Abstract
HER2/neu oncogene is frequently deregulated in cancers, and the (PI3K)-Akt signaling is one of the major pathways in mediating HER2/neu oncogenic signal. p57Kip2, an inhibitor of cyclin-depependent kinases, is pivotal in regulating cell cycle progression, but its upstream regulators remain unclear. Here we show that the HER2-Akt axis is linked to p57Kip2 regulation, and that Akt is a negative regulator of p57Kip2. Ectopic expression of Akt can decrease the expression of p57Kip2, while Akt inhibition leads to p57Kip2 stabilization. Mechanistic studies show that Akt interacts with p57Kip2 and causes cytoplasmic localization of p57Kip2. Akt phosphorylates p57 on Ser 282 or Thr310. Akt activity results in destabilization of p57 by accelerating turnover rate of p57 and enhancing p57 ubiquitination. Importantly, the negative impact of HER2/Akt on p57 stability contributes to HER2-mediated cell proliferation, transformational activity and tumorigenicity. p57 restoration can attenuate these defects caused by HER2. Significantly, Kaplan-Meier analysis of tumor samples demonstrate that in tumors where HER2 expression was observed, high expression levels of p57Kip2 were associated with better overall survival. These data suggest that HER2/Akt is an important negative regulator of p57Kip2, and that p57 restoration in HER2-overexpressing cells can reduce breast tumor growth. Our findings indicate the applicability of employing p57 regulation as a therapeutic intervention in HER2-overexpressing cancers.
Introduction
HER2/neu (human EGF receptor type 2) oncogene amplification or overexpression is frequently found in many cancers, including breast, ovarian, lung, gastric and oral cancers.Citation1 HER2 overexpression is associated with poor survival in breast cancer patients.Citation1 In HER2-overexpressing cancer cells, the phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway is often activated.Citation2 Importantly, Akt activity is elevated in several types of human malignancies, including ovarian, breast, lung and thyroid cancers.Citation3 The kinase activity of Akt is constitutively activated in human cancer as a result of dysregulation of its regulators, including the tumor suppressor phosphatase and tensin homolog (PTEN)Citation4 and the amplification of the catalytic subunit of PI3K.Citation5 We previously showed that HER2 signaling is involved in enhancing ubiquitin-mediated degradation of p27Kip1 (p27), a cyclin-dependent kinase (CDK) inhibitor and in causing p27 cytoplasmic localization.Citation6 HER2 can also activate Akt to phosphorylate p21 at threonine 145, resulting in the cytoplasmic location of p21, another CDK inhibitor, which prevents p21’s growth inhibitory activity.Citation7 Although Akt is known to phosphorylate and regulate several important substrates, many of its substrates remain to be characterized.
The p57Kip2 protein (also known as CDKN1c, hereafter abbreviated as p57) is a cyclin-dependent kinase inhibitor sharing p27Kip1 homology.Citation8 p57 gene is a maternally expressed, paternally imprinted gene located on chromosome 11p15.5.Citation2,Citation9 p57-knockout mice have altered cell proliferation, differentiation and a variety of other abnormalities.Citation10,Citation11 Many of these defective phenotypes are also present in patients with Beckwith-Wiedemann syndrome, a childhood overgrowth syndrome, suggesting that the loss of p57 plays a pivotal role in this syndrome.Citation10,Citation11 As a negative regulator of the cell cycle, p57 is deemed as a potential tumor-suppressor gene.Citation12-Citation14 Although decreased expression of p57 has been found in bladder carcinoma, gastric cancer, pancreatic cancer and many other types of cancer,Citation2 p57-associated mutations are rarely found.Citation13,Citation15 It is possible that the post-transcriptional deregulation of p57 is the cause in the cancers, but it remains to be determined how oncogenic signals can downregulate p57 expression at the protein level.
In this study, we demonstrate that HER2 signaling has a negative impact on p57 and further indicate that HER2 signaling mediator Akt is involved in negatively regulating p57 stability. We characterized the biological role of HER2/Akt in destabilizing p57 and demonstrated that the restoration of p57 proteins could inhibit HER2-mediated cell proliferation and cell transformation. Significantly, we showed that p57 expression reduces tumorigenicity of HER2-overexpressing cells in nude mice. Our studies provide important insights into regulating the oncogenic activity of HER2-overexpressing cancers.
Results
Akt is involved in HER2 signaling-mediated p57 regulation
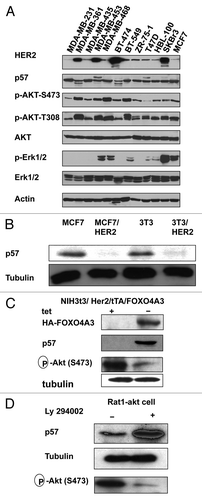
HER2 signaling can positively promote the cell cycle.Citation7 Given that HER2 overexpression can increase cell proliferation,Citation16 HER2 may control the cell cycle by regulating the CDK inhibitor, which affects Cylin-Cdk activity. We determined the expression level of p57 in several breast cancer cell lines with different HER2 statuses. p57 protein levels are lower in HER2-overexpressing cells (). Importantly, while Akt activity is high in HER2-overexpressing cells (such as MDA-MB-361, BT474), p57 CDK inhibitor is lower in this context (). We also examined the expression level of p57 in isogenic cell lines with different HER2 statuses: HER2-overexpressing cells (MCF7/HER2) and non-HER2-overexpressing cells (MCF7). The results showed that p57 protein levels are lower in HER2-overexpressing cells than in non-HER2-overexpressing cells (). Previously, we show that constitutively active FOXO4 [three Akt phosphorylation sites on FOXO4 (Thr32, Ser187 and Ser252) mutated to alanine, FOXO4A3]Citation17 can inhibit endogenous Akt’s kinase activity toward its substrates. We established a cell line, NIH3T3/HER2/tTA-FOXOA3, to examine the level of p57 expression experiment in the absence of tetracycline (tet) (). The data showed that the levels of p57 increased drastically after the FOXOA3 induction (−tet) () with concurrent suppression of Akt activity. Interestingly, inhibiting Akt activity by LY294002 (a PI3K inhibitor) leads to p57 stabilization in Rat1-Akt cells (). These results suggest that p57 is downregulated in the presence of activated Akt.
Figure 1. HER2-Akt signaling regulates p57 stability. (A) HER2 overexpression leads to reduced expression of p57. Indicated equal amounts of cell lysates were were immunoblotted with indicated antibodies. (B) HER2-overexpressin cells have decreased steady-state expression of p57. Indicated amounts of cell lysates were were immunoblotted with indicated antibodies. (C) Immunoblot analysis of p57 in tet-regulated 3T3/HER2/tTA-FOXO4A3 cells. NIH3T3/HER2/tTA-FOXO4A3 cells were treated with tetracycline or left untreated. Equal amounts of cell lysates were immunoblotted with indicated antibodies. (D) Inhibitor of PI3K-Akt pathway causes stabilization of p57. Rat1-Akt cells were treated with LY294002 for 6 h before harvesting. Equal amounts of cell lysates were immunoblotted with indicated antibodies.
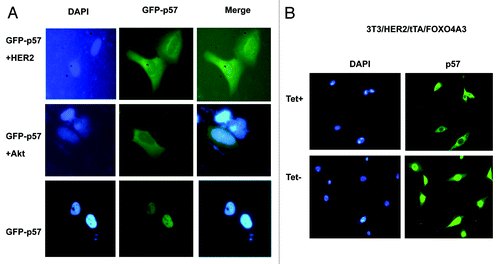
As nuclear exporting is a critical process in protein degradation, we then determined whether HER2-Akt signaling affects p57 subcellular localization. Cells were transfected with GFP-p57 in the presence or absence of HER2 or Akt. GFP-p57 was abundant in the nucleus, while cotransfection with Akt or HER2 promoted more cytoplasmic localization of p57 (). Similar results have been observed in 3T3/HER2/tTA/FOXOA3 cells. While p57 is located in the cytoplasm in Tet-treated cells, p57 is relocated from the cytoplasm to the nucleus in non-treated cells (), supposedly due to inhibiting Akt activity after induced expression of FOXOA3. Together, these results suggest that HER2-Akt activity regulates subcellular localization.
Figure 2. HER2-Akt signaling induces nuclear export of p57. (A) p57 nuclear localization is reduced in the presence of HER2. U2OS cells were transfected with indicated plasmids. Nucleus was stained with DAPI. (B) p57 is localized at the nucleus when Akt is inhibited by the expression of FOXO4A3. Indicated cells treated with or without Tet were stained with Anti-p57 followed with FITC-conjugated secondary Ab. Nucleus was stained with DAPI.
Akt interacts with and phosphorylates p57
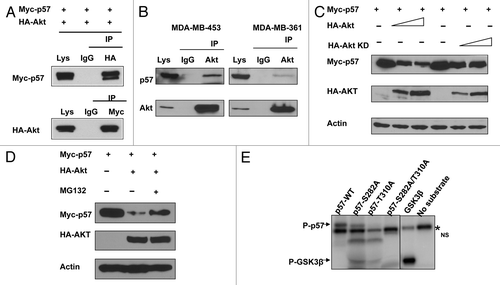
Given that Akt is involved in the stabilization of p57, it is possible that there is an interaction between Akt and p57. To investigate this potential interaction, we analyzed cell lysates cotransfected with Akt and p57. Indeed, exogenous expressed Akt was able to associate with exogenous p57 as assayed by Co-IP (). Endogenous interaction between Akt and p57 is also confirmed (). We then explored whether the Akt signaling pathway is involved in p57 downregulation. p57 levels were decreased when cells were transfected with Akt in a dose-dependent manner (). Akt-mediated p57 downregulation was suppressed by MG132, a proteasome inhibitor, suggesting the involvement of the 26S proteasome (). As an Akt-associated protein, it is possible that p57 is a substrate of Akt. To further investigate if Akt can mediate phosphorylation of p57, we first scanned its sequence using the NetPhos algorithm (www.cbs.dtu.dk/services/NetPhos) and identified potential Akt phosphorylation sites (S282, T310). We therefore mutated the potential serine or threonine phosphorylation site to alanine (S282A or T310A), and found that the phosphorylation of these mutants by Akt was diminished in an in vitro kinase assay, suggesting that S282 or T310 is the Akt phosphorylation site on p57 ().
Figure 3. Akt interacts with and phosphorylates p57. (A) Interaction of Akt with p57. Equal amounts of 293T cell lysates transfected with indicated plasmids were immunoprecipitated (IP) with anti-HA and immunoblotted (IB) with anti-myc. Reciporical IP was also performed. (B) Endogenous interaction between Akt and p57. Equal amounts of indicated cell lysates were also immunoprecipitated with anti-Akt and immunoblotted with anti-p57. (C) Akt decreases steady-state expression of p57 in a dose-dependent manner. (D) Akt-mediated degradation of p57 is proteasome-dependent. 293T cells were co-transfected with the indicated plasmids. Cells were treated with MG132 for six hours before harvesting. Equal amounts of cell lysates were immunoblotted with the indicated antibodies. (E) Akt phosporylates p57 in vitro. Baculovirus-produced active recombinant Akt1 was incubated with indicated equal amounts of bacterially produced and purified recombinant p57 and mutant p57 protein for kinase activity. 32P Autoradiography demonstrated the phosphorylated p57.
Akt signaling negatively regulates p57 protein stability
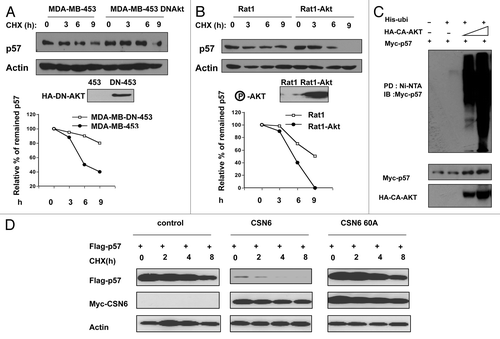
To investigate if Akt downregulates p57 at the post-transcriptional level, we examined the turnover rate of p57 by blocking Akt activity in the presence of the de novo protein synthesis inhibitor, cycloheximide (CHX) (). Expression of dominant-negative Akt (DN-Akt, Akt K179M) in MDA-MB453 cells led to stabilization of p57 (). Consistently, overexpression of Akt in Rat-1 cells increased the turnover rate of p57 when compared with the non-overexpression control group (). Furthermore, we found that Akt increased the ubiquitination level of p57 in a dose-dependent manner (). Our previous data show that HER2-Akt signaling enhances CSN6 stabilization through phosphorylation on Ser60.Citation18,Citation19 As CSN6 also has a role in p57 degradation, we then analyzed the Akt-CSN6 link in p57 turnover by employing an Akt phosphorylation mutant of CSN6 (CSN6S60A). CSN6S60A cannot be phosphorylated by Akt and is less stable when compared with wt CSN6;Citation18,Citation19 therefore, its impact on p57 turnover was compromised when compared with wtCSN6 (), suggesting that Akt-mediated CSN6 phosphorylation is also involved in p57 degradation. Together, these results suggest that Akt activity negatively regulates p57 stability.
Figure 4. HER2-Akt signaling reduces p57 turnover. (A) p57 turnover rate is decreased in DNAkt-overexpressing cells. Indicated cells were treated with cycloheximide (CHX) (100 µg/ml) for the indicated times. Cell lysates were immunoblotted with indicated antibodies. (B) Akt dereases the turnover of p57. Indicated cells were treated with cycloheximide (CHX) (100 µg/ml) for the indicated times. Cell lysates were immunoblotted with indicated antibodies. (C) Akt increases the polyubiquitination level of p57 in a dose-dependent manner. Indicated cells were transfected with indicated plasmids. Cell lysates were pulled down with Ni++ NTA beads and then immunoblotted with anti-Myc. (D) Akt-CSN6 axis is involved in regulating p57 turnover rate. Indicated transfected cells were treated with cycloheximide (CHX) (100 µg/ml) for the indicated times. Cell lysates were immunoblotted with indicated antibodies.
HER2 regulates cell growth, transformation and tumorigenicity through antagonizing p57 activity
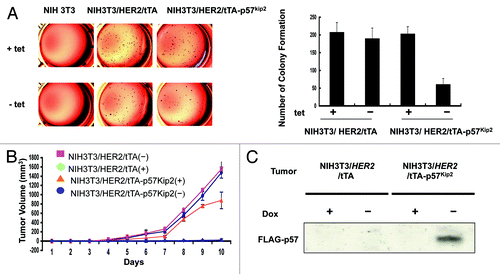
Since HER2 downregulates p57, we reasoned that HER2-p57 axis would have a role in cell proliferation. We established a cell line, NIH3T3/HER2/tTA-p57, to perform a p57 induction experiment in the absence of tet, and found that p57 inhibits CDK activity efficiently after induction as demonstrated by the inhibition of CDK2-associated HH1 kinase activity (). Because p57 inhibits CDK activity, we first tested whether p57 expression (NIH3T3/HER2/tTA-p57 in the absence of tet) could block cell proliferation using a MTT assay in this system. As expected, control NIH3T3/HER2/tTA cells and NIH3T3/HER2/tTA-p57 in the presence of tet all have good cell proliferation rate. In contrast, the cell growth rate of p57-expressing cells (−tet treatment) reduced drastically after the p57 induction (). We then determined the BrdU incorporation rate in these p57-expressing cells. The data showed that the BrdU incorporation rate of p57-expressing cells (−tet treatment) decreased drastically after the p57 induction (), suggesting that induced expression of p57 can dampen cell growth caused by HER2. We sought to further examine the effect of p57 on HER2-mediated cell transformation (). We found that NIH3T3/HER2/tTA-p57 cells with p57 induction showed a decrease in cell colonies with anchorage-independent growth when compared with non-induced cells (). Because p57 suppresses HER2-mediated cell proliferation and anchorage-independent growth, we next examined the impact of p57 on HER2-mediated tumor promotion. We demonstrated that xenografted tumor volume was significantly decreased in p57-induced mice (−dox) compared with control mice (+dox) (). NIH3T3/HER2/tTA cells were used as other controls in this tumorigenicity studies. Further, tumors obtained from this study indicated that indeed p57 was induced in the absense of dox treatment (). Together, these data illustrate that the HER2 oncogenic signals are critical in downregulating p57 during tumorigenesis, thereby promoting cell growth and tumorigenicity. Significantly, p57 restoration can correct the abnormalities mediated by HER2 expression. These data demonstrate the biological consequences of p57’s negative impact on controlling Her2-mediated tumorigenicity.
Figure 5.p57 activity blocks HER2-mediated cell growth. (A) p57 induction hinders HER2-mediated CDK activity. NIH3T3/HER2/tTA-p57 cells were treated without (−) tet. The cell lysates were immunoprecipitated with Anti-Cdk2 and were analyzed cdk2-associated kinase activity toward to HH1. (B) HER2-mediated cell growth was blocked by p57. Indicated cells were treated with tet or without tet. Cell growth was analyzed by MTT assay. (C) p57 induction reduces HER2-mediated BrdU incorporation. Indicated cells were treated with (+) or without (−) tet. The cells were fixed for anti-BrdU staining. (D) BrdU positive cells were counted and presented as a bar graph. Error bars represent standard deviation.
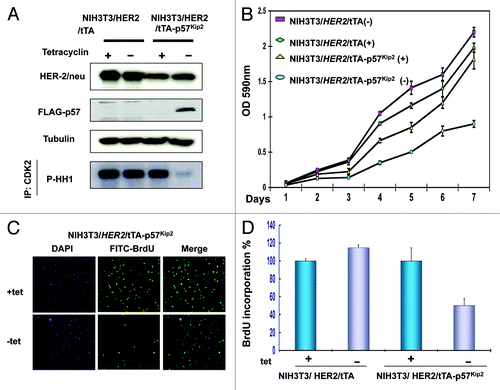
Figure 6. p57 activity hinders HER2-mediated cell transformation and tumorigenicity. (A) p57 induction hampers HER2-mediated cell transformation. Indicated cells were treated with (+) or without (−) tet. The cells were the analyzed for a soft agar colony formation assay. Colony number was counted and presented as a bar graph. Error bars represent standard deviation. (B) p57 induction impeded HER2-mediated tumorigenicity. Cells (1 × 106) were then harvested and s.c. injected into the flank region of female nude mice. Mice were provided with (+) or without (−) tet in the drinking water. Tumor volumes were monitored for 10 d. Change in tumor volume over a 10-d period. Bars, SD (C) p57 induction is responsible for tumor inhibition. Indicated tumor samples were lysed. Equal amounts of lysates were immunoblotted with anti-p57.
Overexpression of HER2 and downregulation of p57 in breast cancer
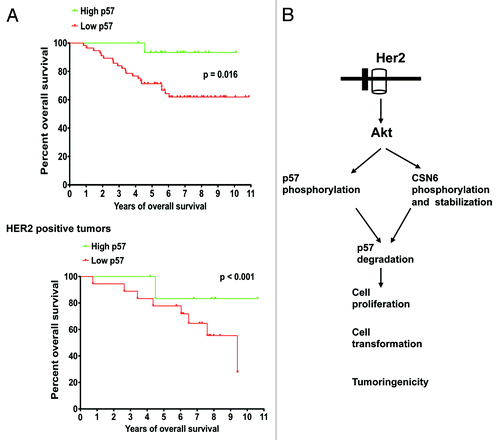
To examine the impact of p57 expression in cancer survival, we assessed the expression level of p57 in a cohort of breast cancer patients with clinical records and correlated p57 expression levels with the overall survival. We found that low levels of p57 expression correlated with poor survival (). To understand the relationship between HER2 and p57 in cancer, we examined the correlation of HER2 and p57 in this cohort of primary human breast cancer samples. In tumors where HER2 expression was observed, Kaplan-Meier analysis showed that high expression levels of p57 were associated with better overall survival (log rank test, p < 0.001) (). On the other hand, low expression levels of p57 were associated with poor overall survival (log rank test, p < 0.001) (). Thus, the expression levels of HER2 and p57 are significant prognostic factors for breast cancer.
Figure 7. Low p57 correlates with poor survival. (A) Kaplan-Meier analysis showed that low expression of p57 was associated with poor overall survival. In HER2-overexpressing tumors, high expression levels of p57 were associated with better overall survival. (B) Model of the HER2/akt-mediated p57 degradation in affecting cell proliferation, cell cycle progression, cell transformation and tumorigenicity. Note the dual role of Akt on CSN6 and p57 phosphorylation.
Discussion
HER2/Akt signaling is highly active in breast cancer. This activation of HER2/Akt signaling has made HER2-Akt axis an attractive target for therapeutic cancer drugs. Nonetheless, although high activation of HER2/Akt is associated with advanced clinical stage and poor prognosis in breast cancer, the impact of aberrant overactivation of HER2/Akt on the cell cycle remains unclear. Here, we discover a critical role for the HER2-Akt axis in controlling homeostasis of CDK inhibitor p57 through regulating its degradation. Our results provide insight into the consequence of activation of the HER2-Akt axis during carcinogenesis and cancer progression.
It is known that Akt phosphorylates p27 and mediates nuclear export.Citation20-Citation22 HER2/Akt signal enhances ubiquitin-mediated p27 degradation.Citation16 Akt downregulation leads to p27 accumulation.Citation23,Citation24 Akt phosphorylates p27 to create a site for 14-3-3 binding, which, in turn, leads to nuclear export.Citation25,Citation26 p27 is then degraded by the Kip1 ubiquitination-promoting complex (KPC) in the cytoplasm.Citation27 In this study, we have shown that HER2/Akt leads to p57 phosphorylation and nuclear export (). How this phosphorylation involves any activity of 14-3-3 or KPC in terms of p57 nuclear export and degradation remains to be investigated.
The downregulation of p57 in several types of cancer demonstrates that this dysregulation plays an important role in human tumorigenesis. Except Beckwith-Wiedemann syndrome (BWS)Citation28,Citation29 and IMAGe syndrome (intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita and genital anomalies),Citation30,Citation31 p57 mutations in cancers are rare,Citation2 suggesting that posttranscriptional regulation or posttranslational silencing are involved in the loss of p57 function in cancers. However, upstream of p57 regulation is not well characterized, although IGFII and TGF-β have been studied. Insulin-like growth factor-II (IGF-II) plays a key role in embryonic growth, and aberrant IGF-II expression is documented in a wide variety of tumors. The expression of p57 is reduced in primary embryo fibroblasts after IGF-II treatment.Citation32 Interestingly, in a mouse study of BWS model, IGFII and p57 indeed act in an antagonistic manner,Citation33 and IGF-II and p57 play antagonistic roles in controlling cell proliferation during normal development. TGF-β also has a role in regulating p57. TGF-β has a variety of signals for numerous cells and sometimes exerts contradictory effects, depending on the cell types and environment. Although TGF-β mainly suppresses cell growth in epithelial cells, it can also promote cell growth in other cells, such as osteoblasts. It was shown that TGF-β causes p57 degradation in osteoblasts, suggesting a new signal for regulating p57.Citation34 Here, our studies about the regulation of p57 by HER2-Akt pathway provide another important insight for the upstream regulation signals of p57 and help us understand more about the role of p57 in HER2-overexpressing cancer.
We previously showed that HER2-Akt signaling enhances CSN6 stabilization.Citation18,Citation19 Interestingly, CSN6, in turn, can facilitate Skp2-medaited p57 ubiquitination and degradation.Citation35 Here, we have added another layer of HER2-Akt signaling in regulating p57 phosphorylation and ubiquitin-mediated degradation. Together, it is clear that Akt has a dual role in controlling the homeostasis of p57 (). When HER2-Akt signaling is highly activated, it will compromise the cell cycle-inhibitory activities of p57, a manifestation of low-level expression of p57 in cancers (). HER2 levels are associated with poor prognosis of several types of human cancer. We have shown that HER2’s negative impact on p57 stability in cancer can compromise patient survival rates, which attests to the significant role of HER2-mediated p57 degradation during tumorigenesis. Obviously, HER2 is an important molecular target for rational cancer therapy. A strategy to inhibit HER2 activity has been the focus in cancer drug discovery. For example, monoclonal antibody to inhibit the activity of HER2 has been employed. Here, we showed that p57 inhibits HER2-activated cell growth. We also found that p57 inhibited the kinase activity of CDK2 potentiated by HER2. Most significantly for clinical application, p57 expression in HER2-overexpressing cells can be regulated in vivo and reduces the tumor volume in a tumor mouse model. Therefore, these data provide compelling evidence that p57 functions as a critical target of HER2 signaling pathway during tumorigenesis. Clearly, the HER2-p57 link is an obvious molecular target for rational cancer therapy. Restoring the p57 may be a useful therapeutic strategy for cancer intervention in cancer with deregulation of HER2/Akt signaling.
Materials and Methods
Cell lines and reagents
HEK-293T (human embryonic kidney cell line), MCF7 and HER18 (human breast cancer cell line) 3T3 and 3T3/HER2, NIH3T3/HER2/tTA/Foxo4A3, NIH3t3/HER2/tTA/p57, Rat1, Rat1-Akt were previously described.Citation18 MDA-MB-231, MDA-MB-361, MDA-MB-435, MDA-MB-453, MDA-MB-468, BT474, BT549, ZR75-1, T47D, HBL-100, SKBR3, MCF7 were obtained from ATCC. NIH3T3/HER2/tTA-cells have been described previously.Citation36 Flag-tagged p57 DNA fragment was cloned into modified pUHD10-3 hygromycin vectorCitation37 and confirmed by sequencing. BrdU, LY294002, MG132, doxoycycline and cycloheximide were obtained from Sigma. Antibodies: HA (Sigma), phosphorylated Akt (Cell Signaling), Flag (M2 monoclonal antibody, Sigma), Erk1 (Sigma), p-ERK1 (Sigma), tubulin (Sigma), p57 (Sigma), ubiquitin (Zymed Laboratories, Inc.). Myc-p57, HA-Akt, DN-Akt, His-ubi were previously described.Citation35 Flag-p57, GFP-p57 were previously described.Citation9,Citation35
Immunoblotting and immunoprecipitation
Total cell lysates were processed as previously described.Citation38 Cell lysates for western blot or immunoprecipitation were lysed with 100–300 μl 1× lysis buffer [0.5 L batch: 7.5g 1 M Tris (Fisher), 15 ml 5M NaCl (Fisher), 0.5 ml NP-40 (USB Corp.), 0.5 ml Triton X-100 (Sigma) and 1 ml 0.5M EDTA (Fisher)] for 20 min at 4°C. Lysates were immunoblotted with indicated antibodies. For immunoprecipitation, cell lysates were prepared and standardized as before and 1 mg protein immunoprecipitated with appropriately diluted antibody in lysis buffer overnight. Antibody was pulled down with 50 μl of either Protein A or G beads. Beads were centrifuged at low speed for 10 min and supernatant discarded. Dried beads were mixed with 2× loading dye and boiled for 5 min. Lysate samples were loaded onto gels and SDS-PAGE was performed as before.
Immunofluorescence
NIH3T3/HER2/tTA/p57 cells were grown on a sterile coverslip in 6-well plate overnight. The cells were fixed in 100% methanol at −20°C for 3 min. Cells were probed with rabbit anti-p57. Alexa Fluor 488 goat anti-rabbit antibody (Molecular Probe) was added to detect p57. DAPI was applied to stain the nuclei. Immunofluorescence was observed by using a confocal microscope as previously described.Citation39 For GFP-p57 detection, cells were observed under a fluorescent microscope. DAPI was applied to stain the nuclei.
In vitro kinase assays
Recombinant p57 substrates were produced by BL-21 E. coli bacteria. Briefly, the bacterial cells were transformed with the pET-Flag-p57 (wild type) and pET-Flag-p57 mutant plasmid and grown in 5 mL LB media overnight and in 500 ml LB media for 5 h. One mM isopropyl β-D-1 thiogalactopyranoside (IPTG) (Fisher) was added in culture media for 3 h. Then the cells were lysed with NETN buffer [20 mM TRIS-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% (vol/vol) Nonidet P-40 plus protease-phosphatase inhibitor mixture] and sonicated.Citation38 The p57 proteins were pulled down by M2 beads and eluted by Flag peptide. Recombinant Akt kinase (Cell Signaling) was incubated with immunopurified p57 substrates in 1× kinase buffer (80 mM Mops, 7.5 mM MgCl2, pH 7.0) with cold ATP and γ32 ATP at 30°C for 15 min. Kinase reactions were analyzed by SDS/PAGE as described before.Citation39 After the SDS/PAGE gel was dried, the radioactivity image was visualized using a phosphoimager cassette (Molecular Dynamics) and a Typhoon Trio variable mode imager.
Turnover assay
The cells were transfected with indicated plasmids and incubated in the 37°C with 5% (vol/vol) CO2 for 24 h. Then cycloheximide was added into the media at 60 μg/ml of final concentration. The cells were harvested at the indicated time points after CHX treatment. The protein levels were analyzed by immunoblotting.
Ubiquitination assay
293T cells cotransfected with indicated plasmids or were used for the experiments. At 24 h post-transfection, cells were treated with 50 μg/ml of MG132 for 6 h. The ubiquitinated proteins were pulled down by the Ni-NTA. The protein complexes were then resolved by SDS-polyacrylamide gel and probed with anti-Myc to observe the ubiquitinated p57.
BrdU incorporations immunofluorescence
Incorporation of BrdU was examined under a fluorescence microscope with fluorescein isothiocyanate-conjugated anti-BrdU as previously described.Citation36 At least 300 cells were counted for BrdU staining. The percentage of BrdU-positive cells was measured. Data shown are from a typical experiment performed in triplicate.
Anchorage-independent soft-agar colony formation assay
NIH3T3/HER2/tTA-p57 cells were treated with 2 mg/ml tet or left untreated and were subjected to these assays. These assays were performed as described.Citation16
Tumor growth in nude mice
Female 4- to 5-week-old nude mice (Charles River Laboratories) were maintained in the animal facility at the University of Texas MD Anderson Cancer Center. NIH3T3/HER2-tTA-p57 cells (1 × 106 cells in 0.2 ml PBS per injection with 2 mg/ml tet for one group, and without tet for other) were injected into the mammary fat pad of mice. NIH3T3/HER2-tTA cells treated with or without doxcycline were used in the experiments as controls. After inoculation, animals were fed drinking water containing 5% sucrose in the presence or absence of 200 mg/ml doxycycline (Sigma).
Human tumor samples
p57 and HER2 expresssion levels were scored from cohort: GSE-20194 consisting of 255 untreated stage I stage III breast cancer patients at MD Anderson Cancer Center.
Acknowledgments
This work was supported by grants in part from the National Institutes of Health (NIH) (R01CA089266), Directed Medical Research Programs (DOD SIDA BC062166 to S.J.Y. and M.H.L.) and Susan G. Komen Breast Cancer Foundation (KG081048). The University of Texas M.D. Anderson Cancer Center is supported by NIH core grant CA16672. We thank Jessica Cromheecke for editing.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987; 235:177 - 82; http://dx.doi.org/10.1126/science.3798106; PMID: 3798106
- Lee MH, Yang HY. Negative regulators of cyclin-dependent kinases and their roles in cancers. Cell Mol Life Sci 2001; 58:1907 - 22; http://dx.doi.org/10.1007/PL00000826; PMID: 11766887
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2:489 - 501; http://dx.doi.org/10.1038/nrc839; PMID: 12094235
- Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci U S A 1998; 95:15587 - 91; http://dx.doi.org/10.1073/pnas.95.26.15587; PMID: 9861013
- Scheid MP, Woodgett JR. PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol 2001; 2:760 - 8; http://dx.doi.org/10.1038/35096067; PMID: 11584303
- Yang HY, Zhou BP, Hung MC, Lee MH. Oncogenic signals of HER-2/neu in regulating the stability of the cyclin-dependent kinase inhibitor p27. J Biol Chem 2000; 275:24735 - 9; http://dx.doi.org/10.1074/jbc.C000147200; PMID: 10859299
- Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol 2001; 3:245 - 52; http://dx.doi.org/10.1038/35060032; PMID: 11231573
- Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994; 78:59 - 66; http://dx.doi.org/10.1016/0092-8674(94)90572-X; PMID: 8033212
- Lee MH, Reynisdóttir I, Massagué J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev 1995; 9:639 - 49; http://dx.doi.org/10.1101/gad.9.6.639; PMID: 7729683
- Yan Y, Frisén J, Lee MH, Massagué J, Barbacid M. Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev 1997; 11:973 - 83; http://dx.doi.org/10.1101/gad.11.8.973; PMID: 9136926
- Zhang P, Liégeois NJ, Wong C, Finegold M, Hou H, Thompson JC, et al. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 1997; 387:151 - 8; http://dx.doi.org/10.1038/387151a0; PMID: 9144284
- Lee MH, Yang HY. Regulators of G1 cyclin-dependent kinases and cancers. Cancer Metastasis Rev 2003; 22:435 - 49; http://dx.doi.org/10.1023/A:1023785332315; PMID: 12884917
- Kavanagh E, Joseph B. The hallmarks of CDKN1C (p57, KIP2) in cancer. Biochim Biophys Acta 2011; 1816:50 - 6; PMID: 21447370
- Guo H, Tian T, Nan KJ, Wang W. p57: A multifunctional protein in cancer (Review). [Review] Int J Oncol 2010; 36:1321 - 9; PMID: 20428755
- Borriello A, Caldarelli I, Bencivenga D, Criscuolo M, Cucciolla V, Tramontano A, et al. p57(Kip2) and cancer: time for a critical appraisal. Mol Cancer Res 2011; 9:1269 - 84; http://dx.doi.org/10.1158/1541-7786.MCR-11-0220; PMID: 21816904
- Yang HY, Shao R, Hung MC, Lee MH. p27 Kip1 inhibits HER2/neu-mediated cell growth and tumorigenesis. Oncogene 2001; 20:3695 - 702; http://dx.doi.org/10.1038/sj.onc.1204472; PMID: 11439332
- Medema RH, Kops GJPL, Bos JL, Burgering BMT. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000; 404:782 - 7; http://dx.doi.org/10.1038/35008115; PMID: 10783894
- Xue Y, Chen J, Choi HH, Phan L, Chou PC, Zhao R, et al. HER2-Akt signaling in regulating COP9 signalsome subunit 6 and p53. Cell Cycle 2012; 11:4181 - 90; http://dx.doi.org/10.4161/cc.22413; PMID: 23095642
- Iyer SV, Iwakuma T. A novel link between the HER2-Akt and MDM2-p53 pathways via CSN6. Cell Cycle 2012; 11:4112; http://dx.doi.org/10.4161/cc.22606; PMID: 23099920
- Viglietto G, Motti ML, Bruni P, Melillo RM, D’Alessio A, Califano D, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med 2002; 8:1136 - 44; http://dx.doi.org/10.1038/nm762; PMID: 12244303
- Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med 2002; 8:1153 - 60; http://dx.doi.org/10.1038/nm761; PMID: 12244302
- Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med 2002; 8:1145 - 52; http://dx.doi.org/10.1038/nm759; PMID: 12244301
- Yang H, Wen YY, Zhao R, Lin YL, Fournier K, Yang HY, et al. DNA damage-induced protein 14-3-3 sigma inhibits protein kinase B/Akt activation and suppresses Akt-activated cancer. Cancer Res 2006; 66:3096 - 105; http://dx.doi.org/10.1158/0008-5472.CAN-05-3620; PMID: 16540659
- Yang H, Zhang Y, Zhao R, Wen YY, Fournier K, Wu HB, et al. Negative cell cycle regulator 14-3-3sigma stabilizes p27 Kip1 by inhibiting the activity of PKB/Akt. Oncogene 2006; 25:4585 - 94; http://dx.doi.org/10.1038/sj.onc.1209481; PMID: 16532026
- Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem 2002; 277:28706 - 13; http://dx.doi.org/10.1074/jbc.M203668200; PMID: 12042314
- Sekimoto T, Fukumoto M, Yoneda Y. 14-3-3 suppresses the nuclear localization of threonine 157-phosphorylated p27(Kip1). EMBO J 2004; 23:1934 - 42; http://dx.doi.org/10.1038/sj.emboj.7600198; PMID: 15057270
- Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, et al. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol 2004; 6:1229 - 35; http://dx.doi.org/10.1038/ncb1194; PMID: 15531880
- Hatada I, Inazawa J, Abe T, Nakayama M, Kaneko Y, Jinno Y, et al. Genomic imprinting of human p57KIP2 and its reduced expression in Wilms’ tumors. Hum Mol Genet 1996; 5:783 - 8; http://dx.doi.org/10.1093/hmg/5.6.783; PMID: 8776593
- Hatada I, Nabetani A, Morisaki H, Xin Z, Ohishi S, Tonoki H, et al. New p57KIP2 mutations in Beckwith-Wiedemann syndrome. Hum Genet 1997; 100:681 - 3; http://dx.doi.org/10.1007/s004390050573; PMID: 9341892
- Arboleda VA, Lee H, Parnaik R, Fleming A, Banerjee A, Ferraz-de-Souza B, et al. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat Genet 2012; 44:788 - 92; http://dx.doi.org/10.1038/ng.2275; PMID: 22634751
- Riccio A, Cubellis MV. Gain of function in CDKN1C. Nat Genet 2012; 44:737 - 8; http://dx.doi.org/10.1038/ng.2336; PMID: 22735584
- Grandjean V, Smith J, Schofield PN, Ferguson-Smith AC. Increased IGF-II protein affects p57kip2 expression in vivo and in vitro: implications for Beckwith-Wiedemann syndrome. Proc Natl Acad Sci U S A 2000; 97:5279 - 84; http://dx.doi.org/10.1073/pnas.080409297; PMID: 10779549
- Caspary T, Cleary MA, Perlman EJ, Zhang P, Elledge SJ, Tilghman SM. Oppositely imprinted genes p57(Kip2) and igf2 interact in a mouse model for Beckwith-Wiedemann syndrome. Genes Dev 1999; 13:3115 - 24; http://dx.doi.org/10.1101/gad.13.23.3115; PMID: 10601037
- Kim M, Nakamoto T, Nishimori S, Tanaka K, Chiba T. A new ubiquitin ligase involved in p57KIP2 proteolysis regulates osteoblast cell differentiation. EMBO Rep 2008; 9:878 - 84; http://dx.doi.org/10.1038/embor.2008.125; PMID: 18660753
- Chen B, Zhao R, Su C, Linan M, Tseng C, Pham L, et al. CDK inhibitor p57Kip2 is negatively regulated by COP9 signalosome subunit 6. Cell Cycle 2012; In press http://dx.doi.org/10.4161/cc.22887
- Yang H, Zhao R, Yang HY, Lee MH. Constitutively active FOXO4 inhibits Akt activity, regulates p27 Kip1 stability, and suppresses HER2-mediated tumorigenicity. Oncogene 2005; 24:1924 - 35; http://dx.doi.org/10.1038/sj.onc.1208352; PMID: 15688030
- Reynisdóttir I, Polyak K, Iavarone A, Massagué J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev 1995; 9:1831 - 45; http://dx.doi.org/10.1101/gad.9.15.1831; PMID: 7649471
- Laronga C, Yang HY, Neal C, Lee MH. Association of the cyclin-dependent kinases and 14-3-3 sigma negatively regulates cell cycle progression. J Biol Chem 2000; 275:23106 - 12; http://dx.doi.org/10.1074/jbc.M905616199; PMID: 10767298
- Gully CP, Velazquez-Torres G, Shin JH, Fuentes-Mattei E, Wang E, Carlock C, et al. Aurora B kinase phosphorylates and instigates degradation of p53. Proc Natl Acad Sci U S A 2012; 109:E1513 - 22; http://dx.doi.org/10.1073/pnas.1110287109; PMID: 22611192