Abstract
Induced pluripotent stem (iPS) cells share some basic properties, such as self-renewal and pluripotency, with cancer cells, and they also appear to share several metabolic alterations that are commonly observed in human tumors. The cancer cells’ glycolytic phenotype, first reported by Otto Warburg, is necessary for the optimal routing of somatic cells to pluripotency. However, how iPS cells establish a Warburg-like metabolic phenotype and whether the metabolic pathways that support the bioenergetics of iPS cells are produced by the same mechanisms that are selected during the tumorigenic process remain largely unexplored. We recently investigated whether the reprogramming-competent metabotype of iPS cells involves changes in the activation/expression status of the H+-ATPase, which is a core component of mitochondrial oxidative phosphorylation that is repressed at both the activity and protein levels in human carcinomas, and of the lipogenic switch, which refers to a marked overexpression and hyperactivity of the acetyl-CoA carboxylase (ACACA) and fatty acid synthase (FASN) lipogenic enzymes that has been observed in nearly all examined cancer types. A comparison of a starting population of mouse embryonic fibroblasts and their iPS cell progeny revealed that somatic cell reprogramming involves a significant increase in the expression of ATPase inhibitor factor 1 (IF1), accompanied by extremely low expression levels of the catalytic β-F1-ATPase subunit. The pharmacological inhibition of ACACA and FASN activities markedly decreases reprogramming efficiency, and ACACA and FASN expression are notably upregulated in iPS cells. Importantly, iPS cells exhibited a significant intracellular accumulation of neutral lipid bodies; however, these bodies may be a reflection of intense lysosomal/autophagocytic activity rather than bona fide lipid droplet formation in iPS cells, as they were largely unresponsive to pharmacological modulation of PPARgamma and FASN activities. The AMPK agonist metformin, which endows somatic cells with a bioenergetic infrastructure that is protected against reprogramming, was found to drastically elongate fibroblast mitochondria, fully reverse the high IF1/β-F1-ATPase ratio and downregulate the ACACA/FASN lipogenic enzymes in iPS cells. The mitochondrial H+-ATP synthase and the ACACA/FASN-driven lipogenic switch are newly characterized as instrumental metabolic events that, by coupling the Warburg effect to anabolic metabolism, enable de-differentiation during the reprogramming of somatic cells to iPS cells.
The enforced aerobic glycolysis that generally accompanies the metabolic reprogramming of cancer cells is also a fundamental phenotypic trait of induced pluripotent stem (iPS) cells. Indeed, the glycolytic phenotype of cancer cells—first reported by Otto Warburg, who suggested that the increased glucose consumption of cancer cells under aerobic conditions might result from an impairment in the bioenergetic activity of their mitochondria—is necessary for the optimal routing of somatic cells to pluripotency.Citation1-Citation9 Somatic mitochondria within iPS cells drastically alter their morphology and functionality to acquire embryonic features, including an immature organelle shape with underdeveloped cristae and low levels of oxidative stress. These changes impact the cellular bioenergetic profile, which is shifted from oxidative phosphorylation (OXPHOS) to glycolysis upon reprogramming, and returns to OXPHOS during subsequent iPS cell differentiation. However, it remains unclear how iPS cells establish a Warburg-like metabolic phenotype and whether the metabolic pathways that support the bioenergetics of iPS cells are a direct or indirect result of the same mechanisms that are selected during the tumorigenic process.
Mitochondrial H+-ATP Synthase-Mediated Energy Adaption: A Shared Role in Carcinogenesis and in Induced Pluripotency
The fuel-sensing enzyme AMP-activated protein kinase is one regulator of bioenergetic metabolism that may account for the activation and/or maintenance of the Warburg effect in iPS cells. Pluripotent stem cells exhibit downregulated expression of the PRKAA1 gene, which encodes the catalytic subunit of AMPK.Citation5 Conversely, the pharmacological activation of AMPK has been shown to establish a metabolic barrier to somatic cell reprogramming that cannot be bypassed even through p53 deficiency, which is a fundamental mechanism used to greatly improve the efficiency of stem cell production.Citation9 In this scenario, we hypothesized that iPS cells might acquire distinctive AMPK-related bioenergetic signatures by employing molecular strategies that similarly induce and maintain the repression of mitochondrial OXPHOS in cancer cells.
On one hand, many tumor cells have developed mechanisms to reduce AMPK activation and, thus, escape from the growth-restraining effects of AMPK.Citation10-Citation12 Accordingly, AMPK underexpression is frequently observed in human carcinomas, and AMPK inactivation promotes carcinogenesis of epithelial cells.Citation13 On the other hand, recent pre-clinical and clinical findings have indicated that the mitochondrial H+-ATP synthase, a reversible engine in the inner mitochondrial membrane that regulates energy conservation by synthesizing or hydrolyzing ATP in response to changes in metabolic cellular conditions, is repressed at both the activity and protein levels in human carcinomas.Citation14,Citation15 The overexpression of the ATPase inhibitor factor 1 (IF1) in both normal and cancer cells limits the activity of H+-ATP synthase and triggers the metabolic switch to an enhanced aerobic glycolysis; the silencing of IF1 has the opposite metabolic effects.Citation16,Citation17 The expression of IF1 is negligible in normal tissues, and IF1 is highly overexpressed in numerous carcinomas, which is sufficient to limit the activity of H+-ATP synthase and promote the acquisition of the Warburg phenotype without any genetic changes. The cellular content of H+-ATP synthase, which directly correlates with OXPHOS activity and inversely correlates with the rate of glucose utilization by aerobic glycolysis,Citation18 can be regulated either by the translational silencing of the mRNA coding for the catalytic β-F1-ATPase subunit via interaction with β-mRNA-binding proteins (e.g., HuR and G3BP1)Citation19-Citation21 or by limiting β-mRNA transcription through hypermethylation of the β-F1-ATPase gene (ATP5B) promoter.Citation22 To explore a scenario in which AMPK is expected to play a central role during the metabolic reprogramming of somatic cells to iPS cells, we delineated a testable working model in which AMPK might interfere with H+-ATP synthase during the short-term (i.e., H+-ATP synthase activity) or long-term (i.e., H+-ATP synthase expression) to repress the somatic mitochondrial OXPHOS, which, in turn, would functionally link the acquisition of the Warburg phenotype with the maintenance of stemness in iPS cells.
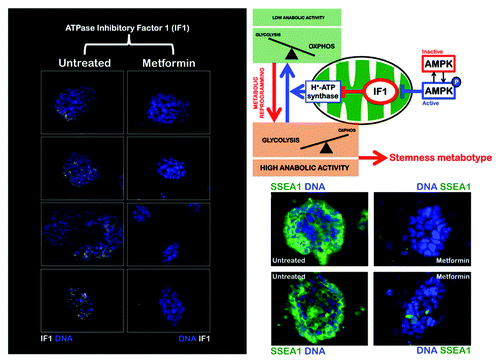
The IF1 ATPase inhibitor is highly expressed in iPS cells
To test this model, we employed iPS cells derived from mouse embryonic fibroblasts (MEFs) and the AMPK agonist metformin. iPS clones were initially selected by morphological criteria, i.e., flat colonies that were composed of small cells with a high nucleus to cytoplasm ratio and prominent nucleoli. The iPS clones were then further evaluated by immunostaining for stemness markers, including alkaline phosphatase (AP), Ssea-1, Oct4, Sox2 and Nanog, as well as for their ability to form bona fide teratomas (data not shown). First, we investigated whether the pharmacological agonism of AMPK activity impacted the expression status of the IF1 ATPase inhibitor. Immunofluorescence microscopy studies revealed abundant IF1 within the small amount of cytoplasm in iPS cells (). It is tempting to suggest that IF1 provides a greater protection of iPS cells against energy dissipation upon H+-ATP synthase reversal for several reasons: (1) IF1 is predominantly compartmentalized inside the mitochondrial matrix; (2) IF1 interacts with the catalytic subunit of H+-ATP synthase, thereby inhibiting the hydrolysis of ATP under conditions that favor the reversion of the enzyme activity; and (3) increased IF1 protein expression is associated with greater H+-ATP synthase binding efficiency.Citation23 However, it should be noted that the binding of IF1 to β-F1-ATPase is regulated not only by the energetic state of the mitochondria, but also by the mass action ratio of these two proteins. Thus, in situations in which mitochondria are scarce and IF1 expression is significantly increased, as we have demonstrated occurs in iPS cells, it is likely that the IF1 protein inhibits both the synthetic and hydrolytic activities of the H+-ATP synthase.Citation23 Therefore, the high mitochondrial content of IF1 in iPS cells may negatively control the activity of OXPHOS, thus mediating and/or ensuring the metabolic shift of iPS cells to enhanced aerobic glycolysis while guaranteeing iPS cell viability by preventing mitochondria from becoming ATP consumers. Importantly, exposure to the AMPK agonist metformin resulted in drastically decreased levels of IF1, accompanied by reductions in the size of iPS colonies (). Although the mechanisms that trigger the upregulation of IF1 in human carcinomas are presently unknown, the present findings provide the first indication that AMPK can operate as an upstream regulator of IF1 in iPS cells. These observations reveal a previously unrecognized AMPK-dependent metabolic axis that may account for the promotion of the metabolic switch in iPS cells by acting directly at the level of mitochondrial OXPHOS activity.
Figure 1. The AMPK agonist metformin suppresses the upregulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATPase in iPS colonies. iPS cells were maintained in an undifferentiated stage on gelatin-coated tissue culture surfaces in the presence of LIF. After 48 h of treatment with vehicle or 10 mmol/L metformin, IF1 protein levels (white staining) were analyzed by immunofluorescent confocal microscopy. DNA was counterstained with Hoechst 33258 (blue). Images are representative of five independent experiments testing two individual iPS clones. Images show also representative images of untreated and metformin-treated iPS colonies that were captured using different channels for SSEA-1 (green) or Hoechst 33258 (blue).
AMPK regulates the expression status of the catalytic β-F1-ATPase subunit in iPS cells
Second, we evaluated whether the pharmacological agonism of AMPK impacted the expression of the catalytic β-F1-ATPase subunit. A specific repression of β-F1-ATPase, which is the rate-limiting component of mitochondrial OXPHOS, occurred in iPS colonies; this phenomenon is also observed in rat hepatocarcinomas and human tumors.Citation15 A comparison of the starting population of MEFs and their iPS cell progeny using immunofluorescence microscopy clearly revealed that somatic cell reprogramming was accompanied by a significant decrease in the cellular content of the catalytic β-F1-ATPase subunit. Thus, whereas the cytoplasmic accumulation of β-F1-ATPase was prominent in MEFs, the β-F1-ATPase content within individual iPS cells was significantly reduced (). Importantly, metformin treatment promoted a significant augmentation of the β-F1-ATPase content within the small cytoplasm of individual iPS cells ().
Figure 2. The AMPK agonist metformin upregulates the expression of the catalytic β-F1-ATPase subunit, the rate-limiting component of mitochondrial OXPHOS in iPS colonies. iPS cells were maintained in an undifferentiated stage on gelatin-coated tissue culture surfaces in the presence of LIF. After 48 h of treatment with vehicle or 10 mmol/L metformin, β-F1-ATPase protein levels (white staining) were analyzed by immunofluorescent confocal microscopy. DNA was counterstained with Hoechst 33258 (blue). Images are representative of five independent experiments testing two individual iPS clones. Figure also shows representative images of untreated and metformin-treated iPS colonies that were captured using different channels for SSEA-1 (green) or Hoechst 33258 (blue).
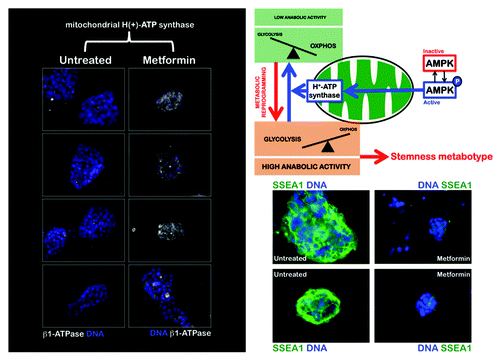
We next wanted to assess whether the alteration of the mitochondrial H+-ATPase-geared switch of energy metabolism impacted the pluripotent status of iPS cells. To this end, we monitored the expression status of the early embryonic antigen, SSEA-1, an initial stemness marker gene absent in parental fibroblasts that becomes activated at early time points during the reprogramming of somatic cells in response to pharmacological agonism of AMPK activity.Citation24,Citation25 iPS cells maintained in an undifferentiated stage on Matrigel-coated dishes in the presence of leukemia inhibitory factor (LIF) were exogenously supplemented with metformin for 48 h. Remarkably, metformin treatment at concentrations that drastically regulated the activity/expression status of H+-ATPase, significantly induced a switch to a SSEA1-negative state in iPS cell colonies ( and ).
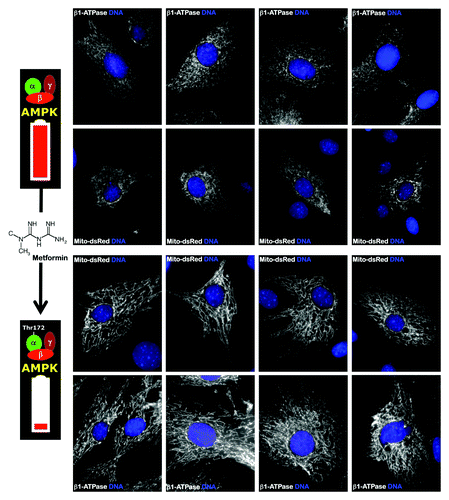
Three independent mechanisms affect the overall bioenergetic activity of H+-ATP synthase in cancer. One mechanism limits the activity of the complex, and the two remaining mechanisms limit the tumor content of the catalytic β-F1-ATPase. The present findings represent the first evidence that the same mechanisms that control the activity and expression of the mitochondrial H+-ATPase in cancer cells also operate in iPS cells. These results provide preliminary mechanistic indications that can explain the abnormal biogenesis and/or bioactivity of mitochondria in iPS cells. Because AMPK activity tightly regulates the activity and/or content of the essential mitochondrial H+-ATPase, it is reasonable to suggest that metabolic switches in iPS cells involve the inhibition of AMPK expression and/or activity to allow the correct reprogramming of the metabolic machinery that is required to fuel stemness and pluripotency. Indeed, our previous findings showed that AMPK agonists endow somatic cells with a bioenergetic infrastructure that is protected against reprogramming,Citation9 and they also showed that this protection could be explained by the upregulation of OXPHOS machinery that is refractory to nuclear reprogramming. Interestingly, treatment of MEFs with the AMPK agonist metformin, which is known to notably reduce the efficiency of reprogramming and to promote mitochondrial biogenesis,Citation26-Citation28 stimulated mitochondrial elongation through a mechanism that apparently involves an unobstructed mitochondrial fusion process ().Citation29,Citation30 Elongated mitochondria have more cristae, increased levels of dimerization and increased H+-ATPase activity, and they also maintain ATP production and are protected from autophagic degradation.Citation29,Citation30 Indeed, metformin treatment counterintuitively recapitulates both the morphological and functional effects of the mitochondrial division inhibitor mdivi-1. Mdivi-1 is a pharmacological inhibitor of mitochondrial fission that rapidly induces the formation of mitochondrial net-like or collapsed perinuclear mitochondrial structures that largely impede somatic cell reprogramming to pluripotency.Citation31 Therefore, the inhibition of mitochondrial division, by either unrestricted fusion or fission inhibition, has similar effects on the reprogramming factor-driven transcriptional network, preventing the epigenetic events that specify the unique phenotype of iPS cells.
Figure 3. The AMPK agonist metformin induces mitochondrial biogenesis and elongation in MEFs. Low-passage MEFs were cultured in the absence or presence of 10 mmol/L metformin for 48 h. Mitochondrial morphology and organization was detected by transient transfection with mito-DsRed or by staining with an antibody against the β1-subunit of the mitochondrial F1-ATPase complex. Figure shows representative images of untreated or metformin-treated MEFs that were captured using different channels for mito-DsRed or β1-ATPase (white) or Hoechst 33258 (blue). Note that metformin-treated MEFs showed significantly more organized and elongated mitochondria.
The Lipogenic Switch and Somatic Cell Reprogramming: The Anabolic Side of the Warburg Effect
While it may be speculated that fast-growing cancer and iPS cells will require more energy than normal cells, it might appear counterintuitive that cancer and iPS cells preferentially use a more primitive and inefficient reaction, aerobic glycolysis, to generate high amounts of energy. One possible reason for this bioenergetic alteration is that other metabolic end products of aerobic glycolysis are required to support the rapid growth and proliferation of these cells.Citation32-Citation36 Rapid cell growth requires the active synthesis of proteins, rRNA and lipids, all of which are switched off by the activation of AMPK. As suggested by Prigione et al.,Citation1 iPS cells may suppress the activation of AMPK, which is a master regulator of energy homeostasis that can switch off biosynthetic pathways to avoid anabolic inhibition, similar to the phenomenon observed in cancer cells.
The de novo biosynthesis of fatty acids is a particularly active anabolic process in proliferating cells, including tumor cells. Indeed, the activation of endogenous fatty acid biogenesis is increasingly recognized as a hallmark of aggressive cancers.Citation36-Citation41 Increased lipid production has been repeatedly linked to an increased need for membrane synthesis during rapid cell proliferation and is considered to be part of a more general metabolic transformation that provides cancer cells with more autonomy and a greater supply of building blocks for growth. However, the role of tumor-associated lipogenesis may extend beyond bulk membrane biosynthesis to meet the needs of rapid cell proliferation. We and others have demonstrated that protection from cell death and the activation of growth factor signaling are just a few of the newly recognized roles of the lipogenic phenotype in cancer cells.Citation42-Citation46 Moreover, shifting lipid acquisition from lipid uptake toward de novo lipogenesis enables cancer cells to dramatically alter their membrane properties and protects tumor cells from both endogenous and exogenous insults.Citation47 As lipogenicity increases, the degree of lipid saturation also decreases, and monounsaturated lipids are less susceptible to lipid peroxidation; together, these observations indicate that the lipogenic switch may protect cancer from free radicals.Citation47 We speculated that iPS cells must create an AMPK-regulated cellular state characterized not only by suppressing the mitochondria-related oxidative stress pathway, but also by evoking a high rate of lipid synthesis to overcome the detrimental effects of toxic free radical generation.
The pivotal lipogenic enzymes acetyl-CoA carboxylase (ACACA) and fatty acid synthase (FASN) are upregulated in iPS cells
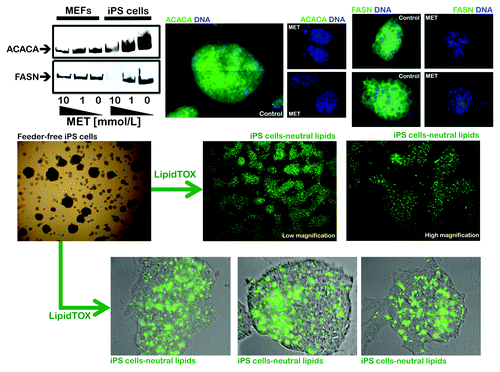
We first assessed whether somatic cell reprogramming significantly impacted the expression levels of the pivotal lipogenic proteins ACACA and FASN. A comparison of the starting population of mouse embryonic fibroblasts (MEFs) and their iPS cell progeny using immunoblotting and immunofluorescence microscopy techniques clearly revealed that somatic cell reprogramming is accompanied by a significant upregulation of the ACACA and FASN proteins (, top). Indeed, cytoplasmic accumulation of ACACA and FASN was highly prominent in bona fide iPS clones. In contrast, unreprogrammed control cultures of MEFs were almost negative for ACACA and FASN staining (data not shown).
Figure 4. Coordinate activation of lipogenic enzymes in iPS cells. Top: Robust and metformin-sensitive expression of the lipogenic enzymes acetyl-CoA carboxylase (ACACA) and fatty acid synthase (FASN) in iPS cells as measured by western blot (left) and immunofluorescence microscopy (right). Note that iPS cells express significantly higher levels of ACACA and FASN than MEFs. Figure shows also representative images of untreated and metformin-treated (10 mmol/L; 48 h) iPS cells that were captured using different channels for ACACA or FASN (green) or Hoechst 33258 (blue). Bottom: The intracellular accumulation of neutral lipids in iPS cells was evaluated with LipidTOX™ Green neutral lipid stain. The significant neutral lipid aggregates within iPS cells might reflect accumulation of organelles of lysosomal/autophagic origin.
iPS cells accumulate neutral lipid bodies
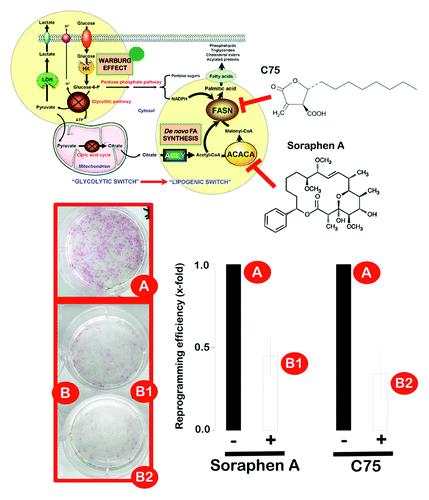
ACACA produces malonyl-CoA, an intermediate metabolite that functions as a substrate for fatty acid synthesis and as a negative regulator of fatty acid oxidation. The ACACA-catalyzed synthesis of malonyl-CoA is the first committed step of fatty acid synthesis and is the most highly regulated step of de novo lipogenesis.Citation48,Citation49 In contrast, FASN is the principal biosynthetic enzyme involved in fatty acid synthesis, which catalyzes the NADPH-dependent condensation of malonyl-CoA and acetyl-CoA to predominantly produce palmitate, a 16-carbon-saturated free fatty acid.Citation50,Citation51 However, it should be acknowledged that the accumulation of fatty acids and neutral lipids in non-adipose cells may be cytotoxic. Excess intracellular free fatty acids can disrupt phospholipid bilayer membrane integrity, alter signaling pathway activity and induce apoptotic cell death. In this scenario, we sought to determine whether somatic cell reprogramming established a lipogenesis/lipolysis-joining point that enabled iPS cells to circumvent endogenous palmitate toxicity while securing palmitate into fat stores to avoid a negative palmitate feedback on FASN function.Citation36,Citation52,Citation53 We performed fluorescent high-content imaging assays using the fluorescent LipidTOX™ Green neutral lipid stain, which has an extremely high affinity for neutral lipid deposits. Compared with unreprogrammed MEFs, iPS cells exhibited intense LipidTOX™ Green staining (, bottom). Although several fixation methods for the study of lipid droplets by immunofluorescence microscopy have been tested,Citation54 fixation with paraformaldehyde, which was performed here, has been described as the method of choice, because cells retain their lipid content, and the structure of cytoplasmic neutral lipid deposits is unaffected by this fixation method.Citation55,Citation56 Together, these assays initially suggested that the somatic reprogramming to stemness activates the expression of pivotal lipogenic enzymes (i.e., ACACA and FASN) and actively promotes the conversion and storage of excess fatty acids to triglycerides, which accumulate as lipid droplet-like neutral lipid depots. As a preliminary test of this counterintuitive hypothesis, bona fide iPS colonies maintained in an undifferentiated state were cultured in the presence of bisphenol A diglycidyl ether (BADGE), a synthetic antagonist of the prime inducer of adipogenesis, PPARgamma, which has been shown to regulate LIF-induced growth and self-renewal of mouse embryonic stem (ES) cells.Citation57-Citation79 BADGE treatment, which is well-known to potently inhibit lipid droplet formation in several experimental systems,Citation60,Citation61 slightly reduced the size of iPS colonies but largely failed to affect the levels of LipidTOX-positive intracellular bodies (data not shown). The pharmacological suppression of the lipogenic activity of FASN using a synthetic, chemically stable inhibitor of FASN that inactivates the β-ketoacyl synthase (3-oxoacyl synthase), enoyl reductase and thioesterase partial activities of FASN,Citation62-Citation66 similarly failed to significantly alter the accumulation of LipidTOX-positive intracellular bodies in iPS cells (data not shown). To address the functional effects of a deficit in de novo fatty acid biogenesis on the generation of iPS cells, we performed experiments using the three-factor (Oct4, Sox2 and Klf4) induction protocol in early-passage MEFs in the absence or presence of ACACA- and FASN-directed blockers. MEFs were first transduced with individual lentiviruses encoding Oct4, Sox2 and Klf4 at a 1:1:1 ratio on day 0, and the transduction was repeated every 12 h for 2 d using the same batches of all three lentiviruses. On day three, after the first transduction, the culture medium was switched to human ES cell growth medium with or without two drugs that have been shown to reverse the lipogenic phenotype in cancer cells: the FASN inhibitor C75 and Soraphen A, a naturally occurring macrocyclic polyketide that functions as a highly potent blocker of ACACA activity by inhibiting the biotin carboxylase domain of ACACA at nanomolar concentrations.Citation67-Citation72 Importantly, the early pharmacological manipulation of endogenous lipogenesis significantly impacted the generation of iPS cells. The treatment of MEFs at the early stages of reprogramming (i.e., before the appearance of iPS colonies) with non-cytotoxic concentrations of Soraphen A or C75, was sufficient to significantly reduce the efficiency of iPS colony formation by more than 50% (). Importantly, the pharmacological inhibition of endogenous lipogenesis after iPS colony appearance did not significantly affect the total number of colonies or the expression status of pluripotency markers in individual colonies (data not shown). These findings suggest that the observed effects of lipogenesis inhibition on reprogramming efficiency were likely due to the impairment of the reprogramming process itself and not to an impairment of iPSC colony survival or growth.
Figure 5. Pharmacological inhibition of endogenous lipogenesis decreases reprogramming efficiency. Early passage MEFs infected with retroviruses encoding Oct4, Sox2 and Klf4 (OSK) were cultured in ES medium in the continuous presence or absence of a non-cytotoxic concentration of soraphen A, C75 or DMSO alone as control, as specified. The numbers of alkaline phosphatase (AP)+ colonies (microphotographs of representative reprogramming experiments are shown) were counted 14 d after the initial infection and were plotted for each condition relative to the controls (x-fold), as specified. The error bars indicate the SEM.
The AMPK agonist metformin suppresses the expression of lipogenic enzymes in iPS cells
Finally, we examined whether the increased expression of the lipogenesis markers described above depended on the activation status of AMPK. We used the AMPK agonist metformin to indirectly explore the putative contribution of the AMPK pathway to the increases in FASN and ACACA expression during the reprogramming of MEFs to iPS cells. At concentrations that suppressed the expression of the early self-renewal marker, SSEA-1, metformin significantly and specifically blocked the increase in FASN and ACACA in iPS cells compared with MEFs (, top). Immunofluorescence analysis using confocal microscopy confirmed that metformin exposure drastically decreased the levels of the ACACA and FASN lipogenic proteins compared with vehicle-treated iPS cells.
The Mitochondrial H+-ATP Synthase and the Lipogenic Switch: New Core Components of Metabolic Reprogramming in iPS Cells
There are conspicuous similarities between the reprogramming of somatic cells to iPS cells and cancer development.Citation73-Citation76 To develop safe iPS-based tissue engineering and cell replacement therapies, much research in the field has focused on the tumorigenic traits of iPS cells. While unraveling how reprogramming strategies may avoid enhancing tumor risk, a better understanding of the molecular details underlying the reprogramming to pluripotency would provide crucial insights into how cancers might arise. Moreover, we can rapidly evaluate whether specific cancer cell traits also necessarily occur in iPS cells, thus indicating further potential routes to a normal and/or cancerous stem cell-like state. Recent studies have provided strong evidence that cancer cells are identical to their undifferentiated ancestors or embryonic stem cells, not only in terms of their metabolism, but also in terms of the molecular pathways they invoke.Citation32-Citation34,Citation73-Citation84
Here, we tested the hypothesis that the H+-ATPase synthase-geared metabolism switch,Citation15 a mitochondria-mediated energy adaptation that is sufficient to promote the acquisition of the Warburg phenotype, is also employed during somatic reprogramming to limit the bioenergetic activity of mitochondria and mediates the shift of iPS cells to enhanced aerobic glycolysis. As in the majority of cancer cells, in which the Warburg phenotype can be acquired without any genetic alteration, the peculiar energy metabolism of iPS cells is a “reversible trait” that is rapidly modulated in response to changes in AMPK activation status. Knoepfler’s group recently confirmed that oncogenic transformation and induced pluripotency are closely related processes.Citation85 Importantly, in their experimental model comparing transformation and cellular reprogramming, they observed that normal fibroblasts must first acquire changes that lead to a downregulation of cell differentiation machinery and a concomitant upregulation of glycolysis and other metabolic pathways; only then do the oncogenic transformation/induced pluripotency pathways diverge depending on other factors, such as the activity of pluripotency genes.Citation85 Thus, the present study adds to the growing body of knowledge that will aid the discovery of metabolic reprogramming-targeted methods for making iPS cells less tumorigenic. Our findings also support further exploration of the specific metabolic pathways that allow pluripotent cells to acquire oncogenic traits. We have recently observed that the AMPK agonist metformin, which endows somatic cells with a bioenergetic infrastructure that is largely refractory to reprogramming,Citation9 also prevents the occurrence of or drastically reduces the size and weight of teratoma-like masses after the transplantation of iPS cells into immunodeficient mice, but that iPS cells implanted into metformin-treated mice retain full pluripotency. The present hypothesis that metformin drastically elongates fibroblast mitochondria and fully reverses the cancer-like high IF1/β-F1-ATPase ratio in iPS cells may explain, at least in part, its ability to efficiently and specifically control the tumorigenic fate of teratoma-initiating iPS cells without interfering with their pluripotency. Further studies will more clearly elucidate the roles of the IF1/β-F1-ATPase ratio and of the H+-ATPase activation status on the creation and/or maintenance of cancer stem cell (CSC) cellular states.
We also tested for the first time whether the activation of de novo fatty acid biogenesis is an instrumental metabolic event that might enable the de-differentiation process during the reprogramming of somatic cells to iPS cells, beyond a cancer-like, energy metabolism switch toward increased glycolysis and decreased mitochondrial OXPHOS (i.e., the “Warburg effect”). Similar to some subsets of cancer cells, iPS cells supercharge lipogenesis by triggering regulatory circuits that activate and provide substrates for the lipogenic enzymes ACACA and FASN. Some aggressive cancers use oncogenic lipid metabolism for cell proliferation and survival, because fatty acid synthesis facilitates a Warburg-like glycolysis instead of mitochondrial OXPHOS for energy production.Citation36,Citation40,Citation86 Oxygen does not serve as the terminal electron acceptor in cells exhibiting Warburg-like glycolytic metabolism. To avoid low NAD+/NADH ratios that would eventually inhibit glycolysis through feedback mechanisms, electrons are incorporated into other molecules, such as lactate, with the concomitant regeneration of NAD+. Thus, de novo fatty acid synthesis enables a rapid regeneration of NAD+ by consuming large amounts of nicotinamide adenine dinucleotide phosphate (NADPH), which is necessary for the lipogenic pathway, because both synthesis and elongation of fatty acids use NADPH as a cofactor. As a result, the cells can continue to catabolize glucose and maintain high rates of glycolysis.Citation87-Citation89
iPS cells are not only metabolically altered to depend on glycolysis and fatty acid synthesis for energy production (i.e., by coupling the Warburg effect with anabolic metabolism) early in the reprogramming process, but also may establish further molecular mechanisms aimed to avert lipotoxicity and palmitate feedback on lipogenic activity. The previously unrecognized ability of iPS cells to accumulate significant amounts of neutral lipid depots recapitulates the ability of fat-addicted HER2-overexpressing cancer cells to promote the conversion and storage of excess fatty acids to triglycerides.Citation52,Citation53 The neutral fat stain BODIPY 493/503 shows that HER2-positive breast cancer cells exhibit approximately 20-fold higher levels of accumulated fat in lipid stores than normal human mammary epithelial cells.Citation52,Citation53 The neutral fat stain LipidTOX similarly revealed that iPS cells exhibit notably higher levels of accumulated fat in intracellular lipid stores. However, these lipid depots were fully refractory to changes in the activation status of the PPARgamma nuclear receptor, which is a central regulator of lipid droplet formation, and of the key lipogenic enzyme, FASN. Indeed, we failed to detect perilipin, a member of the perilipin family of structural lipid droplet proteins that stabilize lipid droplets and control lipolysis by localizing to the surface of intracellular neutral lipid droplets (data not shown).Citation90,Citation91 LipidTOX-positive lipid bodies may represent dense formations of “biological debris” within the iPS cells.Citation92 We are currently investigating whether the striking finding of dense formations co-localized with lipid staining, which were mainly observed in the perinuclear and peripheral area of iPS cells, might indeed reflect an intense lysosomal/autophagocytic activity within iPS cells. In contrast to the situation in some cancer cells, the metabolic reprogramming of iPS cells does not cause products of glycolysis to be stored as triglycerides by upregulating enzymes involved in fatty acid storage, but rather involves the accumulation of glycolytic byproducts, such as lactate and acetate.
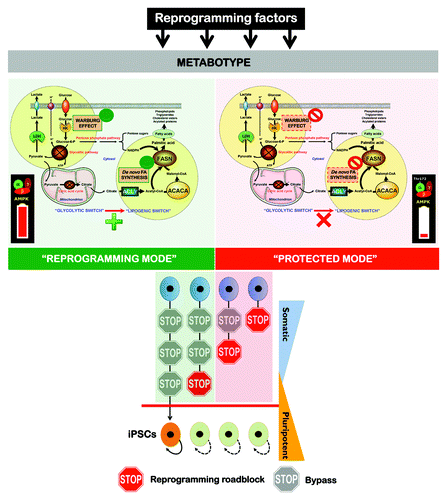
The occurrence of parallel metabolic changes in oncogenesis and the induction of pluripotency supports the notion that cell reprogramming is a naturally occurring phenomenon but not a mere technology.Citation93 Nevertheless, the present results strongly suggested that individual somatic cells’ ability to enter reprogramming at different time points after transgene induction and the length of time required to complete the reprogramming sequence is greatly impacted by the bioenergetic and anabolic status of the cell, namely the AMPK status (). AMPK activation allows fewer cells to undergo the required stochastic epigenetic events and consequently proceed to a fully reprogrammed state of transgene independence. Sustained mitochondrial OXPHOS and the suppression of cell anabolism in response to continuous AMPK activation will, therefore, reduce the number of cells that activate the endogenous expression of essential transcriptional factors that regulate self-renewal and pluripotency (e.g., Nanog and Oct4). We now propose an operating model in which the development of a Warburg-like and anabolic cellular metabotype is a crucial stochastic event that imposes an a priori roadblock on the path of transformation from somatic cells to pluripotent stem cells that does not require the participation of oncogene-driven metabolic changes. In this scenario, the remodeling of energy and biosynthetic metabolism is an active participant (rather than a consequence) in defining cell fate, and both the flexibility and reversibility of such metabolic roadblock, if similarly operational for transition among non-CSC to CSC cellular states, may have important implications for the pharmacological manipulation of the self-renewal and pluripotency underlying CSC-driven tumorigenesis.Citation94-Citation97
Figure 6. The Warburg effect and de novo fatty acid biogenesis: Metabolic reprogramming permits repression of differentiation in iPS cells. The a priori bioenergetic-anabolic signature of somatic cells correlates with their reprogramming efficiencies and with the acquisition of stemness properties. Cells that demonstrate an active glycolysis-lipogenesis axis reprogram more quickly and efficiently than those demonstrating a metabotype closer to the oxidative/non-lipogenic state of normal, non-proliferative somatic cells. We now reveal that, similarly to cancer cells, the Warburg effect in iPS cells can be established by decreasing the activity and expression of β-F1-ATPase, a key subunit of the mitochondrial ATP synthase. Furthermore, iPS-associated metabolic reprogramming also involves an exacerbated activation of ACACA- and FASN-catalyzed de novo-fatty acid synthesis. Activation of H+-ATPase and inhibition of endogenous lipogenesis can endow somatic cells with a metabolic infrastructure protected against reprogramming.
Acknowledgments
We are indebted to Prof. Carlos Lopez-Otin and his team [Departamento de Bioquímica y Biología Molecular, Facultad de Medicina, Instituto Universitario de Oncología (IUOPA), Universidad de Oviedo, Oviedo, Spain] for providing most of the reagents and descriptions of the procedures used in this work. Soraphen A, purified from the myxobacterium S. cellulosum, was provided by Drs. Klaus Gerth and Rolf Jansen (Hemholtz Zentrum für Infektionsforschung GmbH). This work was financially supported by the Instituto de Salud Carlos III [Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria (FIS), grants CP05-00090, PI06-0778 and RD06-0020-0028], the Fundación Científica de la Asociación Española Contra el Cáncer (AECC) and the Ministerio de Ciencia e Innovación (SAF2009-11579, Plan Nacional de I+D+ I, MICINN). A.V.-M. received a Sara Borrell post-doctoral contract (CD08/00283, Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria -FIS-). S.C. received a research fellowship (Formación de Personal Investigador, FPI) from the Ministerio de Ciencia e Innovación (MICINN).
References
- Prigione A, Fauler B, Lurz R, Lehrach H, Adjaye J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 2010; 28:721 - 33; http://dx.doi.org/10.1002/stem.404; PMID: 20201066
- Suhr ST, Chang EA, Tjong J, Alcasid N, Perkins GA, Goissis MD, et al. Mitochondrial rejuvenation after induced pluripotency. PLoS One 2010; 5:e14095; http://dx.doi.org/10.1371/journal.pone.0014095; PMID: 21124794
- Prigione A, Adjaye J. Modulation of mitochondrial biogenesis and bioenergetic metabolism upon in vitro and in vivo differentiation of human ES and iPS cells. Int J Dev Biol 2010; 54:1729 - 41; http://dx.doi.org/10.1387/ijdb.103198ap; PMID: 21305470
- Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CA 4th, Ramalho-Santos J, et al. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One 2011; 6:e20914; http://dx.doi.org/10.1371/journal.pone.0020914; PMID: 21698063
- Prigione A, Lichtner B, Kuhl H, Struys EA, Wamelink M, Lehrach H, et al. Human induced pluripotent stem cells harbor homoplasmic and heteroplasmic mitochondrial DNA mutations while maintaining human embryonic stem cell-like metabolic reprogramming. Stem Cells 2011; 29:1338 - 48; PMID: 21732474
- Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, et al. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 2011; 14:264 - 71; http://dx.doi.org/10.1016/j.cmet.2011.06.011; PMID: 21803296
- Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res 2012; 22:168 - 77; http://dx.doi.org/10.1038/cr.2011.177; PMID: 22064701
- Menendez JA, Vellon L, Oliveras-Ferraros C, Cufí S, Vazquez-Martin A. mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: a roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle 2011; 10:3658 - 77; http://dx.doi.org/10.4161/cc.10.21.18128; PMID: 22052357
- Vazquez-Martin A, Vellon L, Quirós PM, Cufí S, Ruiz de Galarreta E, Oliveras-Ferraros C, et al. Activation of AMP-activated protein kinase (AMPK) provides a metabolic barrier to reprogramming somatic cells into stem cells. Cell Cycle 2012; 11:974 - 89; http://dx.doi.org/10.4161/cc.11.5.19450; PMID: 22333578
- Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 2011; 25:1895 - 908; http://dx.doi.org/10.1101/gad.17420111; PMID: 21937710
- Hardie DG. Adenosine monophosphate-activated protein kinase: a central regulator of metabolism with roles in diabetes, cancer, and viral infection. Cold Spring Harb Symp Quant Biol 2011; 76:155 - 64; http://dx.doi.org/10.1101/sqb.2011.76.010819; PMID: 22071265
- Hardie DG. Sensing of energy and nutrients by AMP-activated protein kinase. Am J Clin Nutr 2011; 93:891S - 6; http://dx.doi.org/10.3945/ajcn.110.001925; PMID: 21325438
- Phoenix KN, Devarakonda CV, Fox MM, Stevens LE, Claffey KP. AMPKα2 suppresses murine embryonic fibroblast transformation and tumorigenesis. Genes Cancer 2012; 3:51 - 62; http://dx.doi.org/10.1177/1947601912452883; PMID: 22893790
- Cuezva JM, Sánchez-Aragó M, Sala S, Blanco-Rivero A, Ortega AD. A message emerging from development: the repression of mitochondrial beta-F1-ATPase expression in cancer. J Bioenerg Biomembr 2007; 39:259 - 65; http://dx.doi.org/10.1007/s10863-007-9087-9; PMID: 17712532
- Sánchez-Aragó M, Formentini L, Cuezva JM. Mitochondria-Mediated Energy Adaption in Cancer: The H(+)-ATP Synthase-Geared Switch of Metabolism in Human Tumors. Antioxid Redox Signal 2012; In press http://dx.doi.org/10.1089/ars.2012.4883; PMID: 22901241
- Sánchez-Cenizo L, Formentini L, Aldea M, Ortega AD, García-Huerta P, Sánchez-Aragó M, et al. Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype. J Biol Chem 2010; 285:25308 - 13; http://dx.doi.org/10.1074/jbc.M110.146480; PMID: 20538613
- Sánchez-Aragó M, Formentini L, García-Bermúdez J, Cuezva JM. IF1 reprograms energy metabolism and signals the oncogenic phenotype in cancer. Cell Cycle 2012; 11:2963 - 4; http://dx.doi.org/10.4161/cc.21387; PMID: 22871729
- Sánchez-Aragó M, Chamorro M, Cuezva JM. Selection of cancer cells with repressed mitochondria triggers colon cancer progression. Carcinogenesis 2010; 31:567 - 76; http://dx.doi.org/10.1093/carcin/bgq012; PMID: 20080835
- Willers IM, Cuezva JM. Post-transcriptional regulation of the mitochondrial H(+)-ATP synthase: a key regulator of the metabolic phenotype in cancer. Biochim Biophys Acta 2011; 1807:543 - 51; http://dx.doi.org/10.1016/j.bbabio.2010.10.016; PMID: 21035425
- Ortega AD, Sala S, Espinosa E, González-Barón M, Cuezva JM. HuR and the bioenergetic signature of breast cancer: a low tumor expression of the RNA-binding protein predicts a higher risk of disease recurrence. Carcinogenesis 2008; 29:2053 - 61; http://dx.doi.org/10.1093/carcin/bgn185; PMID: 18687667
- Ortega AD, Willers IM, Sala S, Cuezva JM. Human G3BP1 interacts with beta-F1-ATPase mRNA and inhibits its translation. J Cell Sci 2010; 123:2685 - 96; http://dx.doi.org/10.1242/jcs.065920; PMID: 20663914
- Li RJ, Zhang GS, Chen YH, Zhu JF, Lu QJ, Gong FJ, et al. Down-regulation of mitochondrial ATPase by hypermethylation mechanism in chronic myeloid leukemia is associated with multidrug resistance. Ann Oncol 2010; 21:1506 - 14; http://dx.doi.org/10.1093/annonc/mdp569; PMID: 20038517
- Faccenda D, Campanella M. Molecular Regulation of the Mitochondrial F(1)F(o)-ATPsynthase: Physiological and Pathological Significance of the Inhibitory Factor 1 (IF(1)). Int J Cell Biol 2012; 2012:367934; http://dx.doi.org/10.1155/2012/367934; PMID: 22966230
- Brambrink T, Foreman R, Welstead GG, Lengner CJ, Wernig M, Suh H, et al. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell 2008; 2:151 - 9; http://dx.doi.org/10.1016/j.stem.2008.01.004; PMID: 18371436
- Cox JL, Rizzino A. Induced pluripotent stem cells: what lies beyond the paradigm shift. Exp Biol Med (Maywood) 2010; 235:148 - 58; http://dx.doi.org/10.1258/ebm.2009.009267; PMID: 20404029
- Kukidome D, Nishikawa T, Sonoda K, Imoto K, Fujisawa K, Yano M, et al. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 2006; 55:120 - 7; http://dx.doi.org/10.2337/diabetes.55.01.06.db05-0943; PMID: 16380484
- Beeson CC, Beeson GC, Schnellmann RG. A high-throughput respirometric assay for mitochondrial biogenesis and toxicity. Anal Biochem 2010; 404:75 - 81; http://dx.doi.org/10.1016/j.ab.2010.04.040; PMID: 20465991
- Oliveras-Ferraros C, Cufí S, Vazquez-Martin A, Menendez OJ, Bosch-Barrera J, Martin-Castillo B, et al. Metformin rescues cell surface major histocompatibility complex class I (MHC-I) deficiency caused by oncogenic transformation. Cell Cycle 2012; 11:865 - 70; http://dx.doi.org/10.4161/cc.11.5.19252; PMID: 22333588
- Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 2011; 13:589 - 98; http://dx.doi.org/10.1038/ncb2220; PMID: 21478857
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 2011; 13:1016 - 23; http://dx.doi.org/10.1038/ncb2329; PMID: 21892142
- Vazquez-Martin A, Cufi S, Corominas-Faja B, Oliveras-Ferraros C, Vellon L, Menendez JA. Mitochondrial fusion by pharmacological manipulation impedes somatic cell reprogramming to pluripotency: new insight into the role of mitophagy in cell stemness. Aging (Albany NY) 2012; 4:393 - 401; PMID: 22713507
- Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 2009; 23:537 - 48; http://dx.doi.org/10.1101/gad.1756509; PMID: 19270154
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029 - 33; http://dx.doi.org/10.1126/science.1160809; PMID: 19460998
- Israël M, Schwartz L. The metabolic advantage of tumor cells. Mol Cancer 2011; 10:70; http://dx.doi.org/10.1186/1476-4598-10-70; PMID: 21649891
- Furuta E, Okuda H, Kobayashi A, Watabe K. Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochim Biophys Acta 2010; 1805:141 - 52; PMID: 20122995
- Menendez JA. Fine-tuning the lipogenic/lipolytic balance to optimize the metabolic requirements of cancer cell growth: molecular mechanisms and therapeutic perspectives. Biochim Biophys Acta 2010; 1801:381 - 91; http://dx.doi.org/10.1016/j.bbalip.2009.09.005; PMID: 19782152
- Menendez JA, Lupu R. Fatty acid synthase-catalyzed de novo fatty acid biosynthesis: from anabolic-energy-storage pathway in normal tissues to jack-of-all-trades in cancer cells. Arch Immunol Ther Exp (Warsz) 2004; 52:414 - 26; PMID: 15577743
- Menendez JA, Lupu R, Colomer R. Targeting fatty acid synthase: potential for therapeutic intervention in her-2/neu-overexpressing breast cancer. Drug News Perspect 2005; 18:375 - 85; http://dx.doi.org/10.1358/dnp.2005.18.6.927929; PMID: 16247515
- Menendez JA, Lupu R. Oncogenic properties of the endogenous fatty acid metabolism: molecular pathology of fatty acid synthase in cancer cells. Curr Opin Clin Nutr Metab Care 2006; 9:346 - 57; http://dx.doi.org/10.1097/01.mco.0000232893.21050.15; PMID: 16778562
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007; 7:763 - 77; http://dx.doi.org/10.1038/nrc2222; PMID: 17882277
- Menendez JA, Vazquez-Martin A, Ortega FJ, Fernandez-Real JM. Fatty acid synthase: association with insulin resistance, type 2 diabetes, and cancer. Clin Chem 2009; 55:425 - 38; http://dx.doi.org/10.1373/clinchem.2008.115352; PMID: 19181734
- Menendez JA, Vellon L, Mehmi I, Oza BP, Ropero S, Colomer R, et al. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc Natl Acad Sci USA 2004; 101:10715 - 20; http://dx.doi.org/10.1073/pnas.0403390101; PMID: 15235125
- Menendez JA, Mehmi I, Verma VA, Teng PK, Lupu R. Pharmacological inhibition of fatty acid synthase (FAS): a novel therapeutic approach for breast cancer chemoprevention through its ability to suppress Her-2/neu (erbB-2) oncogene-induced malignant transformation. Mol Carcinog 2004; 41:164 - 78; http://dx.doi.org/10.1002/mc.20054; PMID: 15390078
- Menendez JA, Oza BP, Atlas E, Verma VA, Mehmi I, Lupu R. Inhibition of tumor-associated fatty acid synthase activity antagonizes estradiol- and tamoxifen-induced agonist transactivation of estrogen receptor (ER) in human endometrial adenocarcinoma cells. Oncogene 2004; 23:4945 - 58; http://dx.doi.org/10.1038/sj.onc.1207476; PMID: 15094777
- Menendez JA, Vellon L, Colomer R, Lupu R. Pharmacological and small interference RNA-mediated inhibition of breast cancer-associated fatty acid synthase (oncogenic antigen-519) synergistically enhances Taxol (paclitaxel)-induced cytotoxicity. Int J Cancer 2005; 115:19 - 35; http://dx.doi.org/10.1002/ijc.20754; PMID: 15657900
- Vazquez-Martin A, Colomer R, Brunet J, Lupu R, Menendez JA. Overexpression of fatty acid synthase gene activates HER1/HER2 tyrosine kinase receptors in human breast epithelial cells. Cell Prolif 2008; 41:59 - 85; http://dx.doi.org/10.1111/j.1365-2184.2007.00498.x; PMID: 18211286
- Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res 2010; 70:8117 - 26; http://dx.doi.org/10.1158/0008-5472.CAN-09-3871; PMID: 20876798
- Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care 2006; 9:358 - 65; http://dx.doi.org/10.1097/01.mco.0000232894.28674.30; PMID: 16778563
- Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, et al. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res 2007; 67:8180 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-07-0389; PMID: 17804731
- Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition 2000; 16:202 - 8; http://dx.doi.org/10.1016/S0899-9007(99)00266-X; PMID: 10705076
- Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res 2006; 66:5977 - 80; http://dx.doi.org/10.1158/0008-5472.CAN-05-4673; PMID: 16778164
- Kourtidis A, Srinivasaiah R, Carkner RD, Brosnan MJ, Conklin DS. Peroxisome proliferator-activated receptor-gamma protects ERBB2-positive breast cancer cells from palmitate toxicity. Breast Cancer Res 2009; 11:R16; http://dx.doi.org/10.1186/bcr2240; PMID: 19298655
- Kourtidis A, Jain R, Carkner RD, Eifert C, Brosnan MJ, Conklin DS. An RNA interference screen identifies metabolic regulators NR1D1 and PBP as novel survival factors for breast cancer cells with the ERBB2 signature. Cancer Res 2010; 70:1783 - 92; http://dx.doi.org/10.1158/0008-5472.CAN-09-1550; PMID: 20160030
- DiDonato D, Brasaemle DL. Fixation methods for the study of lipid droplets by immunofluorescence microscopy. J Histochem Cytochem 2003; 51:773 - 80; http://dx.doi.org/10.1177/002215540305100608; PMID: 12754288
- Grandl M, Schmitz G. Fluorescent high-content imaging allows the discrimination and quantitation of E-LDL-induced lipid droplets and Ox-LDL-generated phospholipidosis in human macrophages. Cytometry A 2010; 77:231 - 42; PMID: 20014301
- Vazquez-Martin A, Ortega-Delgado FJ, Fernandez-Real JM, Menendez JA. The tyrosine kinase receptor HER2 (erbB-2): from oncogenesis to adipogenesis. J Cell Biochem 2008; 105:1147 - 52; http://dx.doi.org/10.1002/jcb.21917; PMID: 18814184
- Wright HM, Clish CB, Mikami T, Hauser S, Yanagi K, Hiramatsu R, et al. A synthetic antagonist for the peroxisome proliferator-activated receptor gamma inhibits adipocyte differentiation. J Biol Chem 2000; 275:1873 - 7; http://dx.doi.org/10.1074/jbc.275.3.1873; PMID: 10636887
- Bishop-Bailey D, Hla T, Warner TD. Bisphenol A diglycidyl ether (BADGE) is a PPARgamma agonist in an ECV304 cell line. Br J Pharmacol 2000; 131:651 - 4; http://dx.doi.org/10.1038/sj.bjp.0703628; PMID: 11030710
- Mo C, Chearwae W, Bright JJ. PPARgamma regulates LIF-induced growth and self-renewal of mouse ES cells through Tyk2-Stat3 pathway. Cell Signal 2010; 22:495 - 500; http://dx.doi.org/10.1016/j.cellsig.2009.11.003; PMID: 19922793
- Inazawa Y, Nakatsu M, Yasugi E, Saeki K, Yuo A. Lipid droplet formation in human myeloid NB4 cells stimulated by all trans retinoic acid and granulocyte colony-stimulating factor: possible involvement of peroxisome proliferator-activated receptor gamma. Cell Struct Funct 2003; 28:487 - 93; http://dx.doi.org/10.1247/csf.28.487; PMID: 14745140
- Yun JW, Shin ES, Cho SY, Kim SH, Kim CW, Lee TR, et al. The effects of BADGE and caffeine on the time-course response of adiponectin and lipid oxidative enzymes in high fat diet-fed C57BL/6J mice: correlation with reduced adiposity and steatosis. Exp Anim 2008; 57:461 - 9; http://dx.doi.org/10.1538/expanim.57.461; PMID: 18946183
- Kuhajda FP, Pizer ES, Li JN, Mani NS, Frehywot GL, Townsend CA. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc Natl Acad Sci USA 2000; 97:3450 - 4; http://dx.doi.org/10.1073/pnas.97.7.3450; PMID: 10716717
- Pizer ES, Thupari J, Han WF, Pinn ML, Chrest FJ, Frehywot GL, et al. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res 2000; 60:213 - 8; PMID: 10667561
- Kuhajda FP, Landree LE, Ronnett GV. The connections between C75 and obesity drug-target pathways. Trends Pharmacol Sci 2005; 26:541 - 4; http://dx.doi.org/10.1016/j.tips.2005.09.002; PMID: 16169094
- Lupu R, Menendez JA. Pharmacological inhibitors of Fatty Acid Synthase (FASN)--catalyzed endogenous fatty acid biogenesis: a new family of anti-cancer agents?. Curr Pharm Biotechnol 2006; 7:483 - 93; http://dx.doi.org/10.2174/138920106779116928; PMID: 17168665
- Bentebibel A, Sebastián D, Herrero L, López-Viñas E, Serra D, Asins G, et al. Novel effect of C75 on carnitine palmitoyltransferase I activity and palmitate oxidation. Biochemistry 2006; 45:4339 - 50; http://dx.doi.org/10.1021/bi052186q; PMID: 16584169
- Gerth K, Bedorf N, Irschik H, Höfle G, Reichenbach H. The soraphens: a family of novel antifungal compounds from Sorangium cellulosum (Myxobacteria). I. Soraphen A1 alpha: fermentation, isolation, biological properties. J Antibiot (Tokyo) 1994; 47:23 - 31; http://dx.doi.org/10.7164/antibiotics.47.23; PMID: 8119858
- Vahlensieck HF, Pridzun L, Reichenbach H, Hinnen A. Identification of the yeast ACC1 gene product (acetyl-CoA carboxylase) as the target of the polyketide fungicide soraphen A. Curr Genet 1994; 25:95 - 100; http://dx.doi.org/10.1007/BF00309532; PMID: 7916271
- Shen Y, Volrath SL, Weatherly SC, Elich TD, Tong L. A mechanism for the potent inhibition of eukaryotic acetyl-coenzyme A carboxylase by soraphen A, a macrocyclic polyketide natural product. Mol Cell 2004; 16:881 - 91; http://dx.doi.org/10.1016/j.molcel.2004.11.034; PMID: 15610732
- Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, et al. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res 2007; 67:8180 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-07-0389; PMID: 17804731
- Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res 2010; 70:8117 - 26; http://dx.doi.org/10.1158/0008-5472.CAN-09-3871; PMID: 20876798
- Jump DB, Torres-Gonzalez M, Olson LK. Soraphen A, an inhibitor of acetyl CoA carboxylase activity, interferes with fatty acid elongation. Biochem Pharmacol 2011; 81:649 - 60; http://dx.doi.org/10.1016/j.bcp.2010.12.014; PMID: 21184748
- Brickman JM, Burdon TG. Pluripotency and tumorigenicity. Nat Genet 2002; 32:557 - 8; http://dx.doi.org/10.1038/ng1202-557; PMID: 12457185
- Knoepfler PS. Deconstructing stem cell tumorigenicity: a roadmap to safe regenerative medicine. Stem Cells 2009; 27:1050 - 6; http://dx.doi.org/10.1002/stem.37; PMID: 19415771
- Blum B, Benvenisty N. The tumorigenicity of human embryonic stem cells. Adv Cancer Res 2008; 100:133 - 58; http://dx.doi.org/10.1016/S0065-230X(08)00005-5; PMID: 18620095
- Ben-David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev Cancer 2011; 11:268 - 77; http://dx.doi.org/10.1038/nrc3034; PMID: 21390058
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis?. Nat Rev Cancer 2004; 4:891 - 9; http://dx.doi.org/10.1038/nrc1478; PMID: 15516961
- Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, et al. Glycolytic enzymes can modulate cellular life span. Cancer Res 2005; 65:177 - 85; PMID: 15665293
- Kondoh H, Lleonart ME, Nakashima Y, Yokode M, Tanaka M, Bernard D, et al. A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxid Redox Signal 2007; 9:293 - 9; http://dx.doi.org/10.1089/ars.2006.1467; PMID: 17184172
- Kondoh H, Lleonart ME, Bernard D, Gil J. Protection from oxidative stress by enhanced glycolysis; a possible mechanism of cellular immortalization. Histol Histopathol 2007; 22:85 - 90; PMID: 17128414
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 2008; 7:11 - 20; http://dx.doi.org/10.1016/j.cmet.2007.10.002; PMID: 18177721
- Kondoh H. Cellular life span and the Warburg effect. Exp Cell Res 2008; 314:1923 - 8; http://dx.doi.org/10.1016/j.yexcr.2008.03.007; PMID: 18410925
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21:297 - 308; http://dx.doi.org/10.1016/j.ccr.2012.02.014; PMID: 22439925
- Riggs JW, Barrilleaux BL, Varlakhanova N, Bush KM, Chan V, Knoepfler PS. Induced Pluripotency and Oncogenic Transformation Are Related Processes. Stem Cells Dev 2012; In press PMID: 22998387
- Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, et al. Metformin: multi-faceted protection against cancer. Oncotarget 2011; 2:896 - 917; PMID: 22203527
- Menendez JA, Colomer R, Lupu R. Why does tumor-associated fatty acid synthase (oncogenic antigen-519) ignore dietary fatty acids?. Med Hypotheses 2005; 64:342 - 9; http://dx.doi.org/10.1016/j.mehy.2004.07.022; PMID: 15607569
- Folmes CD, Nelson TJ, Terzic A. Energy metabolism in nuclear reprogramming. Biomark Med 2011; 5:715 - 29; http://dx.doi.org/10.2217/bmm.11.87; PMID: 22103608
- Folmes CD, Nelson TJ, Dzeja PP, Terzic A. Energy metabolism plasticity enables stemness programs. Ann N Y Acad Sci 2012; 1254:82 - 9; http://dx.doi.org/10.1111/j.1749-6632.2012.06487.x; PMID: 22548573
- Tansey JT, Sztalryd C, Hlavin EM, Kimmel AR, Londos C. The central role of perilipin a in lipid metabolism and adipocyte lipolysis. IUBMB Life 2004; 56:379 - 85; http://dx.doi.org/10.1080/15216540400009968; PMID: 15545214
- Brasaemle DL. Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res 2007; 48:2547 - 59; http://dx.doi.org/10.1194/jlr.R700014-JLR200; PMID: 17878492
- Chen PM, Gombart ZJ, Chen JW. Chloroquine treatment of ARPE-19 cells leads to lysosome dilation and intracellular lipid accumulation: possible implications of lysosomal dysfunction in macular degeneration. Cell Biosci 2011; 1:10; http://dx.doi.org/10.1186/2045-3701-1-10; PMID: 21711726
- Zhang Y, Yao L, Yu X, Ou J, Hui N, Liu S. A poor imitation of a natural process: A call to reconsider the iPSC engineering technique. Cell Cycle 2012; 11; http://dx.doi.org/10.4161/cc.22575; PMID: 23114619
- Menendez JA, Vazquez-Martin A. Rejuvenating regeneration: metformin activates endogenous adult stem cells. Cell Cycle 2012; 11:3521 - 2; http://dx.doi.org/10.4161/cc.21878; PMID: 22935702
- Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Quirantes R, Segura-Carretero A, Micol V, et al. Metformin lowers the threshold for stress-induced senescence: a role for the microRNA-200 family and miR-205. Cell Cycle 2012; 11:1235 - 46; http://dx.doi.org/10.4161/cc.11.6.19665; PMID: 22356767
- Corominas-Faja B, Quirantes-Piné R, Oliveras-Ferraros C, Vazquez-Martin A, Cufí S, Martin-Castillo B, et al. Metabolomic fingerprint reveals that metformin impairs one-carbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging (Albany NY) 2012; 4:480 - 98; PMID: 22837425
- Vazquez-Martin A, Cufí S, Lopez-Bonet E, Corominas-Faja B, Oliveras-Ferraros C, Martin-Castillo B, et al. Metformin limits the tumourigenicity of iPS cells without affecting their pluripotency. Sci Rep 2012; 2:964; http://dx.doi.org/10.1038/srep00964; PMID: 23236586