplification/HER2 protein overexpression within basal-like gene signatures might naturally delineate a novel breast cancer sub-entity that is inherently resistant to trastuzumab.Citation27,Citation28
Gene Signatures in HER2-Positive Breast Cancer: The Association of a Poor Prognosis with HER2-Negative Basal-Like Tumors
Although an ever-growing, deeper understanding of the signal network integration of breast cancer cells is rapidly providing promise for genotype/phenotype-based “personalized breast cancer medicine,”Citation29,Citation30 accurate tools to improve our current understanding of the molecular therapeutic behavior of individual HER2-positive breast carcinomas are lacking.Citation31,Citation32 When employing currently available genomic signatures, patients overexpressing the HER2 oncogene are classified as high-risk individuals that should benefit, in terms of improved survival, from treatment with HER2-targeted agents, including trastuzumab.Citation33-Citation35 However, currently available gene expression signatures cannot identify HER2-positive subgroups of good-trastuzumab responders, who will not experience relapse ab initio, or bad-trastuzumab responders, who will not respond to trastuzumab-based therapies ab initio. By correlating the genetic heterogeneity of HER2-positive tumors with clinical outcome by molecular profiling, Staaf and colleaguesCitation36 provided strong support for the notion that variability of clinical outcome and lack of uniformity in the beneficial effect of trastuzumab in HER2-positive breast cancer patients may be due to the inherent heterogeneity of HER2-positive breast tumors. The identification of a 158-gene prognostic predictor, termed the HER2-derived prognostic predictor (HDPP), significantly improved the stratification of good and poor prognosis for both overall survival and distant metastasis-free survival. This improved stratification was realized not only in their HER2 tumor set, but also in multiple breast cancer data sets, including the Netherlands Cancer Institute data set, in which the HDPP gene signature demonstrated superior prognostic stratification of HER2-positive tumors compared with the MammaprintCitation33 (Agendia) and Oncotype DXCitation34 (Genomic Health) systems.
Notably, the HDPP signature described by Staaf and colleaguesCitation36 performed well not only in the HER2-enriched molecular subtype, but also in the basal-like subtype, and was associated with survival in triple-negative breast carcinomas. By contrast, HDPP showed no prognostic value in luminal (A and B) or normal-like breast cancer subtypes. In agreement with earlier studies, these findings may merely suggest that different gene expression modules or signatures must be applied to predict the outcome of different subgroups of breast tumors. Because the HER2-positive and basal-like breast cancer subtypes have been shown to be associated with the worst prognosis, and the luminal A subtype has been associated with the best prognosis, these findings may only reveal that the ability of the HDPP signature to predict survival is largely attributable to the differential and selective expression of HDPP signature in well-recognized subsets of aggressive breast carcinomas. Furthermore, because proliferation is an important component of most prognostic gene signatures and has been reported to be associated with outcome solely in ER-positive/HER2-negative breast cancer,Citation37-Citation39 the ability of the HDPP signature to add independent prognostic information for ER-negative, lymph node-negative or high-grade clinically aggressive tumors strongly suggests that the recognition of a biological phenomenon beyond HER2 gene amplification and high proliferation status may be crucial to improve the prediction of outcome and the selection of patients for molecularly targeted therapies. The notion that HER2-positive breast tumors do not form a homogenous biological entity, but rather a clinical subset of breast cancer patients, is clearly supported by recent studies addressing the biological homogeneity of HER2-positive tumors at the genomic level.Citation40,Citation41 HER2-positive breast carcinomas are known to display a great diversity of anomalies, which affect most of the genome. Moreover, amplification of the HER2 locus at 17q yields two different patterns that may be related to different breast cancer subsets. Narrow amplicons centered around the HER2 gene that comprise six to 10 genes have been identified in more aggressive ER-negative tumors, whereas large amplicons that start centromeric to HER2 and extend telomeric to the TOP2A gene correlate with ER-positive tumors and better survival.Citation40,Citation41
The above-mentioned findings confirm and further extend recent studies that failed to delineate a HER2-positive breast cancer subgroup exclusively composed of HER2-amplified breast tumors; rather, HER2-positive tumors appear to cluster in other molecular subgroups. In other words, although the majority of HER2-positive breast carcinomas have HER2-enriched gene expression profiles, other intrinsic breast cancer subtypes have HER2-positive tumors within them, suggesting that the HER2-positive clinical category is biologically heterogeneous. In this extremely complex scenario, at least two challenging questions remain to be answered in the context of the inherent breast cancer response to HER2-targeted therapies. The first question refers to the possibility that patients who are clinically HER2-negative within the HER2-enriched breast cancer subtype could benefit from anti-HER2 therapies, such as trastuzumab. Pre-clinical HER2-negative breast cancer models engineered to exhibit a constitutive activation of HER2 by overexpressing the HER3 ligand heregulin preliminarily defined a specific HER2-negative breast cancer patient population for which trastuzumab plus chemotherapy may be extremely efficient.Citation42,Citation43 Accordingly, de Alava and colleaguesCitation44 confirmed that the spectrum of breast cancer patients who may benefit from trastuzumab-based therapies should be widened to include patients with metastatic breast tumors lacking HER2 gene amplification but expressing high levels of EGFR ligands, which leads to HER2 transactivation. In this regard, women with HER2-negative breast cancers who do not meet the established criteria for HER2-positive disease on central review have been found to receive as much clinical benefit from adjuvant trastuzumab as women with HER2 gene-amplified breast cancer.Citation45 The second question refers to the possibility that patients who are clinically HER2-positive within the basal-like breast cancer subtype might delineate a naturally occurring subgroup of breast cancer patients with de novo (primary) resistance to trastuzumab-based therapies. In this regard, it is tempting to suggest that the use of signatures, such as the above-mentioned HDPP, should be part of the decision-making process for the therapeutic management of HER2-positive breast cancer disease. However, as is the case with most breast cancer gene signatures, the performance of the HDPP signature is based on retrospective data from patients who did not receive trastuzumab therapy.Citation46 Therefore, the prognostic value of HDPP may not necessarily be predictive of responsiveness to trastuzumab-based therapies. Indeed, a subgroup with favorable prognosis, as identified by HDPP signature, was demonstrated to display high activation of PI3K, a well-recognized determinant of trastuzumab resistance.Citation47 Interestingly, when applied to a small HER2-positive data set of patients preoperatively treated with trastuzumab and vinorelbine, the HDPP signature correlated with the occurrence of trastuzumab resistance. Although the sample size was insufficient to form any conclusion on the value of HDPP for predicting the response to targeted treatment modalities (e.g., trastuzumab) in HER2-positive patients, it should be noted that when formerly analyzing this data set, Harris and colleaguesCitation48 auspiciously suggested that “HER2-overexpressing tumors with a basal-like phenotype” were more likely to be intrinsically resistant to pre-operative trastuzumab.
Breast Cancer Stem Cells and HER2-Targeted Therapies: From Biology to Therapeutics
Integrating breast cancer molecular taxonomy and mammary-stem cell biology could be the best approach to understanding de novo resistance to HER2-targeted therapies and the increased intrinsic aggressiveness of specific subgroups of HER2-positive breast cancers.Citation49 The HER2 gene appears to play a crucial role in mammary carcinogenesis by actively driving the pool of replenishing cancer-initiating cells or cancer-propagating cells, popularly known as cancer stem cells (CSCs), which have the unique ability to both promote self-renewal and give rise to phenotypically heterogeneous breast cancer progeny.Citation50-Citation53 HER2 overexpression was originally described to actively enhance the cancer-initiating component that drives tumorigenesis, invasion and metastasis by significantly increasing the fraction of breast cancer cells positive for ALDH1 (aldehyde dehydrogenase),Citation54 a marker of normal and cancerous human mammary epithelial cells with stem/progenitor properties.Citation55 Accordingly, a landmark study by Li and colleaguesCitation56 demonstrated that in contrast to chemotherapy treatment, the epidermal growth factor receptor (EGFR)/HER2 dual tyrosine kinase inhibitor (TKI) lapatinib (Tykerb™) decreases the subpopulation of chemotherapy-resistant cancer-initiating cells, which was measured by calculating the percentage of cells with the CD44+/CD24-/low stem/progenitor immunophenotype that exhibited the ability to form mammospheres in vivo as an indication of self-renewal. Magnifico and colleaguesCitation57 have further expanded this notion by demonstrating that HER2-positive breast CSCs, which appear to express the highest levels of HER2 protein regardless of HER2 gene amplification status, are exquisitely sensitive to trastuzumab. This crucial finding (i.e., that the HER2 oncoprotein may be selectively upregulated in breast CSCs but not in bulk breast cancer cell populations) might explain the results of the above-mentioned retrospective studies, which demonstrated that in breast tumors currently classified as HER2-negative [i.e., immunohistochemistry (IHC) 1/2+ or fluorescence in situ hybridization (FISH) negative], HER2-targeting agents may be effective when administered in adjuvant but not advanced settings.Citation45,Citation58
In breast tumors clinically classified as bona fide HER2-positive (i.e., IHC 3+ or FISH positive), HER2 is overexpressed in both breast cancer stem cells and bulk tumor populations. Therefore, these tumors are expected to be sensitive to HER2-targeting agents in both advanced and adjuvant settings to promote tumor regression and reduce recurrence, respectively.Citation58 However, while accumulating evidence indicates that molecularly targeted agents directed against HER2, such as trastuzumab, could effectively target cancer-initiating cells in HER2-positive breast carcinomas, counterintuitively, recent observations have also suggested that clinical resistance to trastuzumab and other HER2-targeted therapies may be driven by breast CSCs. We already recognize that CS-like cells isolated from breast carcinomas intrinsically possess high resistance to hormonal therapy, chemotherapy and radiotherapy.Citation56,Citation59-Citation61 Indeed, CS-like cells are highly prevalent at the invasive edge of breast carcinomas following treatment with conventional therapies.Citation56,Citation62,Citation63 Correspondingly, it could be argued that overexpression of CSC-related biomarkers would promote resistance to trastuzumab by generally enhancing self-renewal/proliferation in progenitor cells and/or inducing stem cell properties, including the epithelial-to-mesenchymal transition (EMT) genetic program. Accordingly, trastuzumab treatment reduces the stem-cell population in trastuzumab-sensitive breast cancer cell lines but not in trastuzumab-unresponsive breast cancer cell lines.Citation49,Citation54 Moreover, a variety of possible mechanisms of escape from trastuzumab, such as the overexpression of the stem cell marker CD44, which leads to the loss or blockade of the trastuzumab-binding site at the HER2 extracellular domain, and the upregulation of stem cell markers such as CXCR4, β1 integrin or Notch-1, which lead to the activation of alternative pathways circumventing HER2 signaling, appear to involve many of the same biomarkers that have been implicated in the biology of CS-like cells.Citation54
The suggestion that HER2 gene-amplified breast cancer stem-like cell populations are unresponsive to trastuzumab ab initio is the ultimate cause underlying the inherent resistance of HER2-positive breast carcinomas to trastuzumab largely lacks the observational and experimental evidence we normally expect of standard predictive factors for breast cancer treatment selection. Indeed, a major challenge now is to define gene signatures and/or gene products and specific breast CSC-like and/or EMT attributes that may specifically inform about the intrinsic sensitivity of HER2-positive breast carcinomas to HER2-targeted therapies. Apparent approaches to provide biomarkers of trastuzumab-refractory breast CSCs include microarray analyses-based detection of stemness-focused gene signatures, which are significantly enriched in ab initio trastuzumab-refractory HER2-positive breast carcinomas. We recently envisioned that the occurrence of SC-like features in trastuzumab-refractory HER2-positive breast carcinomas could be re-derived, not only from clinically well-defined patient cohorts, but also by identifying biological determinants of trastuzumab sensitivity in panels of cancer cell lines that are able to accurately capture the genetic heterogeneity of the HER2-enriched breast cancer subtype.
Stem Cell-Like Features are Overexpressed in Trastuzumab-Resistant HER2-Gene Amplified Breast Cancer Cells: A Genome-Wide Pathway-Focused Analysis
The cell line SKBR3 is a widely used preclinical model of HER2 gene-amplified breast cancer cells that is exquisitely sensitive to trastuzumab.Citation64 Another unique model of HER2-positive breast cancer disease that exhibits de novo cross-resistance to multiple HER2-targeting drugs is the JIMT1 cell line,Citation65 which is a ductal carcinoma pleural metastasis of a 62-y-old patient who did not respond to trastuzumab treatment, despite having a tumor with HER2 gene amplification. Taking advantage of the properties of these two cell lines, we recently performed a genome-wide pathway-focused analysis to evaluate whether the presence of tumor-initiating, breast CS-like genetic signatures might underlie de novo resistance to HER2-targeting agents in HER2 gene-amplified breast carcinomas (Fig. S1). Genome-wide analyses of 44K Agilent's whole human arrays were bioinformatically evaluated by Gene Set Enrichment Analysis (GSEA)-based screening of the KEGG pathway database. GSEA performs competitive analyses of predefined gene sets; these analyses are appropriate for examining relatively heterogeneous biological samples. Four enriched gene sets that could be used to functionally differentiate trastuzumab-responsive SKBR3 cells from trastuzumab-refractory JIMT1 cells were identified. Within the non-small cell lung cancer pathway, the JIMT1 transcriptome exhibited downregulation of genes such as ERBB2 (i.e., the trastuzumab target itself), PLCγ, PI3K and PKB (Akt) (Fig. S1, top). Within the cell cycle (Fig. S1, middle) and ribosome (Fig. S1, bottom) pathways, the trastuzumab-refractory JIMT1 transcriptome displayed co-downregulation of 15 ribosomal protein genes that have been described as haploinsufficient tumor suppressors,Citation66,Citation67 and of numerous genes encoding cyclin-dependent kinases (CDKs). Importantly, trastuzumab-resistant JIMT1 cells exhibited a significant repression of the CDK inhibitor p16INK4a, a condition common to stem cells and many tumor cells that can link sustained induction of EMT and the activation of DNA methylation of genes that are silenced in basal-like breast carcinomas.Citation68 p16INK4a downregulation has been observed in some basal-like breast cancer cell lines and cells that are CD44+CD24-/low, a phenotype associated with stem-like breast cancer cells that is more frequent in ER-negative/p16INK4a-negative breast cancer cell lines than in ER-negative/p16INK4-positive lines.Citation69 Given that p16INK4a depletion has been suggested to reduce the response of ER-negative breast cancer cells to chemotherapy by increasing the percentage of CD44+CD24-/low cells and enhancing the expression of embryonic stem-like genes (e.g., Nanog, Oct4 and Sox2), it is tempting to suggest that, by conferring CSC-like properties, downregulation of p16INK4 expression could also underlie the de novo resistance to trastuzumab in HER2 gene-amplified JIMT1 cells. Within the extracellular matrix(ECM)-receptor interaction pathway (), the JIMT1 transcriptome exhibited upregulation of the CD44 transcript, which is positively associated with stem cell-like characteristics,Citation70 and the expression of gene transcripts encoding structural elements of basal epithelial cells, including the α6 and β1 integrin subunits (ITGA6 and ITGB1)Citation71-Citation76 and the laminin γ2 subunit (LAMC2),Citation77 all of which represent putative stem cell biomarkers.
Figure 1. Trastuzumab-refractory JIMT1 cells are enriched with stem cell markers.
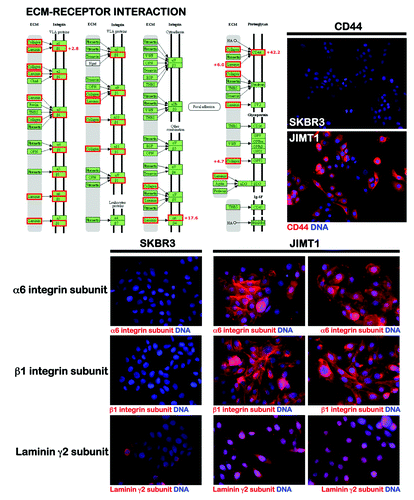
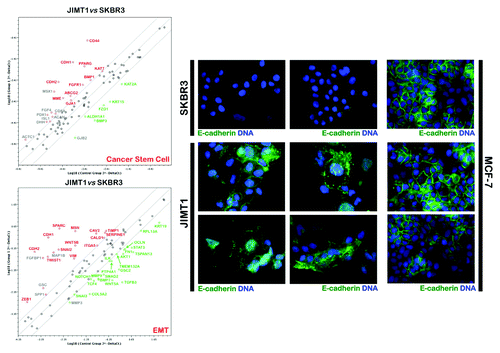
To confirm these findings, we profiled the expression of genes related to the identification, growth and differentiation of stem cells in trastuzumab-sensitive SKBR3 cells and in trastuzumab-refractory JIMT1 cells by quantitative real-time PCR (qRT-PCR) arrays (, left). These analyses not only confirmed the overexpression of the CD44 gene in trastuzumab-refractory JIMT1 cells, but also revealed significantly higher transcriptional activation of the following well-established stem cell-related genes: PPAR-gamma;Citation78,Citation79 the ABCG2 transporter/breast cancer-resistance protein (BCRP), which is a characteristic of stem cells termed “side population cells” that may contribute to the intrinsic resistance and longevity of normal cells and CSCsCitation80-Citation82 and can be identified by their ability to exclude the fluorescent dye Hoechst 33342; the FGFR1 component of the FGF signaling pathway;Citation83,Citation84 and the membrane endopeptidase (MME; CD10) neprilysin, a zinc-dependent metalloprotease that normally regulates the growth of the ductal tree during mammary gland development, because its ability to cleave signaling factors, together with β1 integrin-mediated contact with the basement membrane, is required to maintain the progenitor and stem cell pools in the mammary lineage.Citation85-Citation88 Perhaps more importantly, the two major breast cancer subtypes, based on their gene-expression profiles (i.e., luminal- and basal-type tumors), express markers corresponding to the major differentiation states of epithelial cells in the breast: luminal (EpCAM+) and basal/myoepithelial (CD10+). In this scenario, the fact that trastuzumab-refractory, HER2 gene-amplified JIMT1 cells are positive for the myoepithelial marker CD10, strongly suggests that HER2 gene amplification occurs in basal-like carcinomas that exhibit a certain degree of myoepithelial differentiation. In this regard, the expression of the epithelial marker E-cadherin (CDH1) in JIMT1 cells is consistent with a “mixed-lineage” nature of human basal-like breast cancers and, therefore, might reflect an aberrant/incomplete myoepithelial differentiation, but not necessarily the occurrence of global stem cell/progenitor features in mesenchymal-like phenotypes.Citation90 Notably, immunofluorescence studies have revealed that E-cadherin is associated with disorganized adhesive structures in trastuzumab-unresponsive JIMT1 cells (, right). We previously demonstrated that when grown under conventional culture conditions, JIMT1 cells exhibit a spiky-like appearance in which the cells are surrounded by numerous lamellipodial protrusions.Citation28 This morphology is relatively common in mesenchymal cells but not in cells expressing functional, E-cadherin-based cell-cell adhesion complexes, such as bona fide luminal epithelial-like cells (e.g., MCF-7; , right). In luminal MCF-7 cells, the localization of E-cadherin is restricted to the plasma membrane at areas of cell-cell contact. In “basoluminal” JIMT1 cells, E-cadherin was found across the entire surface of the plasma membrane and was notably distributed in various cytoplasmic locations (e.g., peri-nuclear areas). Indeed, E-cadherin staining was notably diffused at areas of cell-cell contact, and JIMT1 cells normally exhibited reduced and punctuated E-cadherin staining that was not restricted to areas of cell-cell contact but was also observed along cell edges without adjacent cells. These findings, together with the previously reported abundance of filopodia protrusions, strongly suggest that trastuzumab-refractory JIMT1 cells largely lack junctional organization and have disorganized adhesive structures containing E-cadherin. Although E-cadherin is usually assigned a tumor-suppressor role in epithelial cells, under some circumstances, E-cadherin is associated with increased tumorigenesis and tumor dispersion. Our data are consistent with the idea that deregulated E-cadherin signaling can be an important component of trastuzumab-refractory growth of HER2-positive breast carcinomas cells. Indeed, when JIMT1 cells were infected with a lentivirus expressing E-cadherin-targeted small hairpin RNAs, we failed to obtain surviving cell populations with a good depletion of the E-cadherin protein (data not shown) when the cells were exposed to antibiotic selection to produce a polyclonal population of JIMT1 cells with a stable knockdown of E-cadherin; this finding suggests a crucial role for E-cadherin in the biological properties of trastuzumab-unresponsive JIMT1 cells. Although our data on E-cadherin expression may suggest an inability of JIMT1 cells to induce a classic EMT cadherin-switching, it should be noted that the JIMT1 cells were also positive for N-cadherin (CDH2), a classic EMT marker. Therefore, E-cadherin expression appears to be an exception to the EMT rule, because many other aspects of the EMT are operational in trastuzumab-refractory JIMT1 cells (see below). Although forthcoming studies should definitely elucidate how deregulated E-cadherin function may facilitate primary unresponsiveness to trastuzumab in HER2-positive breast carcinomas, our findings provide a glimpse into the poorly recognized importance of E-cadherin in the biology of breast cancer cell populations enriched with SC-like features.
Figure 2. Left: Activation of EMT/CSC-related gene signatures in trastuzumab-refractory JIMT1 cells. Right: Aberrant E-cadherin localization in trastuzumab-refractory JIMT1 cells.
Basal/HER2-Positive Breast Carcinomas: Evidence for a Novel Stratification of Breast Cancer Taxonomy to Predict Primary Resistance to Trastuzumab
The data obtained from trastuzumab-refractory, HER2 gene-amplified JIMT1 cells suggests that when HER2 gene amplification, which generally occurs within differentiated luminal breast cancer phenotypes, occurs in a basal molecular background, it might result in a basal/HER2-positive subtype of breast carcinoma that naturally exhibits an inherent (i.e., primary) resistance to trastuzumab.Citation28 Mechanistically, an intrinsic tumor cell plasticity that is able to efficiently drive the emergence of a breast CSC-related CD44+CD24-/low mesenchymal phenotype might account for the de novo resistance to trastuzumab in this distinct subgroup of basal/HER2-positive breast carcinomas.Citation27,Citation89 Alternatively, EMT-related CD44+CD24-/low mesenchymal subpopulations within basal/HER2-positive breast carcinomas might reflect the intrinsic activation of an inherent mechanism of escape from more epithelial, trastuzumab-responsive CS-like cellular states. Celià-Terrasa and colleaguesCitation91 recently proposed that highly metastatic tumor-initiating cells are enriched for a strong epithelial gene program, whereas tumor cell subpopulations with stable mesenchymal traits are notably depleted in CSCs. In their study, the constitutive overexpression of EMT transcription factors in epithelial, CSC-enriched cancer cell populations fully suppressed their self-renewal and metastatic phenotypes upon the activation of a mesenchymal gene program; conversely, knockdown of EMT factors in the mesenchymal subpopulation caused an increase in CSC-like properties, along with an enhancement of epithelial features. Accordingly, Sarrio and colleaguesCitation90 recently demonstrated that epithelial and mesenchymal subpopulations within basal breast cell lines could exhibit distinct stem cell/progenitor properties. Intriguingly, stem cell/progenitor properties, such as regenerative potential, high aldehyde dehydrogenase 1 activity and the formation of three-dimensional acini-like structures, were found to predominantly reside within the epithelial subpopulation,Citation90 and the acquisition of global stem cell/progenitor features was lost when a mesenchymal-like phenotype was spontaneously generated through an EMT process in epithelial cells. In their study, well-recognized EMT inducers, such as SLUG/SNAIL2 and ZEB1, inhibited luminal differentiation, thus preventing the loss of epithelial progenitor and mesenchymal-like phenotypes within basal breast cell lines.
Taken together, these findings suggest that the occurrence of high levels of EMT-driving transcriptional regulators might allow basal/HER2-positive breast carcinoma cells to “enter” and “exit” trastuzumab-responsive CS-like epithelial states. To preliminarily test this hypothesis, we took advantage of earlier studies that aimed to summarize the molecular and cellular analyses of EMT features across breast cancer cell lines in the publically accessible “Neve breast cancer dataset” ().Citation92,Citation93 Neve and colleaguesCitation94 profiled 51 human breast cancer cell lines with the Affymetrix HG-U133A chip and integrated further comparative genome hybridization (CGH) and proteomic analyses. With some exceptions, cell lines that clustered to basal B (as distinct from basal A or luminal) in Neve’s study corresponded with the group designated as mesenchymal by Charafe-Jaufreet and colleagues,Citation95 who interrogated 31 breast cancer cell lines with Affymetrix U133 Plus 2.0 arrays, and found that the cells clustered into luminal, basal and mesenchymal subgroups. More recently, Prat and colleaguesCitation96,Citation97 analyzed a data set of 52 breast cancer cell lines, including three cell lines that were not included in Neve’s study,Citation94 using a 50-gene supervised risk predictor of breast cancer based on intrinsic subtypes (i.e., luminal A, luminal B, HER2-enriched and basal-like). The major luminal, basal A and basal B subgroups originally identified by Neve and colleaguesCitation94 were again evident, and most of the basal B/mesenchymal cell lines exhibited a gene expression pattern similar to that of the claudin-low tumor subtype [i.e., breast cancer cells with the lowest expression of genes involved in epithelial cell-cell adhesion (E-cadherin and claudins 3, 4 and 7), luminal differentiation (CD24 and EpCAM) and high CD44/CD24 and CD49f/EpCAM mRNA ratios].Citation96,Citation97
Figure 3. Association of the clinical subtypes of breast cancer cell lines and the primary response to trastuzumab based on the relative EMT/CSC features’ enrichment.
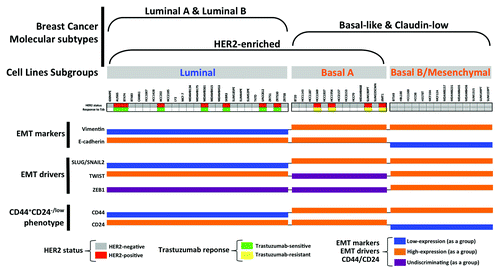
When we organized the 51 cell lines by the subclasses defined in the public Lawrence Berkeley Laboratory (LBL) Breast Cancer Collection,Citation94 the expression status of the most widely used EMT markers (i.e., E-cadherin and vimentin) and EMT transcriptional drivers (i.e., SLUG/SNAI2, TWIST and ZEB1), as well as the CD44/CD24 ratios and most of the HER2 gene-amplified breast carcinomas cell lines (i.e., AU565, BT474, HCC202, MDA-MB-361, SKBR3, UACC812 and ZR7530), were found to belong to the luminal subclass of breast cancer cell lines. This finding was expected because luminal differentiation is a property shared by the majority of HER2-overexpressing breast carcinomas and by the cell lines derived from them. Another expected finding was that the entire subset of mesenchymal-like basal B cell lines lacked HER2 gene amplification. Interestingly, a few HER2 gene-amplified breast cancer cell lines matched not only the luminal subgroup, but also the basal A/basal subgroup of cell lines (i.e., HCC1569, HCC1954 and SUM190T). Moreover, when HER2 gene-amplified breast cancer cell lines were classified as trastuzumab-sensitive and trastuzumab-refractory based on data from the literature, we observed that trastuzumab sensitivity ab initio was notably restricted to the luminal/HER2-positive cell lines, whereas all the basal/HER2-positive cell lines exhibited inherent, primary resistance to trastuzumab. Given the predominance of UT Southwestern-derived HCC breast cancer cells among the basal A subgroup, it could be argued that the correlation between the basal/HER2-positive phenotype and the occurrence of primary resistance to trastuzumab might reflect certain biases in the source of cell lines and/or in the culturing methods. However, the non-HCC-related JIMT1 cells, which were not originally included in the Neve data and have been shown to exhibit a strong correlation with the HER2-positive and basal-like subtypes and an inverse correlation with the luminal A subtype of breast carcinomas,Citation98-Citation100 also belonged to the basal/HER2-positive subgroup of trastuzumab-refractory breast cancer cell lines.
EMT-Like Features are Overexpressed in Trastuzumab-Resistant HER2 Gene-Amplified Breast Cancer Cells
The luminal epithelial marker E-cadherin, which is an important discriminator between basal A cells, which all express approximately median levels of E-cadherin and basal B/mesenchymal cells, which mostly exhibit reduced expression of E-cadherin, was not a good discriminator between trastuzumab-sensitive luminal/HER2-positive breast cancer cells and trastuzumab-resistant basal/HER2-positive cell lines. With the exception of SKBR3 cells, which have a homozygous deletion at 16q22.1 that includes the CDH1 (E-cadherin) gene,Citation100 all the HER2 gene-amplified breast carcinoma cells could be cataloged as E-cadherin-positive, regardless of their luminal or basal status. However, as has been noted for trastuzumab-resistant JIMT1 cells, the examination of E-cadherin expression alone does not inform about its aberrant localization in disorganized adhesive structures. Considering that many epithelial metastases retain E-cadherin expression (i.e., non-EMT cells can establish metastases or MET can occur at the metastatic site), it might be relevant to evaluate the potential relationship between deregulated E-cadherin function in HER2-positive breast carcinomas exhibiting a JIMT1-like “pseudo-epithelial” differentiation and the intrinsic aggressiveness of these tumors. Although vimentin (VIM) expression is not itself proof of EMT, it was absent in the entire subgroup of trastuzumab-responsive luminal/HER2-positive breast cancer cell lines, whereas it was notably expressed in all the trastuzumab-refractory breast cancer cell lines belonging to the basal/HER2 subgroup. Among the EMT transcriptional drivers, high relative expression of ZEB1 was limited to HER2-negative basal B/mesenchymal lines, and little differential expression was observed among the luminal/HER2-positive and basal/HER2-positive breast cancer cell lines. TWIST1 was also highly expressed in HER2-negative basal/mesenchymal cell lines and, unexpectedly, in a significant number of luminal cell lines. The mixed expression of TWIST1 among the basal A group of cell lines further precluded a discriminating role between trastuzumab responders and non-responders. Of note, the EMT transcription factor found to have the highest discriminator score between trastuzumab-sensitive luminal/HER2-positive and trastuzumab-resistant basal/HER2-positive breast cancer cell lines was SLUG/SNAIL2 (). Thus, all the trastuzumab-refractory basal/HER2-positive cells exhibited relatively high expression of SLUG/SNAIL2, whereas all the trastuzumab-sensitive luminal/HER2-positive cells exhibited relatively low expression of SLUG/SNAI2.
The CD44+CD24-/low phenotype, as determined by mRNA expression, has been repeatedly used to define the mesenchymal immunophenotype of human breast CSCsCitation51,Citation52 and was, as expected, found to be a hallmark of the HER2-negative basal B/mesenchymal subgroup of breast cancer cell lines. Although basal A breast cancer cell lines exhibited a certain degree of overexpression of mesenchymal markers, they notably lacked the CD44+CD24-/low mesenchymal phenotype and were positive for both markers (i.e., they exhibited a double-positive CD44+CD24+ phenotype). Breast cancer cell lines maintaining a luminal epithelial phenotype exhibited strong positivity for CD24, in agreement with numerous studies addressing the fact that CD24+ cells are associated with more differentiated tissues or tumors.Citation70 Given the positive association between CD44 and stem cell-like characteristics and the correlation between CD24 expression and differentiated epithelial features,Citation101 the expression status of CD44 can therefore be considered, in addition to the EMT transcriptional factor SLUG/SNAIL2, a good discriminator between trastuzumab-sensitive luminal/HER2-positive (SLUG-/CD44-) and trastuzumab-refractory basal/HER2-positive (SLUG+/CD44+) breast cancer cells. Indeed, our and other groups have previously described the importance of the CD44-hyalyronan pathways for the escape of breast cancer from trastuzumab-based therapies. Dhillon and colleaguesCitation102 observed that long-term exposure of luminal/HER2 BT474 cells to trastuzumab selected for a resistant subpopulation that was enriched for CD44.Citation102 Conversely, small interfering RNA inhibition of CD44 restored trastuzumab sensitivity. We have demonstrated that long-term exposure of luminal/HER2-positive SKBR3 cells to trastuzumab similarly selects for a resistant subpopulation of CD44+ cells.Citation103 Therefore, it is intriguing to note that both inherent (primary) and acquired (secondary) resistance to trastuzumab converge in CD44+CD24+ breast cancer cells with a basal-epithelial phenotype ().
To definitively confirm the hypothesis that trastuzumab-refractory, basal/HER2-positive tumor cells may be intrinsically enriched with EMT features, we profiled the expression of 84 key genes that either exhibit changes in expression during the EMT process or regulate those EMT-related expression changes by using commercially available qRT-PCR arrays with trastuzumab-responsive SKBR3 cells (i.e., luminal/HER2-positive) and trastuzumab-refractory JIMT1 cells (i.e., basal/HER2-positive).Citation104 When a 3-fold or greater difference in mRNA levels was used to determine significant changes in the expression of genes involved in the EMT genetic program, trastuzumab-refractory JIMT1 cells were found to exhibit significantly enhanced expression of developmental EMT drivers that have been repeatedly demonstrated to play pivotal roles in EMT-driven metastatic breast cancer progression, including SNAIL2 (SLUG), TWIST1 and, to a lesser extent, ZEB1 (, left). Moreover, the expression of specific, invasion-promoting EMT target genes, such as plasminogen activator inhibitor type-1 (PAI-1; SERPINE1)Citation105 and the mesenchymal marker VIM, was significantly increased in trastuzumab-refractory JIMT1 cells. In addition to an upregulation of the expression of α(5)-integrin (ITGA5), a fibroblast marker and the natural receptor for fibronectin, whose upregulation generally occurs in parallel with an upregulation of α-smooth muscle actin,Citation106 and caldesmon (CALD1), an actin-linked regulatory protein found in smooth muscle and non-muscle cells that has numerous functions in cell motility exerted via the reorganization of the actin cytoskeleton,Citation107 compared with trastuzumab-responsive SKRB3 cells, trastuzumab-refractory JIMT1 cells were also found to exhibit increased levels of TIMP1, an endogenous inhibitor of matrix metalloproteinases (MMPs) that also functions as a signaling molecule that induces the expression of developmental EMT transcription factors, such as SNAIL2/SLUG, TWIST1 and ZEB,Citation108 and of secreted protein acidic and rich in cysteine (SPARC), a matricellular protein involved in extracellular matrix remodeling and invasion that is directly involved in the acquisition of mesenchymal traits that favor metastatic dissemination.Citation109-Citation111
Suppression of EMT-Like Features Circumvents Primary Resistance to Trastuzumab in Basal/HER2-Positive Breast Cancer Cells
In the above-mentioned scenario, it might be tempting to suggest that primary resistance to trastuzumab can be explained in terms of trastuzumab-unresponsive, EMT-enriched CD44+ CSC-like populations. In other words, an intrinsic enrichment of EMT-like CSC cellular states might explain the intrinsic refractoriness of basal/HER2-positive breast cancer cells to trastuzumab. Although this suggestion might appear counterintuitive, given the widely accepted CD44+CD24-/low mesenchymal immunophenotype of the so-called breast CSCsCitation51,Citation52 and that trastuzumab-resistant basal/HER2-positive cells are positive for both marks (i.e., CD44+CD24+),Citation27 it should be noted that CD24 exhibits a dynamic regulation, as was recently demonstrated by Mayer and colleagues.Citation112 CD44+CD24+ basal cells can readily give rise to CD44+CD24-/low mesenchymal cells and vice versa. Moreover, when we recently explored the spontaneous evolution of the CD44+CD24-/low mesenchymal immunophenotype in trastuzumab-refractory basal/HER2-positive JIMT1 cells, we concluded that the dynamic expression of EMT-related markers was not limited to CD44/CD24, i.e., the number of cells bearing the CD44+CD24-/low mesenchymal immunophenotype switched over time from 10% in early passages to 80% in late passages. This phenotypic switch occurred because the trastuzumab-unresponsive, basal/HER2-positive JIMT1 cell cultures enriched with CD44+CD24-/low mesenchymal cells also exhibited a reduced expression of the HER2 protein and an increased secretion of pro-invasive/metastatic chemokines and metalloproteases.Citation27 Korkaya and colleaguesCitation113 have also reported that generation of trastuzumab resistance, either by knocking down PTEN expression or by long-term exposure to trastuzumab, is mediated in both cases by the expansion of CSC populations that display EMT-like phenotypes and oversecrete pro-invasive/metastatic chemokines (i.e., IL6). Taken together, these findings appear to confirm that either intrinsic or microenvironmental activation of paths to stemness, such as the EMT, directly regulate the responsiveness of CS-like cells to trastuzumab.
We have recently explored the causal relationship between EMT-driven tumor cell plasticity, which can drive the emergence of a CS-related CD44+CD24-/low mesenchymal phenotype, and the maintenance of de novo resistance to trastuzumab in basal/HER2-positive breast cancer cells.Citation114 Lentivirus-delivered small hairpin RNAs were employed to specifically and stably reduce the expression of EMT transcription factors in trastuzumab-refractory basal/HER2-positive cells. Then, cell proliferation assays and pre-clinical nude mice xenograft-based studies were performed to assess the contribution of specific EMT transcription factors to inherent trastuzumab resistance. The specific knockdown of SLUG/SNAIL2 suppressed the CD44+CD24-/low mesenchymal immunophenotype, and the isolation of these cells by magnetic-activated cell sorting confirmed that their intrinsic unresponsiveness to trastuzumab was mediated by transcriptional upregulation of the luminal epithelial marker CD24 in basal/HER2-positive cells, which, in turn, gained sensitivity to the growth-inhibitory effects of trastuzumab following SLUG/SNAIL2 gene depletion. Accordingly, depletion of the SLUG/SNAIL2-driven CD44+CD24-/low mesenchymal subpopulation reduced the tumorigenic potential of basal/HER2-positive JIMT1 cells and switched their trastuzumab-refractory phenotype to a sensitive phenotype when injected into nude mice. Therefore, aberrant expression of the EMT transcription factor SLUG/SNAIL2 appears to bias basal/HER2-positive epithelial cells toward a highly tumorigenic, CSC-like mesenchymal fate that is unresponsive to trastuzumab. In other words, the special proclivity of basal/HER2-positive breast cancer cells to undergo EMT reflects the intrinsic phenotypic plasticity of basal/HER2-positive breast cancer cells to rapidly acquire an invasive differentiation status of trastuzumab-refractory CS-like cells. Alternatively, the CSCs might not be considered distinct cancer cell entities (i.e., normal and tumor non-stem cells can spontaneously convert to a stem-like state and vice versa), because tumor cells can transiently acquire or lose SC-like properties as a consequence of stochastic state transitions, such as EMT regulation.Citation115-Citation119 Trastuzumab-responsive CSCs could potentially give rise to non-CSC cellular states that, by lacking the major attributes of CSCs, could remain “hidden” from trastuzumab activity. In the latter scenario, the predominant CSC cellular state in trastuzumab-responsive luminal/HER2-positive breast carcinomas would be different from that of basal/HER2-positive cell types, which may be characterized as an intermediate CD44+CD24+ stage of EMT in epithelial stem cellsCitation120 that, unlike luminal cell types, may be susceptible to SLUG/SNAIL2-driven acquisition of the bona fide CD44+CD24-/low mesenchymal phenotype. The CSC/non-CSC social structure within HER2-enriched breast carcinomas may be critical for trastuzumab-based treatment decisions in the clinic, because it may primarily dictate both the intrinsic sensitivity to trastuzumab in luminal/HER2-positive breast cancer cells and the intrinsic refractoriness to trastuzumab in basal/HER2+ breast cancer cells. Thus, in addition to the relationship of the trastuzumab-refractory CD44+CD24-/low mesenchymal phenotype of basal/HER2-positive breast carcinoma cells with the developmental origins of the intrinsic breast cancer subtypes (discussed below), it is relevant to note our recent discovery that EMT transcription factors, such as SLUG/SNAIL2, might induce an enhanced phenotypic plasticity that would allow basal/HER2-positive breast cancer cells to dynamically “enter” and “exit” trastuzumab-responsive SC-like states. It remains to be unambiguously defined whether SLUG/SNAIL2-driven CD44+CD24-/low subpopulations reflect an enrichment of trastuzumab-refractory CSC mesenchymal cells or, alternatively, an intrinsic mechanism of escape from more epithelial, trastuzumab-responsive CS-like cellular states.
Suppression of EMT-Like Features Might Counterintuitively Enhance the Occurrence of CS-Like Pluripotent States in Basal/HER2-Positive Breast Cancer Cells
It was recently demonstrated that activation of the EMT genetic program could efficiently suppress major attributes of human epithelial CSCs.Citation90,Citation91 In this regard, it should be noted that reprogramming of the normal cellular fate, such as activating a new pathological developmental program, in the case of cancer tissues, or generating induced pluripotent stem cells (iPSCs) from somatic cells, and cellular plasticity are tightly intertwined; thus, only some tumor or normal cells intrinsically possess the necessary plasticity for tumoral or somatic reprogramming to occur, respectively. In induced pluripotency, the reprogramming factors [the four Yamanaka transcription factors: Oct4, Sox2, Klf4 and c-Myc (OSKM)] are, in principle, all that is required to induce dedifferentiation. However, the extremely low efficiency and kinetics of the process clearly indicate that other molecular aspects of the target cell’s biology also play a crucial role. To generate a CSC cellular state, the capacity for self-renewal and asymmetric division must be re-acquired not only through oncogenic changes that destabilize the normal transcriptome of the target cell, but also through permissive/selective genetic, epigenetic and metabolic alterations that prevent cells from changing their original identity. Therefore, in both scenarios, the loss of a molecular feature that is essential for the maintenance of cell identity may act as a tumor- and iPSC-inducing alteration. One of the master regulators of cellular identity that must be eliminated to allow cells to become amenable to full reprogramming is the degree of EMT differentiation.Citation121 The generation of iPSCs from normal adult fibroblasts requires the proper activation and functioning of the mesenchymal-to-epithelial transition (MET) program, which is orchestrated by suppressing pro-EMT signals from the extracellular milieu (e.g., the EMT activator TGFβ) and activating an epithelial gene program intracellularly.Citation122-Citation127 In fact, the four canonical Yamanaka pluripotency factors transcriptionally impede the EMT process: Sox2/Oct4 suppress the EMT mediator SNAIL; c-Myc downregulates TGF-β1 and TGF-β receptor 2, and Klf4 induces epithelial genes, including E-cadherin.Citation122-Citation124 Moreover, during cellular reprogramming of fibroblasts to iPSCs by OSKM, fully reprogrammed cells can be exclusively observed within cell populations that are positive for the epithelial marker E-cadherin, but they cannot be obtained in the absence of E-cadherin. E-cadherin is upregulated during somatic cell reprogramming, and the established iPSCs possess E-cadherin-mediated cell-cell contacts and, thus, are morphologically indistinguishable from embryonic stem (ES) cells, in which E-cadherin is highly expressed and interference with E-cadherin causes differentiation. Accordingly, it has been shown that (1) the expression of E-cadherin alone can facilitate the reprogramming of adult fibroblasts and the acquisition of pluripotency, even in the absence of exogenous Oct4, and that (2) chemicals reported to upregulate E-cadherin considerably increase reprogramming efficiency, whereas knockdown of endogenous E-cadherin or the abrogation of E-cadherin-mediated cell-cell contact compromises the generation of iPSCs.Citation125-Citation127
We have demonstrated that the steady depletion of the CSC-related CD44+CD24-/low mesenchymal immunophenotype in response to the specific knockdown of EMT drivers, such as SLUG/SNAIL2, efficiently circumvents the primary resistance of basal/HER2-positive cells to trastuzumab.Citation114 However, genetic ablation of the EMT program failed to alter the ability of trastuzumab-sensitized basal/HER2-positive breast cancer cells to form bona fide mammospheres, which are non-adherent, spherical cell clusters enriched in mammary stem/progenitor cells. Our findings, along with the recently revealed requirement of epithelial cellular states to enable the activation of stemness programs in normal and cancer stem cells, strongly favor a functional heterogeneity in the breast CS-like compartment and suggest that, in addition to the widely recognized mesenchymal-like phenotypes of breast CSCs, tumor-initiating cells with an epithelia-like morphology and functionality should also be considered to gain a better understanding of the primary responses of HER2 gene-amplified breast cancer cell to trastuzumab. To preliminarily evaluate the hypothesis that, although trastuzumab effectively targets CSCs, the degree of trastuzumab responsiveness in a clinical setting may counterintuitively be driven by changes in the epithelial/mesenchymal cellular state of CSCs, we performed two sets of experiments. First, to directly explore the mechanisms by which differentiated cancer cells dynamically enter into the so-called cancer stem cell (CSC) cellular state, we investigated whether the expression status of the epithelial marker E-cadherin operates as one of the biological barriers to prevent the “differentiated cancer cell state” from being reversed to a “pluripotent SC-like cancer cell state” (even partially) (unpublished observations). Second, to establish a linkage between the EMT phenomenon and the reprogramming ability of basal/HER2-positive breast cancer cells, we determined whether the gain of epithelial features (e.g., E-cadherin expression) upon the silencing of EMT drivers (e.g., SLUG/SNAIL2, TWIST1) negatively or positively impacted the endogenous expression of pluripotent Yamanaka transcription factors.
The lentiviral introduction of the four canonical reprogramming transcription factors used in the Yamanaka cocktail to E-cadherin-positive MCF-7 breast cancer cells induced the appearance of rare colonies with characteristic embryonic SC-like morphologies (unpublished observations); the immature status of E-cadherin-positive MCF-7/iPSC clones was confirmed by their positivity for widely used pluripotency markers (e.g., Tra-1-60, Tra-1-81, Ssea-1, Ssea-4, Oct4, Sox2 and Nanog). Notably, when we introduced the OSKM transcription factors into E-cadherin knocked-down (KD) MCF-7 cells generated in our laboratory by short hairpin technology, we only observed changes in cell culture morphology that were somewhat analogous to “background colonies.” However, E-cadherin KD MCF-7 cells failed to generate bona fide iPSC clones, suggesting that epithelial breast cancer cells cannot be reprogrammed to pluripotent stem cell states (even partially) by silencing E-cadherin. Notably, HER2 expression, in the absence of HER2 gene amplification, was upregulated in E-cadherin-positive MCF-7/iPSC clones (unpublished observations). Indeed, HER2 expression was also upregulated when fibroblasts, a product of the EMT, were pushed to an E-cadherin-positive stem cell-like state via reprogramming with the OSKM transcription factors (unpublished observations). Furthermore, unpublished observations by Wichas’s group indicate that clinically HER2-negative primary luminal breast carcinomas contain a CS-like subpopulation of cells that express the HER2 protein on the invasive edge of the tumor.Citation128
To assess whether repression of mesenchymal features in basal/HER2-positive breast carcinomas cells significantly alters the expression of endogenously expressed reprogramming transcription factors, we monitored the expression of each individual reprogramming factor in JIMT1 parental cells and in JIMT1 cells engineered to ablate the expression of the EMT transcription factor SLUG/SNAIL2. When we performed real-time PCR analyses to assess Oct4, Sox2, Klf4 and c-Myc mRNA levels, we concluded that the gain of epithelial features (e.g., E-cadherin expression) in response to silencing of the CD44+CD24-/low-driver SLUG/SNAIL2 in basal/HER2-positive cells was accompanied by a significant upregulation of the pluripotency gene Klf4 (). These findings confirm and extend recent studies about the joint link between SLUG, Klf4 and EMT in tumor formation and metastatic progression.Citation129,Citation130 E-cadherin is a transcriptional target of Klf4, and, therefore, Klf4 is required to maintain E-cadherin expression and prevent the EMT in mammary epithelial cells. Indeed, forced expression of Klf4 in pure CD44+CD24-/low mesenchymal breast cancer cells is sufficient to restore E-cadherin expression and suppress migration and invasion.Citation131 The tumor- and metastasis-suppressive role of Klf4 in breast cancer has been recently confirmed by Yori and colleagues,Citation132 who reported that Klf4 expression is lost in a mouse model of HER2-positive breast cancer, even in the context of the MMTV-Neu transgene, thus suggesting a negative selection for the sustained expression of Klf4 in HER2-positive breast cancer cells. Moreover, transient overexpression of Klf4 in a HER2-expressing 4T1 orthotopic mammary cancer model attenuated not only primary tumor growth, but also micrometastases to the lungs and liver via a reduction in the expression of SNAIL.Citation132 However, Yu and colleaguesCitation133 have reported that Klf4 is highly expressed in CSC-enriched populations in mouse primary mammary tumor and breast cancer cell lines. Knockdown of Klf4 significantly decreased the proportion of stem/progenitor cells, suppressing cell migration and invasion, reducing colony formation in vitro and inhibiting tumorigenesis in immunocompromised mice. In this regard, although our findings appear to confirm that the tumor-suppressing activity of the EMT driver SLUG in basal/HER2-positive breast cancer cells operates via a reciprocal regulation of SLUG and the reprogramming factor Klf4, as previously suggested by other groups,Citation129,Citation130,Citation132 our findings further complicate the disputed nature (i.e., tumor suppressive vs. oncogenic) of the EMT/Klf4 duo.
Figure 4. Top: Specific regulation of the reprogramming factor Klf4 in response to the genetic ablation of the EMT factor SLUG/SNAIL2 in trastuzumab-refractory JIMT1 cells. Bottom: Restoration of E-cadherin along the basolateral membrane in response to the genetic ablation of the EMT transcription factors SLUG/SNAIL2 and TWIST1.
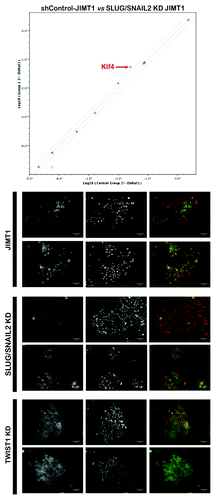
These results delineate the following scenarios: (1) breast cancer cells require the expression of the epithelial (basal/luminal) marker E-cadherin for the de novo generation of pluripotent, HER2-expressing CS-like cellular states, and (2) suppression of the trastuzumab-unresponsive CSC-related CD44+CD24-/low mesenchymal immunophenotype results in the endogenous upregulation of the pluripotent transcription factor Klf4. Therefore, by impeding the activation of the EMT program in basal/HER2-positive cells, the remaining HER2-positive cell population switches to a trastuzumab-responsive, more epithelial phenotype that is apparently enriched with the reprogramming factor Klf4. In response to knockdown of SLUG/SNAIL and the concomitant upregulation of Klf4, basal/HER2-positive JIMT1 cells gained a better junctional organization, and the adhesive structures containing E-cadherin became more structured (). Accordingly, the maturation of adherens junctions was concomitant with a cobblestone or cell-cell clustered morphology and the formation of cell contacts, which was not observed in basal/HER2-positive cells transduced with a control-shRNA. The morphological shift of trastuzumab-unresponsive basal/HER2-positive JIMT1 cells toward the epithelial end of the mesenchymal-epithelial spectrum occurred irrespective of the cell culture density; therefore, this shift was not affected when we limited the establishment of cell-cell connections. Thus, it is reasonable to suggest that outside-in signaling mediated by the upregulation of E-cadherin is not necessary for the morphological and functional change of basal/HER2-positive breast cancer cells. While it is well-accepted that E-cadherin expression is intimately connected to the degree of epitheliality of a cell with respect to its morphology and migratory/invasive abilities, it appears that restoring E-cadherin functionality is intimately connected with the ability of trastuzumab to induce growth-inhibitory and antitumor activity in HER2-positive breast carcinoma cells. This scenario suggests that enhancing the occurrence of trastuzumab-responsive, CS-like epithelial phenotypes could potentially circumvent the primary resistance to trastuzumab observed in basal/HER2-positive breast carcinomas. It is well-established that tumorigenesis and reprogramming are partially promoted by overlapping mechanisms, and research on reprogramming might help unravel the mechanisms of carcinogenesis and vice versa.Citation134-Citation141 However, the scarce knowledge that is currently available regarding the induction of a pluripotent phenotype via the activation of Yamanaka transcription factors in cancer cells indicates, somewhat unexpectedly, that reprogramming decreases the tumorigenicity of cancer cells by promoting their terminal differentiation, thus preventing the expression of aggressive phenotypes (i.e., a loss of mesenchymal lineage markers).Citation142-Citation144 Given that basal/HER2-positive breast cancer cells may express some reprogramming transcription factors at baseline, it might be plausible that by reversing a critical roadblock toward induced pluripotency (i.e., the EMT), e.g., preventing the EMT in epithelial cells cultured with serum can produce iPSCs with only two reprogramming factors,Citation122 basal/HER2-positive breast cancer cells may stably acquire a more appropriate CS-like phenotype in terms of tumor cell “hibernation” and trastuzumab “responsiveness.”
Molecular Forecasting of HER2-Positive Breast Carcinomas Based on CSC States and the EMT
The currently available molecular taxonomy of breast carcinomas appears to be robust, reproducible and relatively easy to apply in the clinic. Conversely, our ability to a priori identify which patients from the major breast cancer subclasses, luminal, basal and HER2 subtypes, are suitable for specific therapies is rather limited. We have recently envisioned that new predictive approaches aimed at better forecasting early responses of HER2-positive breast cancer patients to trastuzumab can be re-derived not only from clinically well-defined patient cohorts, but also by identifying biological determinants of trastuzumab sensitivity in cancer cell lines that capture the genetic heterogeneity of HER2-positive breast carcinomas in a manner that can help us to differentiate at least two biologically distinct subgroups of trastuzumab-responsive and trastuzumab-unresponsive tumors ab initio. Our current approach is based on the molecular detail that the clinical efficacy of trastuzumab is mostly related to its ability to specifically and efficiently target HER2-driven populations of CSCs and that, somewhat counterintuitively, a variety of recently discovered mechanisms of escape from trastuzumab appear to involve many of the same well-accepted biomarkers implicated in the biology of CS-like, tumor-initiating cells. How can we reconcile these findings to improve our ability to a priori identify which patients of the HER2 subtype are suitable for trastuzumab-based therapy ab initio? In the traditional, one-way hierarchy of CSCs, in which all cancer cells should descend from special subpopulations of self-renewing CSCs, HER2-positive CSCs can occur solely by self-renewal; trastuzumab treatment, by impeding the self-renewal property of HER2-positive CSCs and their intrinsic resistance to conventional treatments, is expected to induce tumor shrinkage and further reduce the rate of breast cancer recurrence when used in combination with traditional anti-breast cancer therapies. In the new, alternate model that we present here, more differentiated, non-stem CS might revert to trastuzumab-refractory CS-like cells due to the intrinsic or microenvironmentally driven activation of paths-to-stemness, similar to the CS-related EMT phenomenon. However, this model can also operate in a reverse mode in which stochastic transitions of trastuzumab-responsive CSC-like cellular states might give rise to non-CSC cellular states that, by lacking major attributes of CSCs, remain molecularly obscured from trastuzumab activity.
Trastuzumab is an anti-CSC drug
If the effectiveness of HER2-targeted drugs, including trastuzumab, largely depends on their ability to target breast CSCs, we can no longer underestimate the cellular state and focus solely on cellular phenotype when defining HER2-positive CSCs. A better understanding of the CSC/non-CSC social structure within HER2-overexpressing breast carcinomas should be viewed as a critical aspect for trastuzumab-based treatment decisions in the clinic. When deciphering the biological significance of CSC features and EMT-related phenomena for reinterpreting the genetic heterogeneity that differentiates trastuzumab-responders from non-responders among HER2-positive breast carcinoma cell lines, we concluded that novel predictive approaches aimed at better forecasting early tumor responses to trastuzumab should incorporate biological determinants causally underlying the intrinsic flexibility of HER2-positive CSCs to “enter” or “exit” trastuzumab-sensitive states. An accurate integration of CSC cellular state- and EMT-related biomarkers could better refine the currently available breast cancer molecular taxonomy of HER2-enriched breast carcinomas to distinguish, at a minimum, two intrinsically distinct HER2-positive breast cancer subtypes (i.e., luminal/HER2-positive and basal/HER2-positive) based on their prognostic and/or trastuzumab-responsiveness features. Critical to this new sub-classification of the HER2-enriched breast cancer subtype, is the notion that the effectiveness of anticancer therapies, such as trastuzumab, which are able to target CSCs, can be greatly impacted by the de novo generation of either CSCs (because non-CSCs would regenerate CSCs after cessation of trastuzumab therapy and lead to renewed tumor growth) or well-camouflaged CSC-like stages that escape trastuzumab therapy. Therefore, to be effective, anti-HER2 breast carcinoma approaches will need to combine agents that are selectively toxic to HER2-positive CSCs (i.e., the trastuzumab antibody itself) by targeting the bulk, non-CSC populations within HER2-positive breast carcinomas (e.g., taxane- or anthracycline-based chemotherapy), as well as by inhibiting EMT-related transitions from non-CSC to CSC states and/or from a trastuzumab-responsive CSC (epithelial) state to a non-responsive (mesenchymal) state (e.g., salinomycin, mTOR inhibitors, CD44-targeting antibodies, polyamine metabolism inhibitors and AMPK agonists such as metformin).Citation89,Citation103,Citation145-Citation155
CSCs “are made, not born”: Incorporating “mesenchymal transition signatures” to predict the anti-CSC efficacy of trastuzumab
Assuming that CSCs “are made, not born,” and that CSCs are flexible entities that can transform their trastuzumab-sensitivity-state, we must determine how to integrate CSC- and/or EMT-related biomarkers with currently available clinic-pathological variables to determine their prognostic value in HER2-positive breast carcinomas treated with trastuzumab-based therapy. While we acknowledge that an accurate development and validation of CSC/EMT-based trastuzumab-associated predictive signatures requires prospectively controlled clinical trials, it is worth noting that Liu and colleaguesCitation156 have recently confirmed that a 17-gene signature from enriched HER2 mammary CSCs can accurately predict clinical outcome for human HER2-positive/ER-negative breast carcinomas. Of note, their 17-gene HER2-CSCs-enriched signature, which was generated on the basis of differentially expressed genes in CSC vs. non-CSC fractions and trained on one HER2-positive breast cancer cohort, revealed that patients with HER2-positive/ER-negative tumors enriched with CSC features were significantly resistant to chemotherapy but responded well to chemotherapy plus trastuzumab. Thus, it again appears that the intrinsic enrichment of CSC features is associated with an effective response to trastuzumab in HER2-positive breast carcinomas. Although Giordano and colleaguesCitation157 recently revealed that gene transcripts of EMT-inducing transcription factors and CSC features are elevated in a significant percentage of HER2-positive metastatic breast cancer patients, they did not determine whether EMT and/or CSC features had prognostic value in patients treated with trastuzumab-based therapy. Therefore, additional studies are needed to unambiguously determine if the application of predictive CSCs/EMT-related tests can inform which HER2-positive tumors are suitable for trastuzumab-based regimens. A set of genes comprising an “EMT core signature” has been recently derived after triggering the EMT and observing the resulting shared changes in gene expression.Citation158 Another multi-cancer gene expression signature involving several EMT markers that are coordinately overexpressed in only a subset of malignant samples that have exceeded a particular cancer-staging threshold was also recently identified.Citation159 The latter signature, which includes 20 known EMT-associated genes, suggests a fibroblastic nature after passing through a SLUG/SNAIL2-based EMT, and, notably, E-cadherin is not downregulated, at least at the mRNA level. Anastassiou and colleaguesCitation160 have confirmed that their precise multi-cancer EMT signature is present in numerous publicly available data sets and in human cancer cell xenografts in vivo but not in stromal cells. Because Anastassiou’s group acknowledges that their “cancer mesenchymal transition signature” may initiate from spontaneously initiated CSCs states,Citation117,Citation160 it might be tempting to suggest that such a mesenchymal transition signature could be used as a predictive signature that merits validation both prospectively and in samples from completed trastuzumab-based clinical trials.Citation161 Moreover, prospective trastuzumab therapy-based clinical trials might be designed with predictive EMT-related marker validation in mind, following rigorous design,Citation161 because although it is plausible that not all EMT markers included in the mesenchymal transition signature will meet all surrogacy criteria, their use could provide crucial information on the occurrence of primary sensitivity/resistance to trastuzumab. In this regard, the neoadjuvant setting constitutes an ideal scenario for the validation of such markers because we can easily relate early-CSC/EMT biomarker response to trastuzumab outcome.Citation162 However, a mesenchymal transition signature may likely reflect a reversible process in which the mesenchymal markers in CSC/non-CSC cancer cells may dynamically appear and disappear depending on intrinsic and/or microenvironmental signals. Consequently, the potential use of mesenchymal transition-like predictive signatures would have high selectivity but not sensitivity to predict the trastuzumab unresponsiveness of HER2-positive breast carcinomas ab initio (e.g., the presence of the signature would likely imply inherent resistance to trastuzumab, but the lack of the signature in a particular extracted sample from a HER2-positive breast carcinoma might not imply a trastuzumab-sensitive tumor state, because the signature may have appeared earlier).
Apart from their direct implication in the EMT phenomenon, EMT transcriptional drivers (i.e., SNAIL factors) can also regulate other cellular processes related to cell proliferation and survival, thus contributing to resistance to pro-apoptotic stresses.Citation163-Citation165 In epithelial cells, promoting cell survival can be a primary function of EMT factors such as SLUG/SNAIL2 rather than being only acquired concomitantly with the EMT.Citation166 It is noteworthy that, in processes that do not require a complete EMT and in which cells retain some cell-cell junctions and do not acquire full mesenchymal characteristics at any point, as appears to occur in mixed basal/HER2-positive breast carcinoma cells, E-cadherin is redistributed in a manner that is no longer restricted to adherens junctions. Moreover, the redistribution of E-cadherin is not accompanied by a reduction in the levels of E-cadherin protein, even in cells with high concentrations of nuclear SLUG/SNAIL2. In this scenario, SLUG/SNAIL2 does not function as a repressor of E-cadherin but rather rescues the cells from apoptosis by repressing pro-apoptotic factors that otherwise would promote cell death during a partial EMT.Citation163-Citation166 Although it has been suggested that members of the SNAIL family could act primarily as survival factors and inducers of cell movement rather than inducers of the EMT,Citation167-Citation169 a more likely scenario is that SLUG/SNAIL2’s survival function can precede its role in inducing a full EMT.Citation166 The latter notion is supported by our recent findings that the SLUG/SNAIL2-mediated pro-survival/resistance function is associated, at least in part, with EMT and stemness in basal/HER2-positive breast cancer cells inherently resistant to trastuzumab.Citation114 Although we acknowledge that further studies in large tumor series using highly specific anti-SLUG/SNAIL2 antibodies are required to achieve a better understanding of the clinical relevance of EMT/CSC-related factors in the primary response of HER2-positive breast carcinomas to HER2-targeted therapies, the potential use of SLUG/SNAIL2-based predictive signatures would certainly have high selectivity and sensitivity for predicting the trastuzumab unresponsiveness of HER2-positive breast carcinomas, because these signatures can function as surrogates of two HER2-positive breast cancer subtypes (e.g., basal/HER2 vs. luminal/HER2), which distinctly benefit from trastuzumab-based therapies ab initio.
Corollary: It is Time to Refine HER2-Positive Breast Cancer Molecular Taxonomy
Candidate predictive classifiers for trastuzumab responsiveness ab initio need to be re-derived from well-characterized patient cohorts defined by virtue of clinical and biological variables. The best method and most accurate biomarkers for the identification of CSC states within distinct molecular subtypes need to be better explored, because we urgently need to translate the CSC concept to clinical practice, particularly when using drugs with anti-CSC activity, such as the anti-HER2 monoclonal antibody trastuzumab. In this regard, it must be taken into consideration that whenever breast cancer CSC-associated markers (CD44+CD24-/low) are present in tumors, they most likely identify the intrinsic ability of the tumor cell of origin to dynamically originate CSC/EMT cellular states (). Previous studies have already demonstrated an association between basal-like carcinomas and the widely accepted breast CSC phenotype, CD44+CD24-/low.Citation52,Citation70,Citation170-Citation172 Within basal-like tumors, Ricardo and colleaguesCitation172 have demonstrated a tendency toward reduced patient survival [disease-free survival (DFS) and overall survival (OS)] when carcinomas exhibit a predominant CD44+CD24-/low CSC-like phenotype. Although HER2-overexpressing tumors, as a group, are unlikely to exhibit increased expression of CD44+CD24-/low markers, it would be of interest to evaluate whether rapidly interchangeable CD44+CD24+/CD44+CD24-/low phenotypes could help to distinguish different HER2 subtypes in terms of trastuzumab responsiveness ab initio.
Figure 5. Refining HER2-positive breast cancer molecular taxonomy based on EMT/CSC features: From trastuzumab-responsive luminal/HER2 breast carcinomas to trastuzumab-refractory basal/HER2 breast carcinomas.
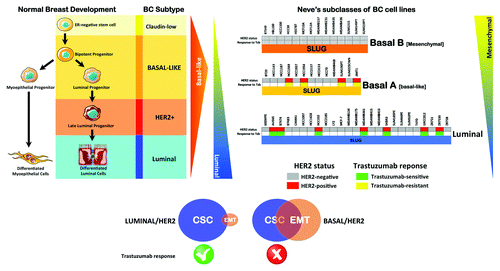
Gene expression microarrays have almost exclusively been able to discriminate HER2 oncogene-amplified tumors and basal-like tumors as separate clusters.Citation1,Citation3,Citation4 The inverse association is not complete, because basal cytokeratin expression and HER2 gene amplification have been observed concomitantly in tumorsCitation173,Citation174 and in the trastuzumab-unresponsive, basal/HER2-positive JIMT1 cell line.Citation98 Indeed, the presence of a basal/HER2-positive phenotype enriched with an intrinsic plasticity to “enter” may certainly explain the pioneering finding by Harris and colleaguesCitation48 that a specific HER2-positive tumor phenotype that was more likely to express genes associated with the basal-like phenotype, including higher expression of basal keratins (CK5, CK14, CK15 and CK17), was more frequent in the non-responding group of patients receiving pre-operative trastuzumab than in the responding group. Supporting the notion that the “basal phenotype” based on basal cytokeratins 5 and 14 immunostaining patterns may comprise more than one breast cancer biological entity, Laakso and colleaguesCitation173 demonstrated that the amplification of HER2 can be found almost exclusively in the so-called “basoluminal” group, which exhibited shorter relapse-free survival than the basal tumor group. Notably, when the basoluminal tumor group was sub-stratified by HER2 status, the authors confirmed that the difference between basal and basoluminal tumors was not due to more frequent HER2 amplification in the basoluminal group.Citation173 Although there was a trend toward faster progression of basoluminal/HER2-positive breast carcinomas, it became obvious that basal and basoluminal tumors differed in their natural biological aggressiveness. Can we propose a basal-cytokeratin surrogate definition of HER2-positive, breast carcinomas to predict the response to trastuzumab therapy? Liu and colleagues,Citation175 when initially characterizing the prognostic features of basal-like breast carcinomas without the influence of hormone receptor status in a series of hormone receptor-negative breast tumors, confirmed the existence of a small group of hormone-receptor-negative tumors that express HER2 and basal markers simultaneously. Moreover, and in contrast to previous studies, the subtype with the poorest outcome was not basal-like or HER2-positive but rather a basal/HER2-positive phenotype.Citation175 Bagaria and colleaguesCitation176 have recently followed a similar strategy to characterize the prognostic features of three different immunohistochemically defined subgroups of HER2-positive breast carcinomas. When “luminal-HER2+” (ER-positive and basal CK-negative), “HER2+” (ER-negative and basal CK-negative) and “basal/HER2+” (ER-negative and basal CK-positive) were correlated with clinicopathological features and overall survival, the authors concluded that the basal/HER2+ subtype was associated with the worst prognosis after adjusting for age, tumor size, lymph node status and adjuvant treatment. Although the authors acknowledged that the use of adjuvant trastuzumab and chemotherapy did not greatly differ between patients with HER2+ and basal/HER2+ tumors, the fact that patients with basal/HER2+ tumors displayed worse survival appears to suggest that the basal/HER2+ subtype perhaps may not only have prognostic value but may also have utility as a predictive marker of trastuzumab resistance. Our own laboratory has shown that a subgroup of HER2-overexpressing breast carcinomas overlaps in the histological features of the basal-like phenotype.Citation28 We observed that in some cases, HER2-positive breast carcinomas stained positively for basal epithelium cytokeratin markers (i.e., CK5/6), often displaying checkerboard-type intratumoral heterogeneity, as previously described by Laakso and colleagues in “basoluminal” HER2+ breast carcinomas,Citation173 and sometimes displaying a uniform or almost uniform CK5/6-positive staining that mimicked the pattern of pure basal-like breast carcinomas. Indeed, the subgroup of basal/HER2-positive breast carcinomas displayed histological features of basal-like tumors (i.e., poor differentiation, high-grade, geographic necrosis, pushing margins of invasion, syncytial arrangement of tumor cells, ribbon- or festoon-like architecture, squamous metaplasia, stromal lymphocytic infiltrates, high mitotic index and p53 positivity). Preliminary analyses in our own series of “luminal-HER2+,” “HER2+” and “basal/HER2” appear to definitely corroborate that the occurrence of the basal-like phenotype in HER2-overexpressing breast carcinomas, as defined by the immunohistochemical expression of basal CKs, is associated with an aggressive phenotype that significantly shortens the overall survival of HER2-positive breast cancer patients (unpublished observations).
Given that predictive studies are a specific form of prognostic study in which the outcome of interest is the response to treatment, and the application of the test is to guide the suitability of patients for specific therapy, these limitations make the identification of predictive signatures more difficult than the identification of prognostic factors and require prospectively controlled clinical trials.Citation161 Beyond confirming that the basal-like phenotype in HER2-overexpressing breast cancer can predict worse overall survival, we are currently evaluating whether the simple immunohistochemical identification of the basal/HER2-positive subtype alone may be sufficient to predict primary resistance to trastuzumab, which may have crucial implications for the current use of targeted therapies in patients originally identified as suitable for trastuzumab-based therapies based on their HER2 gene-amplified phenotype. We have already determined that the SNAIL factors can negatively regulate the expression of cytokeratins associated with luminal epithelial cells;Citation177,Citation178 therefore, we can identify a significant overlap between mesenchymal transition-like signatures and the occurrence of the basal-like immunophenotype in HER2-positive breast carcinomas. If forthcoming studies definitely confirm that an accurate integration of CSC cellular states and EMT-related biomarkers with the currently available breast cancer molecular taxonomy may significantly improve our ability to a priori inform whether patients belonging to HER2 subtypes that are differentially enriched with a “mesenchymal transition signature” (i.e., basal/HER2-positive vs. luminal/HER2-positive) would distinctly benefit from trastuzumab-based therapy ab initio, larger retrospective studies that include hundreds of samples from each of the major subtypes, as defined by nodal status and a small number of markers (ER/PR/HER2 and basal cytokeratins), should be conducted to generate both a more definitive HER2-positive breast cancer taxonomy and also better predictive molecular forecasting signatures to be validated both prospectively and in samples from trastuzumab-based clinical trials.
Additional material
Download Zip (9.6 MB)Acknowledgments
This work was financially supported through funding from the Instituto de Salud Carlos III [Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria (FIS), Spain, Grants CP05–00090 and PI06–0778 and RD06–0020–0028]; the Fundación Científica de la Asociación Española Contra el Cáncer (AECC, Spain); and the Ministerio de Ciencia e Innovación (SAF2009–11579, Plan Nacional de I+D+ I, MICINN, Spain). A.V.-M. received the Sara Borrell post-doctoral contract (CD08/00283, Ministerio de Sanidad y Consumo, FIS, Spain). S.C. received a Research Fellowship (Formación de Personal Investigador, FPI) from the Ministerio de Ciencia e Innovación (MICINN, Spain).
References
- Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience?. Nat Rev Clin Oncol 2010; 7:683 - 92; http://dx.doi.org/10.1038/nrclinonc.2010.154; PMID: 20877296
- Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature 2000; 406:747 - 52; http://dx.doi.org/10.1038/35021093; PMID: 10963602
- Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 2001; 98:10869 - 74; http://dx.doi.org/10.1073/pnas.191367098; PMID: 11553815
- Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA 2003; 100:8418 - 23; http://dx.doi.org/10.1073/pnas.0932692100; PMID: 12829800
- Dawood S, Broglio K, Buzdar AU, Hortobagyi GN, Giordano SH. Prognosis of women with metastatic breast cancer by HER2 status and trastuzumab treatment: an institutional-based review. J Clin Oncol 2010; 28:92 - 8; http://dx.doi.org/10.1200/JCO.2008.19.9844; PMID: 19933921
- Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, et al, Herceptin Adjuvant (HERA) Trial Study Team. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med 2005; 353:1659 - 72; http://dx.doi.org/10.1056/NEJMoa052306; PMID: 16236737
- Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr., Davidson NE, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 2005; 353:1673 - 84; http://dx.doi.org/10.1056/NEJMoa052122; PMID: 16236738
- Hortobagyi GN. Trastuzumab in the treatment of breast cancer. N Engl J Med 2005; 353:1734 - 6; http://dx.doi.org/10.1056/NEJMe058196; PMID: 16236745
- Gonzalez-Angulo AM, Hortobágyi GN, Esteva FJ. Adjuvant therapy with trastuzumab for HER-2/neu-positive breast cancer. Oncologist 2006; 11:857 - 67; http://dx.doi.org/10.1634/theoncologist.11-8-857; PMID: 16951389
- Gonzalez-Angulo AM, Litton JK, Broglio KR, Meric-Bernstam F, Rakkhit R, Cardoso F, et al. High risk of recurrence for patients with breast cancer who have human epidermal growth factor receptor 2-positive, node-negative tumors 1 cm or smaller. J Clin Oncol 2009; 27:5700 - 6; http://dx.doi.org/10.1200/JCO.2009.23.2025; PMID: 19884543
- Chavez-MacGregor M, Gonzalez-Angulo AM. HER2-neu positivity in patients with small and node-negative breast cancer (pT1a,b,N0,M0): a high risk group?. Clin Adv Hematol Oncol 2009; 7:591 - 8; PMID: 20020671
- Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol 2002; 20:719 - 26; http://dx.doi.org/10.1200/JCO.20.3.719; PMID: 11821453
- Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol 2006; 3:269 - 80; http://dx.doi.org/10.1038/ncponc0509; PMID: 16683005
- Nahta R, Esteva FJ. HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Res 2006; 8:215; http://dx.doi.org/10.1186/bcr1612; PMID: 17096862
- Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol 2007; 18:977 - 84; http://dx.doi.org/10.1093/annonc/mdl475; PMID: 17229773
- Nahta R, Esteva FJ. Trastuzumab: triumphs and tribulations. Oncogene 2007; 26:3637 - 43; http://dx.doi.org/10.1038/sj.onc.1210379; PMID: 17530017
- Spector NL, Blackwell KL. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol 2009; 27:5838 - 47; http://dx.doi.org/10.1200/JCO.2009.22.1507; PMID: 19884552
- Esteva FJ, Yu D, Hung MC, Hortobagyi GN. Molecular predictors of response to trastuzumab and lapatinib in breast cancer. Nat Rev Clin Oncol 2010; 7:98 - 107; http://dx.doi.org/10.1038/nrclinonc.2009.216; PMID: 20027191
- Pusztai L. Molecular heterogeneity of breast cancer: implications for treatment and clinical trial design. Breast Cancer Res 2009; 11:Suppl 1 S4; http://dx.doi.org/10.1186/bcr2265; PMID: 20030879
- Roukos DH. Beyond HER2 and trastuzumab: heterogeneity, systems biology, and cancer origin research may guide the future for personalized treatment of very early but aggressive breast cancer. J Clin Oncol 2010; 28:e279 - 80, author reply e282-3; http://dx.doi.org/10.1200/JCO.2009.27.7061; PMID: 20406920
- Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006; 295:2492 - 502; http://dx.doi.org/10.1001/jama.295.21.2492; PMID: 16757721
- Millikan RC, Newman B, Tse CK, Moorman PG, Conway K, Dressler LG, et al. Epidemiology of basal-like breast cancer. Breast Cancer Res Treat 2008; 109:123 - 39; http://dx.doi.org/10.1007/s10549-007-9632-6; PMID: 17578664
- Perou CM, Børresen-Dale AL. Systems biology and genomics of breast cancer. Cold Spring Harb Perspect Biol 2011; 3; http://dx.doi.org/10.1101/cshperspect.a003293; PMID: 21047916
- Rakha EA, Ellis IO. Triple-negative/basal-like breast cancer: review. [review] Pathology 2009; 41:40 - 7; http://dx.doi.org/10.1080/00313020802563510; PMID: 19089739
- Rakha EA, Elsheikh SE, Aleskandarany MA, Habashi HO, Green AR, Powe DG, et al. Triple-negative breast cancer: distinguishing between basal and nonbasal subtypes. Clin Cancer Res 2009; 15:2302 - 10; http://dx.doi.org/10.1158/1078-0432.CCR-08-2132; PMID: 19318481
- Badve S, Dabbs DJ, Schnitt SJ, Baehner FL, Decker T, Eusebi V, et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Mod Pathol 2011; 24:157 - 67; http://dx.doi.org/10.1038/modpathol.2010.200; PMID: 21076464
- Oliveras-Ferraros C, Vazquez-Martin A, Martin-Castillo B, Cufí S, Del Barco S, Lopez-Bonet E, et al. Dynamic emergence of the mesenchymal CD44(pos)CD24(neg/low) phenotype in HER2-gene amplified breast cancer cells with de novo resistance to trastuzumab (Herceptin). Biochem Biophys Res Commun 2010; 397:27 - 33; http://dx.doi.org/10.1016/j.bbrc.2010.05.041; PMID: 20470755
- Oliveras-Ferraros C, Vazquez-Martin A, Martin-Castilló B, Pérez-Martínez MC, Cufí S, Del Barco S, et al. Pathway-focused proteomic signatures in HER2-overexpressing breast cancer with a basal-like phenotype: new insights into de novo resistance to trastuzumab (Herceptin). Int J Oncol 2010; 37:669 - 78; PMID: 20664936
- Gonzalez-Angulo AM, Hennessy BT, Mills GB. Future of personalized medicine in oncology: a systems biology approach. J Clin Oncol 2010; 28:2777 - 83; http://dx.doi.org/10.1200/JCO.2009.27.0777; PMID: 20406928
- Ellsworth RE, Decewicz DJ, Shriver CD, Ellsworth DL. Breast cancer in the personal genomics era. Curr Genomics 2010; 11:146 - 61; http://dx.doi.org/10.2174/138920210791110951; PMID: 21037853
- Ross JS, Slodkowska EA, Symmans WF, Pusztai L, Ravdin PM, Hortobagyi GN. The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist 2009; 14:320 - 68; http://dx.doi.org/10.1634/theoncologist.2008-0230; PMID: 19346299
- Roukos DH. Trastuzumab and beyond: sequencing cancer genomes and predicting molecular networks. Pharmacogenomics J 2011; 11:81 - 92; http://dx.doi.org/10.1038/tpj.2010.81; PMID: 20975737
- van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002; 347:1999 - 2009; http://dx.doi.org/10.1056/NEJMoa021967; PMID: 12490681
- Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004; 351:2817 - 26; http://dx.doi.org/10.1056/NEJMoa041588; PMID: 15591335
- Eichhorn PJ, Baselga J. HER2 signatures in breast cancer: ready to go to print?. J Clin Oncol 2010; 28:1809 - 10; http://dx.doi.org/10.1200/JCO.2009.26.7146; PMID: 20231675
- Staaf J, Ringnér M, Vallon-Christersson J, Jönsson G, Bendahl PO, Holm K, et al. Identification of subtypes in human epidermal growth factor receptor 2--positive breast cancer reveals a gene signature prognostic of outcome. J Clin Oncol 2010; 28:1813 - 20; http://dx.doi.org/10.1200/JCO.2009.22.8775; PMID: 20231686
- Wirapati P, Sotiriou C, Kunkel S, Farmer P, Pradervand S, Haibe-Kains B, et al. Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res 2008; 10:R65; http://dx.doi.org/10.1186/bcr2124; PMID: 18662380
- Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, Bontempi G, et al. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res 2008; 14:5158 - 65; http://dx.doi.org/10.1158/1078-0432.CCR-07-4756; PMID: 18698033
- Sotiriou C, Pusztai L. Gene-expression signatures in breast cancer. N Engl J Med 2009; 360:790 - 800; http://dx.doi.org/10.1056/NEJMra0801289; PMID: 19228622
- Staaf J, Jönsson G, Ringnér M, Vallon-Christersson J, Grabau D, Arason A, et al. High-resolution genomic and expression analyses of copy number alterations in HER2-amplified breast cancer. Breast Cancer Res 2010; 12:R25; http://dx.doi.org/10.1186/bcr2568; PMID: 20459607
- Theillet C. What do we learn from HER2-positive breast cancer genomic profiles?. Breast Cancer Res 2010; 12:107; http://dx.doi.org/10.1186/bcr2571; PMID: 20519027
- Menendez JA, Mehmi I, Lupu R. Trastuzumab in combination with heregulin-activated Her-2 (erbB-2) triggers a receptor-enhanced chemosensitivity effect in the absence of Her-2 overexpression. J Clin Oncol 2006; 24:3735 - 46; http://dx.doi.org/10.1200/JCO.2005.04.3489; PMID: 16847284
- Arteaga CL. Can trastuzumab be effective against tumors with low HER2/Neu (ErbB2) receptors?. J Clin Oncol 2006; 24:3722 - 5; http://dx.doi.org/10.1200/JCO.2006.06.5268; PMID: 16847283
- de Alava E, Ocaña A, Abad M, Montero JC, Esparís-Ogando A, Rodríguez CA, et al. Neuregulin expression modulates clinical response to trastuzumab in patients with metastatic breast cancer. J Clin Oncol 2007; 25:2656 - 63; http://dx.doi.org/10.1200/JCO.2006.08.6850; PMID: 17602072
- Paik S, Kim C, Wolmark N. HER2 status and benefit from adjuvant trastuzumab in breast cancer. N Engl J Med 2008; 358:1409 - 11; http://dx.doi.org/10.1056/NEJMc0801440; PMID: 18367751
- Esteva FJ, Yu D, Hung MC, Hortobagyi GN. Molecular predictors of response to trastuzumab and lapatinib in breast cancer. Nat Rev Clin Oncol 2010; 7:98 - 107; http://dx.doi.org/10.1038/nrclinonc.2009.216; PMID: 20027191
- Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007; 12:395 - 402; http://dx.doi.org/10.1016/j.ccr.2007.08.030; PMID: 17936563
- Harris LN, You F, Schnitt SJ, Witkiewicz A, Lu X, Sgroi D, et al. Predictors of resistance to preoperative trastuzumab and vinorelbine for HER2-positive early breast cancer. Clin Cancer Res 2007; 13:1198 - 207; http://dx.doi.org/10.1158/1078-0432.CCR-06-1304; PMID: 17317830
- Bedard PL, Cardoso F, Piccart-Gebhart MJ. Stemming resistance to HER-2 targeted therapy. J Mammary Gland Biol Neoplasia 2009; 14:55 - 66; http://dx.doi.org/10.1007/s10911-009-9116-x; PMID: 19259796
- Jones RJ, Matsui WH, Smith BD. Cancer stem cells: are we missing the target?. J Natl Cancer Inst 2004; 96:583 - 5; http://dx.doi.org/10.1093/jnci/djh095; PMID: 15100335
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100:3983 - 8; http://dx.doi.org/10.1073/pnas.0530291100; PMID: 12629218
- Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res 2008; 10:R25; http://dx.doi.org/10.1186/bcr1982; PMID: 18366788
- Kakarala M, Wicha MS. Implications of the cancer stem-cell hypothesis for breast cancer prevention and therapy. J Clin Oncol 2008; 26:2813 - 20; http://dx.doi.org/10.1200/JCO.2008.16.3931; PMID: 18539959
- Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008; 27:6120 - 30; http://dx.doi.org/10.1038/onc.2008.207; PMID: 18591932
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007; 1:555 - 67; http://dx.doi.org/10.1016/j.stem.2007.08.014; PMID: 18371393
- Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008; 100:672 - 9; http://dx.doi.org/10.1093/jnci/djn123; PMID: 18445819
- Magnifico A, Albano L, Campaner S, Delia D, Castiglioni F, Gasparini P, et al. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. Clin Cancer Res 2009; 15:2010 - 21; http://dx.doi.org/10.1158/1078-0432.CCR-08-1327; PMID: 19276287
- Liu S, Wicha MS. Targeting breast cancer stem cells. J Clin Oncol 2010; 28:4006 - 12; http://dx.doi.org/10.1200/JCO.2009.27.5388; PMID: 20498387
- Debeb BG, Xu W, Woodward WA. Radiation resistance of breast cancer stem cells: understanding the clinical framework. J Mammary Gland Biol Neoplasia 2009; 14:11 - 7; http://dx.doi.org/10.1007/s10911-009-9114-z; PMID: 19252973
- Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol 2008; 26:2839 - 45; http://dx.doi.org/10.1200/JCO.2007.15.1829; PMID: 18539962
- Nguyen NP, Almeida FS, Chi A, Nguyen LM, Cohen D, Karlsson U, et al. Molecular biology of breast cancer stem cells: potential clinical applications. Cancer Treat Rev 2010; 36:485 - 91; http://dx.doi.org/10.1016/j.ctrv.2010.02.016; PMID: 20231058
- Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med 2007; 356:217 - 26; http://dx.doi.org/10.1056/NEJMoa063994; PMID: 17229949
- Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA 2009; 106:13820 - 5; http://dx.doi.org/10.1073/pnas.0905718106; PMID: 19666588
- Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res 2002; 62:4132 - 41; PMID: 12124352
- Oliveras-Ferraros C, Vazquez-Martin A, Cufí S, Torres-Garcia VZ, Sauri-Nadal T, Barco SD, et al. Inhibitor of Apoptosis (IAP) survivin is indispensable for survival of HER2 gene-amplified breast cancer cells with primary resistance to HER1/2-targeted therapies. Biochem Biophys Res Commun 2011; 407:412 - 9; http://dx.doi.org/10.1016/j.bbrc.2011.03.039; PMID: 21402055
- Amsterdam A, Sadler KC, Lai K, Farrington S, Bronson RT, Lees JA, et al. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol 2004; 2:E139; http://dx.doi.org/10.1371/journal.pbio.0020139; PMID: 15138505
- Deisenroth C, Zhang Y. Ribosome biogenesis surveillance: probing the ribosomal protein-Mdm2-p53 pathway. Oncogene 2010; 29:4253 - 60; http://dx.doi.org/10.1038/onc.2010.189; PMID: 20498634
- Dumont N, Wilson MB, Crawford YG, Reynolds PA, Sigaroudinia M, Tlsty TD. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc Natl Acad Sci USA 2008; 105:14867 - 72; http://dx.doi.org/10.1073/pnas.0807146105; PMID: 18806226
- Arima Y, Hayashi N, Hayashi H, Sasaki M, Kai K, Sugihara E, et al. Loss of p16 expression is associated with the stem cell characteristics of surface markers and therapeutic resistance in estrogen receptor-negative breast cancer. Int J Cancer 2012; 130:2568 - 79; http://dx.doi.org/10.1002/ijc.26271; PMID: 21717460
- Park SY, Lee HE, Li H, Shipitsin M, Gelman R, Polyak K. Heterogeneity for stem cell-related markers according to tumor subtype and histologic stage in breast cancer. Clin Cancer Res 2010; 16:876 - 87; http://dx.doi.org/10.1158/1078-0432.CCR-09-1532; PMID: 20103682
- Ali HR, Dawson SJ, Blows FM, Provenzano E, Pharoah PD, Caldas C. Cancer stem cell markers in breast cancer: pathological, clinical and prognostic significance. Breast Cancer Res 2011; 13:R118; http://dx.doi.org/10.1186/bcr3061; PMID: 22112299
- Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010; 6:421 - 32; http://dx.doi.org/10.1016/j.stem.2010.02.018; PMID: 20452317
- Cariati M, Naderi A, Brown JP, Smalley MJ, Pinder SE, Caldas C, et al. Alpha-6 integrin is necessary for the tumourigenicity of a stem cell-like subpopulation within the MCF7 breast cancer cell line. Int J Cancer 2008; 122:298 - 304; http://dx.doi.org/10.1002/ijc.23103; PMID: 17935134
- Jo M, Eastman BM, Webb DL, Stoletov K, Klemke R, Gonias SL. Cell signaling by urokinase-type plasminogen activator receptor induces stem cell-like properties in breast cancer cells. Cancer Res 2010; 70:8948 - 58; http://dx.doi.org/10.1158/0008-5472.CAN-10-1936; PMID: 20940399
- Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, et al. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res 2008; 68:3243 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-07-5480; PMID: 18451150
- Vassilopoulos A, Wang RH, Petrovas C, Ambrozak D, Koup R, Deng CX. Identification and characterization of cancer initiating cells from BRCA1 related mammary tumors using markers for normal mammary stem cells. Int J Biol Sci 2008; 4:133 - 42; http://dx.doi.org/10.7150/ijbs.4.133; PMID: 18461147
- Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer 2005; 5:744 - 9; http://dx.doi.org/10.1038/nrc1694; PMID: 16148886
- Reka AK, Kurapati H, Narala VR, Bommer G, Chen J, Standiford TJ, et al. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor metastasis by antagonizing Smad3-mediated epithelial-mesenchymal transition. Mol Cancer Ther 2010; 9:3221 - 32; http://dx.doi.org/10.1158/1535-7163.MCT-10-0570; PMID: 21159608
- Papi A, Guarnieri T, Storci G, Santini D, Ceccarelli C, Taffurelli M, et al. Nuclear receptors agonists exert opposing effects on the inflammation dependent survival of breast cancer stem cells. Cell Death Differ 2012; 19:1208 - 19; http://dx.doi.org/10.1038/cdd.2011.207; PMID: 22261616
- An Y, Ongkeko WM. ABCG2: the key to chemoresistance in cancer stem cells?. Expert Opin Drug Metab Toxicol 2009; 5:1529 - 42; http://dx.doi.org/10.1517/17425250903228834; PMID: 19708828
- Ding XW, Wu JH, Jiang CP. ABCG2: a potential marker of stem cells and novel target in stem cell and cancer therapy. Life Sci 2010; 86:631 - 7; http://dx.doi.org/10.1016/j.lfs.2010.02.012; PMID: 20159023
- Natarajan K, Xie Y, Baer MR, Ross DD. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem Pharmacol 2012; 83:1084 - 103; http://dx.doi.org/10.1016/j.bcp.2012.01.002; PMID: 22248732
- Bendall SC, Stewart MH, Menendez P, George D, Vijayaragavan K, Werbowetski-Ogilvie T, et al. IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature 2007; 448:1015 - 21; http://dx.doi.org/10.1038/nature06027; PMID: 17625568
- Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res 2008; 10:R10; http://dx.doi.org/10.1186/bcr1855; PMID: 18241344
- Bachelard-Cascales E, Chapellier M, Delay E, Pochon G, Voeltzel T, Puisieux A, et al. The CD10 enzyme is a key player to identify and regulate human mammary stem cells. Stem Cells 2010; 28:1081 - 8; http://dx.doi.org/10.1002/stem.435; PMID: 20506111
- Maguer-Satta V, Besançon R, Bachelard-Cascales E. Concise review: neutral endopeptidase (CD10): a multifaceted environment actor in stem cells, physiological mechanisms, and cancer. Stem Cells 2011; 29:389 - 96; http://dx.doi.org/10.1002/stem.592; PMID: 21425402
- Maguer-Satta V, Chapellier M, Delay E, Bachelard-Cascales E. CD10: a tool to crack the role of stem cells in breast cancer. Proc Natl Acad Sci USA 2011; 108:E1264 - , author reply E1265; http://dx.doi.org/10.1073/pnas.1116567108; PMID: 22109559
- Keller PJ, Arendt LM, Skibinski A, Logvinenko T, Klebba I, Dong S, et al. Defining the cellular precursors to human breast cancer. Proc Natl Acad Sci USA 2012; 109:2772 - 7; http://dx.doi.org/10.1073/pnas.1017626108; PMID: 21940501
- Cufi S, Corominas-Faja B, Vazquez-Martin A, Oliveras-Ferraros C, Dorca J, Bosch-Barrera J, et al. Metformin-induced preferential killing of breast cancer initiating CD44+CD24-/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget 2012; 3:395 - 8; PMID: 22565037
- Sarrio D, Franklin CK, Mackay A, Reis-Filho JS, Isacke CM. Epithelial and mesenchymal subpopulations within normal basal breast cell lines exhibit distinct stem cell/progenitor properties. Stem Cells 2012; 30:292 - 303; http://dx.doi.org/10.1002/stem.791; PMID: 22102611
- Celià-Terrassa T, Meca-Cortés O, Mateo F, de Paz AM, Rubio N, Arnal-Estapé A, et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest 2012; 122:1849 - 68; http://dx.doi.org/10.1172/JCI59218; PMID: 22505459
- Blick T, Widodo E, Hugo H, Waltham M, Lenburg ME, Neve RM, et al. Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin Exp Metastasis 2008; 25:629 - 42; http://dx.doi.org/10.1007/s10585-008-9170-6; PMID: 18461285
- Blick T, Hugo H, Widodo E, Waltham M, Pinto C, Mani SA, et al. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J Mammary Gland Biol Neoplasia 2010; 15:235 - 52; http://dx.doi.org/10.1007/s10911-010-9175-z; PMID: 20521089
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10:515 - 27; http://dx.doi.org/10.1016/j.ccr.2006.10.008; PMID: 17157791
- Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adélaïde J, Cervera N, et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 2006; 25:2273 - 84; http://dx.doi.org/10.1038/sj.onc.1209254; PMID: 16288205
- Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res 2010; 12:R68; http://dx.doi.org/10.1186/bcr2635; PMID: 20813035
- Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol 2011; 5:5 - 23; http://dx.doi.org/10.1016/j.molonc.2010.11.003; PMID: 21147047
- Tanner M, Kapanen AI, Junttila T, Raheem O, Grenman S, Elo J, et al. Characterization of a novel cell line established from a patient with Herceptin-resistant breast cancer. Mol Cancer Ther 2004; 3:1585 - 92; PMID: 15634652
- Nagy P, Friedländer E, Tanner M, Kapanen AI, Carraway KL, Isola J, et al. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res 2005; 65:473 - 82; PMID: 15695389
- Jönsson G, Staaf J, Olsson E, Heidenblad M, Vallon-Christersson J, Osoegawa K, et al. High-resolution genomic profiles of breast cancer cell lines assessed by tiling BAC array comparative genomic hybridization. Genes Chromosomes Cancer 2007; 46:543 - 58; http://dx.doi.org/10.1002/gcc.20438; PMID: 17334996
- Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007; 11:259 - 73; http://dx.doi.org/10.1016/j.ccr.2007.01.013; PMID: 17349583
- Dhillon J, Astanehe A, Lee C, Fotovati A, Hu K, Dunn SE. The expression of activated Y-box binding protein-1 serine 102 mediates trastuzumab resistance in breast cancer cells by increasing CD44+ cells. Oncogene 2010; 29:6294 - 300; http://dx.doi.org/10.1038/onc.2010.365; PMID: 20802512
- Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res Treat 2011; 126:355 - 64; http://dx.doi.org/10.1007/s10549-010-0924-x; PMID: 20458531
- Oliveras-Ferraros C, Vazquez-Martin A, Cufí S, Queralt B, Báez L, Guardeño R, et al. Stem cell property epithelial-to-mesenchymal transition is a core transcriptional network for predicting cetuximab (Erbitux™) efficacy in KRAS wild-type tumor cells. J Cell Biochem 2011; 112:10 - 29; http://dx.doi.org/10.1002/jcb.22952; PMID: 21104905
- Freytag J, Wilkins-Port CE, Higgins CE, Higgins SP, Samarakoon R, Higgins PJ. PAI-1 mediates the TGF-beta1+EGF-induced “scatter” response in transformed human keratinocytes. J Invest Dermatol 2010; 130:2179 - 90; http://dx.doi.org/10.1038/jid.2010.106; PMID: 20428185
- White LR, Blanchette JB, Ren L, Awn A, Trpkov K, Muruve DA. The characterization of alpha5-integrin expression on tubular epithelium during renal injury. Am J Physiol Renal Physiol 2007; 292:F567 - 76; http://dx.doi.org/10.1152/ajprenal.00212.2006; PMID: 17018844
- Mayanagi T, Sobue K. Diversification of caldesmon-linked actin cytoskeleton in cell motility. Cell Adh Migr 2011; 5:150 - 9; http://dx.doi.org/10.4161/cam.5.2.14398; PMID: 21350330
- Jung YS, Liu XW, Chirco R, Warner RB, Fridman R, Kim HR. TIMP-1 induces an EMT-like phenotypic conversion in MDCK cells independent of its MMP-inhibitory domain. PLoS One 2012; 7:e38773; http://dx.doi.org/10.1371/journal.pone.0038773; PMID: 22701711
- Sarrió D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res 2008; 68:989 - 97; http://dx.doi.org/10.1158/0008-5472.CAN-07-2017; PMID: 18281472
- Girotti MR, Fernández M, López JA, Camafeita E, Fernández EA, Albar JP, et al. SPARC promotes cathepsin B-mediated melanoma invasiveness through a collagen I/α2β1 integrin axis. J Invest Dermatol 2011; 131:2438 - 47; http://dx.doi.org/10.1038/jid.2011.239; PMID: 21850018
- Fenouille N, Tichet M, Dufies M, Pottier A, Mogha A, Soo JK, et al. The epithelial-mesenchymal transition (EMT) regulatory factor SLUG (SNAI2) is a downstream target of SPARC and AKT in promoting melanoma cell invasion. PLoS One 2012; 7:e40378; http://dx.doi.org/10.1371/journal.pone.0040378; PMID: 22911700
- Meyer MJ, Fleming JM, Ali MA, Pesesky MW, Ginsburg E, Vonderhaar BK. Dynamic regulation of CD24 and the invasive, CD44posCD24neg phenotype in breast cancer cell lines. Breast Cancer Res 2009; 11:R82; http://dx.doi.org/10.1186/bcr2449; PMID: 19906290
- Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell 2012; 47:570 - 84; http://dx.doi.org/10.1016/j.molcel.2012.06.014; PMID: 22819326
- Oliveras-Ferraros C, Corominas-Faja B, Cufí S, Vazquez-Martin A, Martin-Castillo B, Iglesias JM, et al. Epithelial-to-mesenchymal transition (EMT) confers primary resistance to trastuzumab (Herceptin). Cell Cycle 2012; 11:4020 - 32; http://dx.doi.org/10.4161/cc.22225; PMID: 22992620
- Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010; 29:4741 - 51; http://dx.doi.org/10.1038/onc.2010.215; PMID: 20531305
- Floor S, van Staveren WC, Larsimont D, Dumont JE, Maenhaut C. Cancer cells in epithelial-to-mesenchymal transition and tumor-propagating-cancer stem cells: distinct, overlapping or same populations. Oncogene 2011; 30:4609 - 21; http://dx.doi.org/10.1038/onc.2011.184; PMID: 21643013
- Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA 2011; 108:7950 - 5; http://dx.doi.org/10.1073/pnas.1102454108; PMID: 21498687
- Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011; 146:633 - 44; http://dx.doi.org/10.1016/j.cell.2011.07.026; PMID: 21854987
- Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell 2012; 10:717 - 28; http://dx.doi.org/10.1016/j.stem.2012.05.007; PMID: 22704512
- Jordan NV, Johnson GL, Abell AN. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle 2011; 10:2865 - 73; http://dx.doi.org/10.4161/cc.10.17.17188; PMID: 21862874
- Abollo-Jiménez F, Jiménez R, Cobaleda C. Physiological cellular reprogramming and cancer. Semin Cancer Biol 2010; 20:98 - 106; http://dx.doi.org/10.1016/j.semcancer.2010.02.002; PMID: 20188173
- Li R, Liang J, Ni S, Zhou T, Qing X, Li H, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010; 7:51 - 63; http://dx.doi.org/10.1016/j.stem.2010.04.014; PMID: 20621050
- Polo JM, Hochedlinger K. When fibroblasts MET iPSCs. Cell Stem Cell 2010; 7:5 - 6; http://dx.doi.org/10.1016/j.stem.2010.05.018; PMID: 20621040
- Samavarchi-Tehrani P, Golipour A, David L, Sung HK, Beyer TA, Datti A, et al. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010; 7:64 - 77; http://dx.doi.org/10.1016/j.stem.2010.04.015; PMID: 20621051
- Chen T, Yuan D, Wei B, Jiang J, Kang J, Ling K, et al. E-cadherin-mediated cell-cell contact is critical for induced pluripotent stem cell generation. Stem Cells 2010; 28:1315 - 25; http://dx.doi.org/10.1002/stem.456; PMID: 20521328
- Redmer T, Diecke S, Grigoryan T, Quiroga-Negreira A, Birchmeier W, Besser D. E-cadherin is crucial for embryonic stem cell pluripotency and can replace OCT4 during somatic cell reprogramming. EMBO Rep 2011; 12:720 - 6; http://dx.doi.org/10.1038/embor.2011.88; PMID: 21617704
- Lowry WE. E-cadherin, a new mixer in the Yamanaka cocktail. EMBO Rep 2011; 12:613 - 4; http://dx.doi.org/10.1038/embor.2011.117; PMID: 21701504
- Tuma RS. Cancer stem cell hypothesis and trastuzumab in HER2-negative tumors. J Natl Cancer Inst 2012; 104:968 - 9; http://dx.doi.org/10.1093/jnci/djs307; PMID: 22745473
- Liu YN, Abou-Kheir W, Yin JJ, Fang L, Hynes P, Casey O, et al. Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor β-initiated prostate cancer epithelial-mesenchymal transition. Mol Cell Biol 2012; 32:941 - 53; http://dx.doi.org/10.1128/MCB.06306-11; PMID: 22203039
- Lin ZS, Chu HC, Yen YC, Lewis BC, Chen YW. Krüppel-like factor 4, a tumor suppressor in hepatocellular carcinoma cells reverts epithelial mesenchymal transition by suppressing slug expression. PLoS One 2012; 7:e43593; http://dx.doi.org/10.1371/journal.pone.0043593; PMID: 22937066
- Yori JL, Johnson E, Zhou G, Jain MK, Keri RA. Kruppel-like factor 4 inhibits epithelial-to-mesenchymal transition through regulation of E-cadherin gene expression. J Biol Chem 2010; 285:16854 - 63; http://dx.doi.org/10.1074/jbc.M110.114546; PMID: 20356845
- Yori JL, Seachrist DD, Johnson E, Lozada KL, Abdul-Karim FW, Chodosh LA, et al. Krüppel-like factor 4 inhibits tumorigenic progression and metastasis in a mouse model of breast cancer. Neoplasia 2011; 13:601 - 10; PMID: 21750654
- Yu F, Li J, Chen H, Fu J, Ray S, Huang S, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene 2011; 30:2161 - 72; http://dx.doi.org/10.1038/onc.2010.591; PMID: 21242971
- Daley GQ. Common themes of dedifferentiation in somatic cell reprogramming and cancer. Cold Spring Harb Symp Quant Biol 2008; 73:171 - 4; http://dx.doi.org/10.1101/sqb.2008.73.041; PMID: 19150965
- Werbowetski-Ogilvie TE, Bhatia M. Pluripotent human stem cell lines: what we can learn about cancer initiation. Trends Mol Med 2008; 14:323 - 32; http://dx.doi.org/10.1016/j.molmed.2008.06.005; PMID: 18635398
- Blum B, Benvenisty N. The tumorigenicity of human embryonic stem cells. Adv Cancer Res 2008; 100:133 - 58; http://dx.doi.org/10.1016/S0065-230X(08)00005-5; PMID: 18620095
- Blum B, Benvenisty N. The tumorigenicity of diploid and aneuploid human pluripotent stem cells. Cell Cycle 2009; 8:3822 - 30; http://dx.doi.org/10.4161/cc.8.23.10067; PMID: 19887907
- Wobus AM. The Janus face of pluripotent stem cells--connection between pluripotency and tumourigenicity. Bioessays 2010; 32:993 - 1002; http://dx.doi.org/10.1002/bies.201000065; PMID: 21105293
- Castellanos A, Vicente-Dueñas C, Campos-Sánchez E, Cruz JJ, García-Criado FJ, García-Cenador MB, et al. Cancer as a reprogramming-like disease: implications in tumor development and treatment. Semin Cancer Biol 2010; 20:93 - 7; http://dx.doi.org/10.1016/j.semcancer.2010.02.001; PMID: 20188174
- Ben-David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev Cancer 2011; 11:268 - 77; http://dx.doi.org/10.1038/nrc3034; PMID: 21390058
- Ramos-Mejia V, Fraga MF, Menendez P. iPSCs from cancer cells: challenges and opportunities. Trends Mol Med 2012; 18:245 - 7; http://dx.doi.org/10.1016/j.molmed.2012.04.001; PMID: 22521522
- Zhang X, Cruz FD, Terry M, Remotti F, Matushansky I. Terminal differentiation and loss of tumorigenicity of human cancers via pluripotency-based reprogramming. Oncogene 2012; In press http://dx.doi.org/10.1038/onc.2012.237; PMID: 22777357
- Lang JY, Shi Y, Chin YE. Reprogramming cancer cells: back to the future. Oncogene 2012; In press http://dx.doi.org/10.1038/onc.2012.349; PMID: 22869153
- Dewi D, Ishii H, Haraguchi N, Nishikawa S, Kano Y, Fukusumi T, et al. Reprogramming of gastrointestinal cancer cells. Cancer Sci 2012; 103:393 - 9; http://dx.doi.org/10.1111/j.1349-7006.2011.02184.x; PMID: 22151786
- Oak PS, Kopp F, Thakur C, Ellwart JW, Rapp UR, Ullrich A, et al. Combinatorial treatment of mammospheres with trastuzumab and salinomycin efficiently targets HER2-positive cancer cells and cancer stem cells. Int J Cancer 2012; 131:2808 - 19; http://dx.doi.org/10.1002/ijc.27595; PMID: 22511343
- Zhu Y, Zhang X, Liu Y, Zhang S, Liu J, Ma Y, et al. Antitumor effect of the mTOR inhibitor everolimus in combination with trastuzumab on human breast cancer stem cells in vitro and in vivo. Tumour Biol 2012; 33:1349 - 62; http://dx.doi.org/10.1007/s13277-012-0383-6; PMID: 22492237
- Masuko K, Okazaki S, Satoh M, Tanaka G, Ikeda T, Torii R, et al. Anti-tumor effect against human cancer xenografts by a fully human monoclonal antibody to a variant 8-epitope of CD44R1 expressed on cancer stem cells. PLoS One 2012; 7:e29728; http://dx.doi.org/10.1371/journal.pone.0029728; PMID: 22272243
- Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metformin against TGFβ-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle 2010; 9:4461 - 8; http://dx.doi.org/10.4161/cc.9.22.14048; PMID: 21088486
- Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Lopez-Bonet E, et al. The anti-diabetic drug metformin suppresses the metastasis-associated protein CD24 in MDA-MB-468 triple-negative breast cancer cells. Oncol Rep 2011; 25:135 - 40; PMID: 21109968
- Cirenajwis H, Smiljanic S, Honeth G, Hegardt C, Marton LJ, Oredsson SM. Reduction of the putative CD44+CD24- breast cancer stem cell population by targeting the polyamine metabolic pathway with PG11047. Anticancer Drugs 2010; 21:897 - 906; http://dx.doi.org/10.1097/CAD.0b013e32833f2f77; PMID: 20838207
- Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle 2010; 9:1057 - 64; http://dx.doi.org/10.4161/cc.9.6.10994; PMID: 20305377
- Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle 2010; 9:3807 - 14; http://dx.doi.org/10.4161/cc.9.18.13131; PMID: 20890129
- Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, et al. Metformin: multi-faceted protection against cancer. Oncotarget 2011; 2:896 - 917; PMID: 22203527
- Vazquez-Martin A, López-Bonetc E, Cufí S, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, et al. Repositioning chloroquine and metformin to eliminate cancer stem cell traits in pre-malignant lesions. Drug Resist Updat 2011; 14:212 - 23; PMID: 21600837
- Oliveras-Ferraros C, Cufí S, Vazquez-Martin A, Torres-Garcia VZ, Del Barco S, Martin-Castillo B, et al. Micro(mi)RNA expression profile of breast cancer epithelial cells treated with the anti-diabetic drug metformin: induction of the tumor suppressor miRNA let-7a and suppression of the TGFβ-induced oncomiR miRNA-181a. Cell Cycle 2011; 10:1144 - 51; http://dx.doi.org/10.4161/cc.10.7.15210; PMID: 21368581
- Liu JC, Voisin V, Bader GD, Deng T, Pusztai L, Symmans WF, et al. Seventeen-gene signature from enriched Her2/Neu mammary tumor-initiating cells predicts clinical outcome for human HER2+:ERα- breast cancer. Proc Natl Acad Sci USA 2012; 109:5832 - 7; http://dx.doi.org/10.1073/pnas.1201105109; PMID: 22460789
- Giordano A, Gao H, Anfossi S, Cohen E, Mego M, Lee BN, et al. Epithelial-Mesenchymal Transition and Stem Cell Markers in Patients with HER2-Positive Metastatic Breast Cancer. Mol Cancer Ther 2012; 11:2526 - 34; http://dx.doi.org/10.1158/1535-7163.MCT-12-0460; PMID: 22973057
- Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA 2010; 107:15449 - 54; http://dx.doi.org/10.1073/pnas.1004900107; PMID: 20713713
- Kim H, Watkinson J, Varadan V, Anastassiou D. Multi-cancer computational analysis reveals invasion-associated variant of desmoplastic reaction involving INHBA, THBS2 and COL11A1. BMC Med Genomics 2010; 3:51; http://dx.doi.org/10.1186/1755-8794-3-51; PMID: 21047417
- Anastassiou D, Rumjantseva V, Cheng W, Huang J, Canoll PD, Yamashiro DJ, et al. Human cancer cells express Slug-based epithelial-mesenchymal transition gene expression signature obtained in vivo. BMC Cancer 2011; 11:529; http://dx.doi.org/10.1186/1471-2407-11-529; PMID: 22208948
- Brenton JD, Carey LA, Ahmed AA, Caldas C. Molecular classification and molecular forecasting of breast cancer: ready for clinical application?. J Clin Oncol 2005; 23:7350 - 60; http://dx.doi.org/10.1200/JCO.2005.03.3845; PMID: 16145060
- Generali D, Berruti A, Foroni C, Bazzola L, Andreis D, Allevi G, et al. Molecular oncology and the neoadjuvant setting: the perfect blend for treatment personalization and clinical trial design. J Natl Cancer Inst Monogr 2011; 2011:67 - 70; http://dx.doi.org/10.1093/jncimonographs/lgr029; PMID: 22043044
- Inoue A, Seidel MG, Wu W, Kamizono S, Ferrando AA, Bronson RT, et al. Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell 2002; 2:279 - 88; http://dx.doi.org/10.1016/S1535-6108(02)00155-1; PMID: 12398892
- Kajita M, McClinic KN, Wade PA. Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol Cell Biol 2004; 24:7559 - 66; http://dx.doi.org/10.1128/MCB.24.17.7559-7566.2004; PMID: 15314165
- Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM, et al. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell 2005; 123:641 - 53; http://dx.doi.org/10.1016/j.cell.2005.09.029; PMID: 16286009
- Leroy P, Mostov KE. Slug is required for cell survival during partial epithelial-mesenchymal transition of HGF-induced tubulogenesis. Mol Biol Cell 2007; 18:1943 - 52; http://dx.doi.org/10.1091/mbc.E06-09-0823; PMID: 17344479
- Valdés F, Alvarez AM, Locascio A, Vega S, Herrera B, Fernández M, et al. The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor Beta in fetal rat hepatocytes. Mol Cancer Res 2002; 1:68 - 78; PMID: 12496370
- Vega S, Morales AV, Ocaña OH, Valdés F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev 2004; 18:1131 - 43; http://dx.doi.org/10.1101/gad.294104; PMID: 15155580
- Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development 2005; 132:3151 - 61; http://dx.doi.org/10.1242/dev.01907; PMID: 15983400
- Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, et al. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res 2006; 8:R59; http://dx.doi.org/10.1186/bcr1610; PMID: 17062128
- Honeth G, Bendahl PO, Ringnér M, Saal LH, Gruvberger-Saal SK, Lövgren K, et al. The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res 2008; 10:R53; http://dx.doi.org/10.1186/bcr2108; PMID: 18559090
- Ricardo S, Vieira AF, Gerhard R, Leitão D, Pinto R, Cameselle-Teijeiro JF, et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Pathol 2011; 64:937 - 46; http://dx.doi.org/10.1136/jcp.2011.090456; PMID: 21680574
- Laakso M, Loman N, Borg A, Isola J. Cytokeratin 5/14-positive breast cancer: true basal phenotype confined to BRCA1 tumors. Mod Pathol 2005; 18:1321 - 8; http://dx.doi.org/10.1038/modpathol.3800456; PMID: 15990899
- van de Rijn M, Perou CM, Tibshirani R, Haas P, Kallioniemi O, Kononen J, et al. Expression of cytokeratins 17 and 5 identifies a group of breast carcinomas with poor clinical outcome. Am J Pathol 2002; 161:1991 - 6; http://dx.doi.org/10.1016/S0002-9440(10)64476-8; PMID: 12466114
- Liu H, Fan Q, Zhang Z, Li X, Yu H, Meng F. Basal-HER2 phenotype shows poorer survival than basal-like phenotype in hormone receptor-negative invasive breast cancers. Hum Pathol 2008; 39:167 - 74; http://dx.doi.org/10.1016/j.humpath.2007.06.012; PMID: 18045647
- Bagaria SP, Ray PS, Wang J, Kropcho L, Chung A, Sim MS, et al. Prognostic value of basal phenotype in HER2-overexpressing breast cancer. Ann Surg Oncol 2012; 19:935 - 40; http://dx.doi.org/10.1245/s10434-011-2032-5; PMID: 21879270
- Tripathi MK, Misra S, Chaudhuri G. Negative regulation of the expressions of cytokeratins 8 and 19 by SLUG repressor protein in human breast cells. Biochem Biophys Res Commun 2005; 329:508 - 15; http://dx.doi.org/10.1016/j.bbrc.2005.02.006; PMID: 15737616
- Li XY, Zhou X, Rowe RG, Hu Y, Schlaepfer DD, Ilić D, et al. Snail1 controls epithelial-mesenchymal lineage commitment in focal adhesion kinase-null embryonic cells. J Cell Biol 2011; 195:729 - 38; http://dx.doi.org/10.1083/jcb.201105103; PMID: 22105351