Abstract
The mammalian target of rapamycin (mTOR) kinase is a master regulator of protein synthesis that couples nutrient sensing to cell growth, and deregulation of this pathway is associated with tumorigenesis. p53, and its less investigated family member p73, have been shown to interact closely with mTOR pathways through the transcriptional regulation of different target genes. To investigate the metabolic changes that occur upon inhibition of the mTOR pathway and the role of p73 in this response primary mouse embryonic fibroblast from control and TAp73−/− were treated with the macrocyclic lactone rapamycin. Extensive gas chromatography/mass spectrometry (GC/MS) and liquid chromatography/mass spectrometry (LC/MS/MS) analysis were used to obtain a rapamycin-dependent global metabolome profile from control or TAp73−/− cells. In total 289 metabolites involved in selective pathways were identified; 39 biochemical metabolites were found to be significantly altered, many of which are known to be associated with the cellular stress response.
Introduction
The TOR pathway is an evolutionarily conserved pathway that plays critical roles in the regulation of cell proliferation, survival, and energy metabolism.Citation1,Citation2 TOR is a serine/threonine kinase highly conserved from yeast to humans. It forms two complexes in cells, TORC1 and TORC2 (PDK2), with distinctive physiological functions. As a main upstream kinase of the TORC1, AMPK is activated in response to glucose starvation and low intracellular ATP levels, which, in turn, phosphorylates and activates the TSC2 protein.Citation3 TSC2 exerts GTPase activity to negatively regulate GTP-binding protein Rheb, which activates TORC1, thus downregulating TORC1 signaling.Citation4,Citation5 mTORC1 is comprised of raptor, mTOR, and mLST8, and the strength of association of raptor with mTOR is regulated by nutrient and growth signals.Citation6,Citation7 Downstream mTORC1 signals to translation by regulating the association of the cap-binding protein, eIF4E, with its inhibitors the 4EBPs and also through activation of S6K1 and 2 that phosphorylate rpS6 and eIF4B.Citation6-Citation8 Signaling via this pathway results in the enhanced translation of mRNAs involved in ribosomal biogenesis, mitochondrial biogenesis, and oxygen consumption. Thus, activated TORC1 promotes protein synthesis, increases cell mass, and stimulates cell growth.Citation7
The p53 protein, a regulator of cell death,Citation9-Citation13 has been shown to interact closely with the mTOR pathways through the transcriptional regulation of different target genes.Citation14-Citation16 Indeed, p53 negatively regulates the TOR signaling, creating a metabolic network that allows cells to inhibit cell growth and division to avoid the acquisition of errors during these processes under stress conditions. p73, together with p63,Citation17-Citation21 is a structural and functional homolog of the tumor suppressor transcription factor p53, which activates transcription from p53-responsive promoters and, hence, induces cell cycle arrest and apoptosis.Citation22,Citation23 In contrast p63 is more specialized in epithelial developmentCitation24,Citation25 and homeostasisCitation26 but has clear effects on cancer progression and metastasis.Citation27 The Trp73 gene contains two promoters that drive the expression of two major groups of p73 isoforms with opposing cellular actions: the TAp73 isoforms contain the p73 transactivation domain (TA) and exhibit proapoptotic activities,Citation28,Citation29 whereas the DNp73 isoforms lacking the N-terminal TA domain are able to transcriptionally repress TAp73 and p53 as well as showing anti-apoptotic properties.Citation30 We contributed to elucidate the respective stabilityCitation31 and degradation patterns via the Ub E3 ligases ITCH,Citation32,Citation33 FBXO45,Citation34 antizyme pathway,Citation35 and PIR.Citation36 We generated selective knockout mice deficient for either TAp73Citation37,Citation38 or DNp73Citation39 isoforms, allowing the analysis of specific in vivo functions of the individual variants. At birth, total p73-deficient mice manifest hippocampal dysgenesis due to massive apoptosis (ca. 40%) of sympathetic neurons in superior cervical ganglion.Citation40,Citation41 After 1–2 months, they display hypersecretion of cerebrospinal fluid, resulting in hydrocephalus as well as runting and abnormal social and reproductive behavior because of defects in pheromone detection;Citation40 in addition, p73−/− mice show immunological defects with chronic infections and inflammation. The neuronal defects manifested by the p73-KO mice have uncovered a master role for p73 in the developing neuronal cells as well as in mature nervous system in the long-term maintenance of adult neurons. Phenotypical characterization of this animal model revealed that predisposition to spontaneous and carcinogen-induced tumorigenesis is increased by specific TAp73 loss in vivo. In vitro, p73 shows a direct ability to regulate neuronal differentiation.Citation42-Citation44 In addition a second major phenotype of the TAp73−/− selective knockout animals is infertility due to genomic instability of the oocyte, further revealing a role for TAp73 as a tumor suppressor in maintaining the fidelity of the genome.Citation23,Citation37,Citation45 Although p73 shares some biological functions with p53, the contribution of p73 activity on the regulation of cell metabolism and energy production has not been fully investigated yet.
Compounds that decrease the interaction of raptor with mTOR (e.g., the macrocyclic lactone rapamycin) inhibit protein synthesis rates, and several new classes of drugs (rapalogs) have been developed that target the mTOR pathway.Citation46 Importantly, these rapalogs are providing, and are being further developed, for new treatments for some types of cancers. Studies have been performed to assess the global changes that occur in the translatome upon exposure to rapamycin;Citation7 however, there has been no analysis of the metabolic changes that result following exposure to such agents. Given the use of these compounds as anticancer agents and the importance that changes in the metabolome have on the development of the cancerous cell, it is timely to assess the global effects of rapamycin on the metabolome. Both normal control and TAp73−/− fibroblasts were used in order to explore the selective role of TAp73−/− in controlling cellular metabolism regulated by mTOR pathway. In total, of 289 biochemical metabolites involved in very different metabolic pathways, 39 metabolites were found to be significantly affected by rapamycin treatment; many of these are associated to cellular stress response.
Results
Genotype has a minor effect on both time- and treatment-mediated biochemical changes
Activated TORC1 promotes protein translation, increases cell mass, and promotes cell growth through the phosphorylation and regulation of two major substrates, the 4EBP1 protein (eIF4E binding protein) and the S6 kinase.Citation7,Citation8 Thus, mTOR inhibition was confirmed using phospho-S6K1 and phospho-4EBP as markers of mTOR activity. Rapamycin treatment suppressed the phosphorylation of the mTOR substrate S6K1 during starvation, and both in a dose-dependent (Fig. S1A) and time-dependent manner (Fig. S1B). In addition, phosphorylation of the 4EBP substrate decreased in the MEFs treated with rapamycin at different concentrations compared with the untreated cells. Rapamycin was then used at 50 nM concentrations for further studies. Three cell culture conditions were compared: (1) starved cells, collected at a 4 h time point, (2) untreated cells, and (3) cells treated with rapamycin. Each sample was collected at both 24 and 48 h time points, with 5–8 individual biological replicates for each group. In order to determine whether there were cytotoxic effects associated with presence of rapamycin, the induction of apoptosis was examined by FACS analysis. The data show that there was no significant differences in cell death between rapamycin-treated and untreated cells within the time frame used. Similarly, there were no significant differences in the cell cycle profile (Fig. S1C).
Global biochemical profiles, by either gas chromatography/mass spectrometry (GC/MS) or liquid chromatography/mass spectrometry (LC/MS/MS), were performed as described,Citation47 comparing between control vs. TAp73−/− MEFs. Overall, a total of 289 metabolites were identified (; Table S1), 39 of which () were found significantly altered by exposure to rapamycin.
Table 1. Metabolic changes
Based on random chance alone and a P value cut-off of p ≤ 0.05, it could be expected that from among the 289 biochemicals identified in the MEFs from this study, 14 to 15 compounds would achieve statistical significance. The number of biochemical metabolites showing significant changes between wt and TAp73−/− cells at each treatment and time point in this study ranged from 8 to 21 compounds depending upon the comparison. This low number of genotype-based differences suggests that, under the present experimental conditions, TAp73−/− cells had little to no effect on the various treatments at any given time point. A heatmap representing the similar patterns in biochemical changes between wt and TAp73−/− cells is shown in .
Table 2. Genotype in general has only minor effects on both time- and treatment-mediated biochemical changes
The increased levels of amino acids and their metabolites in the rapamycin-treated samples, regardless of genotype, would support the role of rapamycin as an inhibitor of protein synthesis.
Glycolytic intermediates are increased by rapamycin treatment
Glucose () and the majority of the glycolytic intermediates, such as glucose-6-phosphate (G6P, ), fructose-6-phosphate (), fructose 1,6-diphosphate (presented as an isobar with glucose 1,6-diphosphate, ), dihydroxyacetone phosphate (DHAP, ), and 3-phosphoglycerate (), were increased in both wt and TAp73−/− MEFs following rapamycin treatment as compared with untreated samples. This change in glycolytic intermediates was most profound at the 48 h time point. In contrast, lactate was decreased in both wt and TAp73−/− cells treated with rapamycin at 48 h as compared with their relative untreated samples ().
Figure 1. Glycolytic metabolites. Glycolysis is the almost universal pathway that converts glucose into pyruvate. In aerobic organisms the pyruvate passes into the mitochondria where it is completely oxidized by O2 into CO2 and H2O and its potential energy largely conserved as ATP. In the absence of sufficient oxygen, the pyruvate is reduced by the NADH to lactate in animals. The effect of rapamycin exposure for 24 or 48 h is shown on glycolytic metabolites: glucose (A), glucose-6-phosphate (B), fructose-6-phosphate (C), isobar 3-diphosphate, glucose-1,6-diphosphate, myo-inositol-1 (D), dihydroxyacetone phosphate (E), 3-phosphpoglycerate (F), lactate (G). See “Materials and Methods” for technical protocols. Relative values are indicated over control. Left panels: blue, show wild-type MEFs; right panels: red, show TAp73−/− MEFs. WT, wild-type; MEFs; KO, TAp73 knockout MEFs; U, untreated; R, rapamycin; S, starvation.
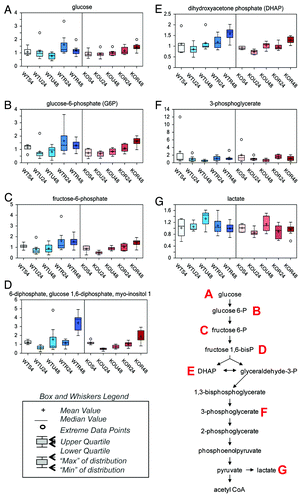
These changes could suggest either a block in glycolysis or increased glucose uptake and metabolism.
Rapamycin increases the pentose phosphate pathway activity at 48 h
To gain further insight into the fate of glucose following rapamycin treatment, we investigated changes in TCA cycle and pentose phosphate pathway intermediates. Although rapamycin had little effect on TCA cycle activity, it appeared to have a great impact on pentose phosphate pathway activity. Following the phosphorylation of glucose to G6P, G6P either continues through glycolysis or is shunted into the pentose phosphate pathway. In the pentose phosphate pathway, G6P is converted to 6-phosphogluconate. Conversion of G6P to 6-phosphogluconate results in increased NADPH levels and potentially also increases nucleotide biosynthesis through the subsequent generation of ribose 5-phosphate. Thus, increased pentose phosphate pathway activity may occur to circumvent periods of increased oxidative stress or during periods of increased anabolic/pro-growth activity. In our samples, the levels of the pentose phosphate pathway intermediates, ribulose-5-phosphate (presented as an isobar with xylulose 5-phosphate, ), ribose 5-phosphate (), and ribose () were all significantly increased after 48 h treatment with rapamycin in both wt and TAp73−/− cells. These changes in pentose phosphate pathway intermediates may suggest that rapamycin treatment, regardless of genotype, results in increased glucose uptake. The glucose could then be phosphorylated and shunted it into the pentose phosphate pathway due to possible increased oxidative stress or for anabolic purposes following rapamycin treatment.
Figure 2. Pentose phosphate pathway. The pentose phosphate pathway is the major source for the NADPH required for anabolic processes and is also responsible for the production of ribose-5-phosphate, which is an important part of nucleic acids. Finally the pathway can also be used to produce glyceraldehyde-3-phosphate, which can then be fed into the Krebs cycle and electron transport chain allowing for the harvest of energy. The effect of rapamycin exposure for 24 or 48 h is shown on isobar ribulose-5-phosphate, xylulose-5-phosphate (A), ribose-5-phosphate (B), ribose (C). See for color and legend code.
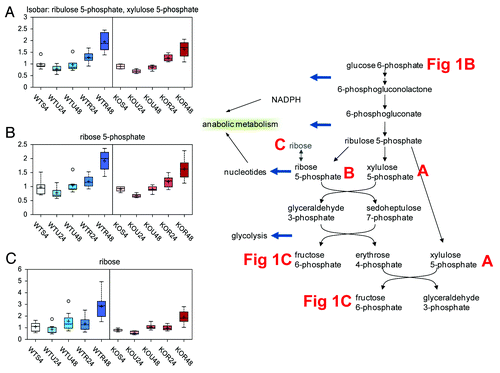
Since the pentose phosphate pathway can feed back into glycolysis at fructose 6-phosphate, this may explain why the subsequent glycolytic intermediates were also increased after 48 h treatment with rapamycin in both wt and TAp73−/− cells as compared with the other treatments and time points.
Rapamycin affects methionine metabolism
Methionine is metabolized to the major methyl donor S-adenosylmethionine (SAM). Utilization of SAM for methylation events results in increased S-adenosylhomocysteine (SAH), which is further metabolized to homocysteine. Homocysteine can either go through the transulfuration pathway to produce cysteine for either taurine or glutathione biosynthesis, or through the transmethylation pathway by reacting with 5-methyltetrahydrofolate (5MeTHF), resulting in methionine and THF. Regulation of the methionine metabolic pathway is based on the availability of methionine and cysteine. If both amino acids are present in adequate quantities, SAM accumulates and is a positive effector on cystathionine synthase, encouraging the production of cysteine and α-ketobutyrate (both of which are glucogenic). However, if methionine is scarce, SAM will form only in small quantities, thus limiting cystathionine synthase activity. In this study, methionine levels were significantly elevated by rapamycin treatment (), while rapamycin did not appear to impact SAM or SAH levels (, respectively). In contrast, lower levels of 5MeTHF, homocysteine, and cysteine () were detected in both wt and TAp73−/− cells treated with rapamycin, generally more evident at the 48 h rapamycin time point. Although these taurine biosynthesis-associated biochemicals (cysteine sulfinic acid, hypotaurine, and taurine) changed over time, the levels of these biochemicals at a given time point showed little difference between rapamycin-treated and untreated in both wt and TAp73−/− cells (Fig. S2A–C). Thus the lower cysteine levels in cells treated for 48 h with rapamycin, both in wt and TAp73−/− MEFs, do not appear to be due to diversion of cysteine to taurine biosynthesis.
Figure 3. Methionine metabolism. Methionine metabolism consists of 2 pathways, the methionine cycle and the transsulfuration sequence. These pathways share 3 common reactions with both, including the conversion of methionine to S-adenosylmethionine (SAM), the utilization of SAM in diverse transmethylation reactions yielding a methylated product plus S-adenosylhomocysteine (SAH), and the cleavage of SAH to yield homocysteine and adenosine. The transulfuration reactions that produce cysteine from homocysteine and serine also produce α-ketobutyrate, the latter being converted first to propionyl-CoA and then via a 3-step process to succinyl-CoA. The effect of rapamycin exposure for 24 or 48 h is shown on methionine (A), SAM (B), SAH (C), 5-methyltetrahydrofolate (D), homocysteine (E), cysteine (F). See for color and legend code.
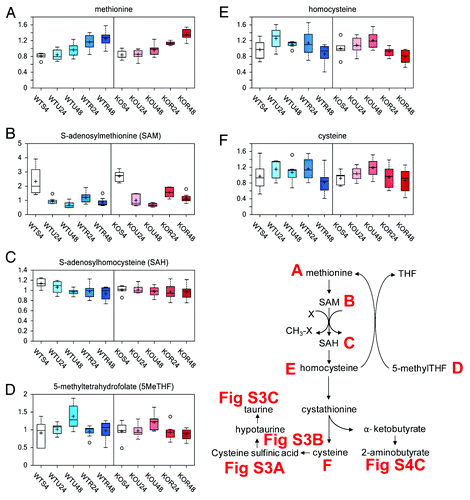
The described changes in methionine metabolism suggest that 48 h rapamycin treatment may be increasing transmethylation of homocysteine to methionine. Effect of rapamycin on transulfuration is reflected by components of the glutathione metabolic pathways.
Rapamycin treatment impacts glutathione synthesis
Glutathione is the major antioxidant pathway in cells. Although changes in reduced glutathione (GSH) and oxidized glutathione (GSSG) did not provide a clear indication of altered redox status with rapamycin (Fig. S3A and B), additional markers provide support for an oxidative stress following treatment with rapamycin, suggesting a possible increase in glutathione utilization in rapamycin-treated wt and TAp73−/− cells, with higher levels of cysteine-glutathione disulphide () and biochemicals associated with glutathione turnover such as cysteinylglycine (), gamma-glutamyl amino acids (), and 5-oxoproline () as compared with untreated cells. In addition, during periods of increased glutathione biosynthesis (such as increased oxidative stress), cysteine levels can become depleted or greatly decreased (). Depletion or decreased cysteine levels can then subsequently result in gamma-glutamylcysteine synthetase reacting with 2-aminobutyrate (Fig. S3C) or alanine (Fig. S3D). The replacement of cysteine with 2-aminobutyrate or alanine results in synthesis of ophthalmate or norophthalmate, respectively. Both ophthalmate () and norophthalmate () were significantly increased in both wt and TAp73−/− cells treated for 48 h with rapamycin, as compared with the other time points and treatments. Higher levels of ophthalmate and likely norophthalmate are indicative of increased oxidative stress.
Figure 4. Glutathione pathway. Glutathione can be found in the cell in oxidized (glutathione disulphide, GSSC) and reduced (glutathione, GSH) form. Reduced glutathione can be either directly formed from glutathione disulfide as the result of activity of glutathione reductase or from conjugation of L-cysteinyl-glycine with (L)-glutamic acid catalyzed by gamma-glutamyltranspeptidase (GGT). L-Cysteinyl-glycine is formed as a result of glutathione conjugation to the L-amino acid moiety catalyzed by GGT. This reaction results in formation of gamma-(L)-glutamyl-aminoacid which is converted by gamma-glutamylcyclotransferase to the 5-oxo-(L)-proline and L-amino acid. 5-Oxo-(L)-proline is then reduced to (L)-glutamic acid. Glutathione synthetase (GCS and GS) catalyzes subsequent conjugation of gamma-(L)-glutamyl-(L)-cysteine and glycine to form glutathione. The effect of rapamycin exposure for 24 or 48 h is shown on cysteine-glutathione disulphide (A), cysteinylglycine (B), gamma-glutamylleucine (C), gamma-glutamylisoleucine (D), 5-oxoproline (E), cysteine (F), ophthalmate (G), norophthalmate (H). See for color and legend code.
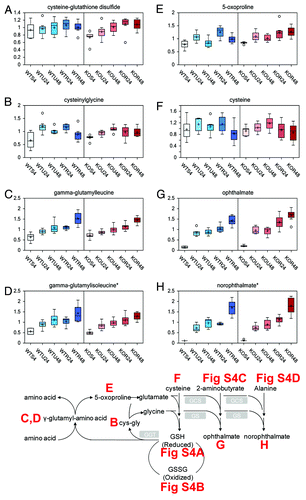
The changes in glutathione metabolism suggest that rapamycin treatment, especially after 48 h, results in a higher oxidative environment regardless of genotype. Further investigation into rapamycin and oxidative stress would be warranted.
Starvation appears to decrease eicosanoid biosynthesis, while TAp73−/− may regulate prostaglandin D2 synthase
The essential fatty acids linoleate ([18:2n6], ) and gamma-linolenate (18:3n6) (presented as an isobar with α-linolenate ([18:3n3], ) were significantly higher in starved cells as compared with untreated and rapamycin cells, regardless of genotype. These essential fatty acids can be further metabolized to the free fatty acid arachidonate (20:4n6). The majority of inflammation-associated prostaglandins are formed from arachidonate (lone exception is prostaglandin E1 formed from the essential fatty acid dihomo-linolenate). Although arachidonate was in general not significantly different among the different treatments (), the majority of the prostaglandins (prostaglandin E1, E2, I2, and 6-keto prostaglandin F1 α) were significantly higher in the untreated and rapamycin cells as compared with starved cells regardless of time or genotype (Fig S4A–D). The lone exception was prostaglandin D2 (), which was generally much lower in the TAp73−/− cells as compared with wt cells. This may suggest a role for TAp73 in the regulation of prostaglandin D2 synthase expression or activity.
Figure 5. Eicosanoid biosynthesis and prostaglandins. Two main pathways are involved in the biosynthesis of eicosanoids. The prostaglandins and thromboxanes are synthesized by the cyclic pathway, the leukotrienes by the linear pathway. Numerous stimuli activate the phospholipase A2 (PLA2), which hydrolyzes arachidonic acid from membrane phospholipids. The prostaglandins are identified as PG. Prostaglandin PGI2 is also known as prostacyclin. PGE2 is synthesized from PGH2 via the action of one of several PGE synthases (PTGES). PTGDS synthases converts PGD2 from PGH2. PGI2 is synthesized from PGH2 via the action of CYP8A. The effect of rapamycin exposure for 24 or 48 h is shown on linoleate (18:2n6) (A), isobar linolenate (18:3n3) α, or gamma (B), arachidonate (20:4n6) (C), PGD2 (D). See for color and legend code.
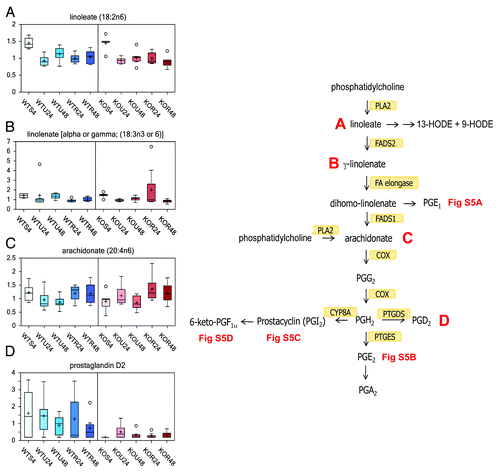
The changes in biochemicals associated with prostaglandin biosynthesis suggest a role for starvation as an inhibitor of inflammatory prostaglandin biosynthesis.
Discussion
The TOR intracellular signal transduction pathway is conserved in eukaryotes from yeast to mammals,Citation48 here mTOR integrates signals (nutrients, hormones, growth factors, stressors) through a complex signaling network by increasing protein synthesis and inhibiting autophagy, which, in turn, results in driving cellular mass growth.Citation49-Citation53 Rapamycin is an inhibitor of the mTOR protein kinase that has long been employed for immunosuppression and more recently as an anticancer treatment. Despite a very large number of studies on rapamycin and the increasing use of rapalogues in cancer chemotherapy, its effects on the global metabolome profile had not yet been investigated.
To assess whether rapamycin directly influenced the cell metabolism, we generated MEFs from E14.5 embryos, passaged them in vitro according to standard 3T3 protocol, left them to starvation for 4 h or treated with rapamycin, and, by analyzing 289 metabolites, we obtained a rapamycin-dependent global metabolome profile. Both rapamycin and starvation resulted in a large number of changes when compared with untreated and starved conditions. Some of the changes associated with the treatment of MEFs with rapamycin included a possible increase in the oxidative environment. The glycolytic intermediates, such as glucose-6-phosphate, fructose-6-phosphate, fructose 1,6-diphosphate, dihydroxyacetone phosphate, and 3-phosphoglycerate, were increased in time, especially at 48 h, indicating either a block in glycolysis or increased glucose uptake and metabolism. However, a greater impact was observed on pentose phosphate pathway activity, resulting in increased levels of 6-phosphogluconate and NADPH. This could suggest an increased nucleotide biosynthesis through the generation of ribose 5-phosphate.
Changes in methionine and glutathione metabolism and increased pentose phosphate pathway, especially at the rapamycin 48 h time point, support an increased oxidative environment in the rapamycin-treated cells. The cause of this higher oxidative environment by rapamycin was not clear from the data. Rapamycin caused an increase in the steady-state methionine levels, while SAM was reduced, suggesting a possible increased transmethylation of homocysteine to methionine. The metabolite profiling suggested also a possible increase in glutathione utilization, with higher levels of cysteine-glutathione disulphide. The changes in glutathione metabolism support the possibility of a higher oxidative environment. Finally, one of the changes associated with starvation was an apparent decrease in inflammation, while TAp73 may regulate the formation of prostaglandin D2.
The genotype of the MEFs had little to no effect on the various treatments at any given time point. At least under the experimental condition used in the present experiment, the metabolic effect of rapamycin was independent of the genotype of TAp73. Indeed, this result was somehow unexpected, as there are several reports linking p73 to mTOR.Citation54,Citation55 Indeed, using a p73 gene signature by integrating whole-genome chromatin immunoprecipitation and expression profiling in a rhabdomyosarcoma cell line, there was enrichment for direct or indirect inhibitors of mTOR.Citation54 This was further refined, as the p73 genomic binding profile demonstrates that rapamycin selectively increased p73 occupancy at a subset of its binding, with rapamycin acting as an inducer of p73.Citation55 Moreover, TAp73 is able to regulate the activity of the mitochondrial complex 4 subunit cox4i1,Citation38 while it is highly probable that it regulates other metabolic pathways, similarly to p53 regulating glutaminase-2Citation56,Citation57 and the serine biosynthesis,Citation58,Citation59 resulting in significant metabolic alterations. Moreover, like rapamycin, which is known to suppress senescence, p53 inhibits the mTOR pathway, thus causing quiescence.Citation60-Citation62 One possible explanation for our negative metabolome results is that embryonic fibroblasts are not a major tissue where TAp73 exerts its function under physiological circumstances; in addition, the developmental role of p73 might differ from its role in cancer biology.
In the present report, we show the ability of rapamycin to significantly alter a significant set of biochemical metabolites, evaluated by GC/MS and LC/MS/MS, affecting the biochemical status of the cell in the glycolytic intermediates and the pentose phosphate pathway. The changes on the methionine and glutathione metabolites suggest a higher oxidative environment.
Materials and Methods
Mice and MEF preparation
Generation and genotyping of TAp73-knockout mice were previously described.Citation37 Animals were treated in accordance with the NIH “Guide for Care and Use of Laboratory Animals”, as approved by the Ontario Cancer Institute Animal Care Committee. Mice were bred and subject to procedures under the project license released from the Home Office, in agreement with MRC requirements.
Fibroblasts were isolated from E13.5 WT and TAp73−/− littermate embryos and cultured according to 3T3 protocol in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum, 2 mM L-glutamine and 1% penicillin/streptomycin. At passage 2, MEF both wt and TAp73−/− were treated with 50 nM rapamacyn for 24 and 48 h. All experiments were performed on littermates.
Western blot
Proteins were extracted with RIPA buffer containing cocktail inhibitors (Roche), and concentration was determined using a Bradford dye-based assay (Biorad). Total protein (30 μg) was subjected to SDS-PAGE followed by immunoblotting with appropriate antibodies at the recommended dilutions. The blots were then incubated with peroxidase linked secondary antibodies followed by enhanced-chemiluminescent detection using Super Signal chemiluminescence kit (Thermo scientific). Antibodies: S6K1 (Cell Signaling, 1:1000), P-S6K1 (Cell Signaling, 1:1000), 4E-BP (Cell Signaling, 1:1000), P-4E-BP (Cell Signaling, 1:1000), Tubulin (Santa Cruz, 1:1000).
Flow cytometry analysis
Flow cytometry was performed as described in.Citation27 Briefly, wt and TAp73−/− MEFs were seeded at 3 × 105 cells per 100 mm plate. Cultures were then either left untreated or treated with rapamycin. At 24 and 48 h after treatment, cells were harvested with 0.025% trypsin for 3 min at 37 °C and then, after addition of 10% FCS in PBS, the cells were centrifuged, washed with PBS, and fixed in 70% cold ethanol. The harvesting of cells using these conditions led to a high yield of undamaged cells. After fixation, cells were washed with PBS, treated for 15 min at 37 °C with RNase (100 μg/ml) and then stained with propidium iodide (10 μg/ml in the dark for 30 min). The samples were then analyzed using a FACScan flow cytometer (Becton Dickinson).
Metabolomic analysis
Samples were immediately stored at -80°C, and, at the time of analysis, were extracted and prepared for analysis using a standard metabolic solvent extraction method, as described in ref Citation47. Briefly, the extracted samples were split into equal parts for analysis by gas chromatography/mass spectrometry (GC/MS) or liquid chromatography/mass spectrometry (LC/MS/MS) platforms. Also included were several technical replicate samples created from a homogeneous pool containing a small amount of all study samples. Global biochemical profiles were compared between wild-type and TAp73−/− knockout mouse embryonic fibroblasts. Three culture conditions were compared in this study: (1) starved cells collected at a 4 h time point, (2) untreated cells, and (3) cells treated with rapamycin each collected at both 24 and 48 h time points; 5–8 independent biological replicates were analyzed per each point.
The LC/MS portion of the platform was based on a Waters ACQUITY UPLC and a Thermo-Finnigan LTQ mass spectrometer, which consisted of an electrospray ionization source and linear ion-trap mass analyzer. The sample extract was split into two aliquots, dried, and then reconstituted in acidic or basic LC-compatible solvents, each of which contained 11 or more injection standards at fixed concentrations. One aliquot was analyzed using acidic positive ion optimized conditions and the other using basic negative ion optimized conditions in two independent injections using separate dedicated columns. Extracts reconstituted in acidic conditions were gradient eluted using water and methanol both containing 0.1% Formic acid, while the basic extracts, which also used water/methanol, contained 6.5 mM ammonium bicarbonate. The MS analysis alternated between MS and data-dependent MS2 scans using dynamic exclusion.
The samples destined for GC/MS analysis were re-dried under vacuum desiccation for a minimum of 24 h prior to being derivatized under dried nitrogen using bistrimethyl-silyl-triflouroacetamide (BSTFA). The GC column was 5% phenyl and the temperature ramp is from 40° to 300°C in a 16 min period. Samples were analyzed on a Thermo-Finnigan Trace DSQ fast-scanning single-quadrupole mass spectrometer using electron impact ionization. The instrument was tuned and calibrated for mass resolution and mass accuracy on a daily basis. The information output from the raw data files was automatically extracted.
For ions with counts greater than 2 million, an accurate mass measurement could be performed. Accurate mass measurements could be made on the parent ion as well as fragments. The typical mass error was less than 5 ppm. Ions with less than 2 million counts require a greater amount of effort to characterize. Fragmentation spectra (MS/MS) were typically generated in a data-dependent manner, but, if necessary, targeted MS/MS could be employed, such as in the case of lower level signals. Compounds were identified by comparison with library entries of purified standards or recurrent unknown entities. Identification of known chemical entities was based on comparison with metabolic library entries of purified standards. The combination of chromatographic properties and mass spectra gave an indication of a match to the specific compound or an isobaric entity.
Data quality: Instrument and process variability
Instrument variability was determined by calculating the median relative standard deviation (RSD) for the internal standards that were added to each sample prior to injection into the mass spectrometers. Overall process variability was determined by calculating the median RSD for all endogenous metabolites (i.e., non-instrument standards) present in 100% of the matrix samples, which are technical replicates of pooled client samples.
Metabolite summary and significantly altered biochemicals
The metabolic analysis, as from above, comprises a total of 289 named biochemicals. Following normalization to total protein determined by Bradford assay, log transformation, and imputation, with minimum observed values was performed for each compound. Welch’s two-sample t tests were then used to identify biochemicals that differed significantly between experimental groups. A summary of the numbers of biochemicals that achieved statistical significance (P ≤ 0.05), as well as those approaching significance (0.05 < P < 0.1), is shown as a table and a supplementary table in the “Results”.
An estimate of the false discovery rate (q value) is calculated to take into account the multiple comparisons that normally occur in metabolic-based studies. For example, when analyzing 200 compounds, we would expect to see about 10 compounds meeting the P ≤ 0.05 cut-off by random chance. The q value describes the false discovery rate; a low q value (q < 0.10) is an indication of high confidence in a result. While a higher q value indicates diminished confidence, it does not necessarily rule out the significance of a result.
| Abbreviations: | ||
| mTOR | = | mammalian target of rapamycin |
| wt | = | wild-type mice |
| MEF | = | mouse embryonic fibroblasts |
| TCA | = | tricarboxylic acid cycle |
| G6P | = | glucose-6-phosphate |
| SAM | = | S-adenosylmethionine |
| DHAP | = | dihydroxyacetone phosphate |
| SAH | = | S-adenosylhomocysteine |
| THF | = | tetrahydrofolate |
| GSH | = | reduced glutathione |
| GSSG | = | oxidized glutathione |
| 4EBP1 | = | eIF4E binding protein |
| GC/MS | = | gas chromatography/Mass Spectrometry |
| LC/MS/MS | = | liquid chromatography/mass spectrometry |
| 5MeTHF | = | 5-methyltetrahydrofolate |
Additional material
Download Zip (2 MB)Acknowledgments
This work has been supported by the Medical Research Council, UK; grants from “Alleanza contro il Cancro” (ACC12), MIUR/PRIN (20078P7T3K_001)/FIRB (RBIP06LCA9_0023, RBIP06LCA9_0C), AIRC (2008–2010_33–08) (#5471) (2011-IG11955), AIRC 5xmille (#9979), Italian Human ProteomeNet RBRN07BMCT, MIUR/PRIN (2008MRLSNZ_004), Telethon Grant (GGPO9133), to GM Research described in this article was also supported in part by Min. Salute (Ricerca oncologica 26/07) and IDI-IRCCS (RF06 c.73, RF07 c.57, RF08 c.15, RF07 c.57) to GM. Work was supported by Ministry of Education and Science of the Russian Federation (11.G34.31.0069).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/cc/article/25450
Related Research Data
References
- Markert EK, Levine AJ, Vazquez A. Proliferation and tissue remodeling in cancer: the hallmarks revisited. Cell Death Dis 2012; 3:e397; http://dx.doi.org/10.1038/cddis.2012.140; PMID: 23034332
- Lyssiotis CA, Vander-Heiden MG, Muñoz-Pinedo C, Emerling BM. Emerging concepts: linking hypoxic signaling and cancer metabolism. Cell Death Dis 2012; 3:e303; http://dx.doi.org/10.1038/cddis.2012.41; PMID: 22552280
- Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004; 6:91 - 9; http://dx.doi.org/10.1016/j.ccr.2004.06.007; PMID: 15261145
- Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet 2005; 37:19 - 24; http://dx.doi.org/10.1038/ng1494; PMID: 15624019
- Inoki K, Guan KL. Tuberous sclerosis complex, implication from a rare genetic disease to common cancer treatment. Hum Mol Genet 2009; 18:R1 R94 - 100; http://dx.doi.org/10.1093/hmg/ddp032; PMID: 19297407
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 2006; 6:729 - 34; http://dx.doi.org/10.1038/nrc1974; PMID: 16915295
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012; 485:109 - 13; http://dx.doi.org/10.1038/nature11083; PMID: 22552098
- Shahbazian D, Parsyan A, Petroulakis E, Topisirovic I, Martineau Y, Gibbs BF, et al. Control of cell survival and proliferation by mammalian eukaryotic initiation factor 4B. Mol Cell Biol 2010; 30:1478 - 85; http://dx.doi.org/10.1128/MCB.01218-09; PMID: 20086100
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19:107 - 20; http://dx.doi.org/10.1038/cdd.2011.96; PMID: 21760595
- Vandenabeele P, Melino G. The flick of a switch: which death program to choose?. Cell Death Differ 2012; 19:1093 - 5; http://dx.doi.org/10.1038/cdd.2012.65; PMID: 22684093
- Marcel V, Dichtel-Danjoy ML, Sagne C, Hafsi H, Ma D, Ortiz-Cuaran S, et al. Biological functions of p53 isoforms through evolution: lessons from animal and cellular models. Cell Death Differ 2011; 18:1815 - 24; http://dx.doi.org/10.1038/cdd.2011.120; PMID: 21941372
- Marcel V, Petit I, Murray-Zmijewski F, Goullet de Rugy T, Fernandes K, Meuray V, et al. Diverse p63 and p73 isoforms regulate Δ133p53 expression through modulation of the internal TP53 promoter activity. Cell Death Differ 2012; 19:816 - 26; http://dx.doi.org/10.1038/cdd.2011.152; PMID: 22075982
- Nayak G, Cooper GM. p53 is a major component of the transcriptional and apoptotic program regulated by PI 3-kinase/Akt/GSK3 signaling. Cell Death Dis 2012; 3:e400; http://dx.doi.org/10.1038/cddis.2012.138; PMID: 23059819
- Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene 2013; http://dx.doi.org/10.1038/onc.2012.640; PMID: 23416979
- Tucci P. Caloric restriction: is mammalian life extension linked to p53?. Aging (Albany NY) 2012; 4:525 - 34; PMID: 22983298
- Melino G. Journal club. A cancer biologist weighs up p53, metabolism and cancer. Nature 2010; 466:905; http://dx.doi.org/10.1038/466905d; PMID: 20725003
- Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ 2011; 18:1487 - 99; http://dx.doi.org/10.1038/cdd.2011.81; PMID: 21760596
- Shalom-Feuerstein R, Lena AM, Zhou H, De La Forest Divonne S, Van Bokhoven H, Candi E, et al. ΔNp63 is an ectodermal gatekeeper of epidermal morphogenesis. Cell Death Differ 2011; 18:887 - 96; http://dx.doi.org/10.1038/cdd.2010.159; PMID: 21127502
- Ben-Sahra I, Dirat B, Laurent K, Puissant A, Auberger P, Budanov A, et al. Sestrin2 integrates Akt and mTOR signaling to protect cells against energetic stress-induced death. Cell Death Differ 2013; 20:611 - 9; http://dx.doi.org/10.1038/cdd.2012.157; PMID: 23238567
- Salah Z, Bar-mag T, Kohn Y, Pichiorri F, Palumbo T, Melino G, et al. Tumor suppressor WWOX binds to ΔNp63α and sensitizes cancer cells to chemotherapy. Cell Death Dis 2013; 4:e480; http://dx.doi.org/10.1038/cddis.2013.6; PMID: 23370280
- Leonard MK, Kommagani R, Payal V, Mayo LD, Shamma HN, Kadakia MP. ΔNp63α regulates keratinocyte proliferation by controlling PTEN expression and localization. Cell Death Differ 2011; 18:1924 - 33; http://dx.doi.org/10.1038/cdd.2011.73; PMID: 21637289
- Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol 2011; 12:259 - 65; http://dx.doi.org/10.1038/nrm3086; PMID: 21427767
- Tomasini R, Mak TW, Melino G. The impact of p53 and p73 on aneuploidy and cancer. Trends Cell Biol 2008; 18:244 - 52; http://dx.doi.org/10.1016/j.tcb.2008.03.003; PMID: 18406616
- Notari M, Hu Y, Koch S, Lu M, Ratnayaka I, Zhong S, et al. Inhibitor of apoptosis-stimulating protein of p53 (iASPP) prevents senescence and is required for epithelial stratification. Proc Natl Acad Sci U S A 2011; 108:16645 - 50; http://dx.doi.org/10.1073/pnas.1102292108; PMID: 21930934
- Candi E, Rufini A, Terrinoni A, Giamboi-Miraglia A, Lena AM, Mantovani R, et al. DeltaNp63 regulates thymic development through enhanced expression of FgfR2 and Jag2. Proc Natl Acad Sci U S A 2007; 104:11999 - 2004; http://dx.doi.org/10.1073/pnas.0703458104; PMID: 17626181
- Rivetti di Val Cervo P, Lena AM, Nicoloso M, Rossi S, Mancini M, Zhou H, et al. p63-microRNA feedback in keratinocyte senescence. Proc Natl Acad Sci U S A 2012; 109:1133 - 8; http://dx.doi.org/10.1073/pnas.1112257109; PMID: 22228303
- Tucci P, Agostini M, Grespi F, Markert EK, Terrinoni A, Vousden KH, et al. Loss of p63 and its microRNA-205 target results in enhanced cell migration and metastasis in prostate cancer. Proc Natl Acad Sci U S A 2012; 109:15312 - 7; http://dx.doi.org/10.1073/pnas.1110977109; PMID: 22949650
- Müller M, Schilling T, Sayan AE, Kairat A, Lorenz K, Schulze-Bergkamen H, et al. TAp73/Delta Np73 influences apoptotic response, chemosensitivity and prognosis in hepatocellular carcinoma. Cell Death Differ 2005; 12:1564 - 77; http://dx.doi.org/10.1038/sj.cdd.4401774; PMID: 16195739
- Wang J, Liu YX, Hande MP, Wong AC, Jin YJ, Yin Y. TAp73 is a downstream target of p53 in controlling the cellular defense against stress. J Biol Chem 2007; 282:29152 - 62; http://dx.doi.org/10.1074/jbc.M703408200; PMID: 17693405
- Grob TJ, Novak U, Maisse C, Barcaroli D, Lüthi AU, Pirnia F, et al. Human delta Np73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death Differ 2001; 8:1213 - 23; http://dx.doi.org/10.1038/sj.cdd.4400962; PMID: 11753569
- Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG Jr., Levrero M, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 1999; 399:806 - 9; http://dx.doi.org/10.1038/21690; PMID: 10391249
- Rossi M, De Laurenzi V, Munarriz E, Green DR, Liu Y-C, Vousden KH, et al. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J 2005; 24:836 - 48; http://dx.doi.org/10.1038/sj.emboj.7600444; PMID: 15678106
- Rossi M, Aqeilan RI, Neale M, Candi E, Salomoni P, Knight RA, et al. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc Natl Acad Sci U S A 2006; 103:12753 - 8; http://dx.doi.org/10.1073/pnas.0603449103; PMID: 16908849
- Peschiaroli A, Scialpi F, Bernassola F, Pagano M, Melino G. The F-box protein FBXO45 promotes the proteasome-dependent degradation of p73. Oncogene 2009; 28:3157 - 66; http://dx.doi.org/10.1038/onc.2009.177; PMID: 19581926
- Dulloo I, Gopolan G, Melino G, Sabapathy S. The DeltaNp73 oncogene is degraded in a c-Jun-dependent manner upon genotoxic stress through the antizyme-mediated pathway. Proc Natl Acad Sci USA 2010; 107:4902 - 7; http://dx.doi.org/10.1073/pnas.0906782107; PMID: 20185758
- Sayan BS, Yang AL, Conforti F, Tucci P, Piro MC, Browne GJ, et al. Differential control of TAp73 and DeltaNp73 protein stability by the ring finger ubiquitin ligase PIR2. Proc Natl Acad Sci U S A 2010; 107:12877 - 82; http://dx.doi.org/10.1073/pnas.0911828107; PMID: 20615966
- Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 2008; 22:2677 - 91; http://dx.doi.org/10.1101/gad.1695308; PMID: 18805989
- Rufini A, Niklison-Chirou MV, Inoue S, Tomasini R, Harris IS, Marino A, et al. TAp73 depletion accelerates aging through metabolic dysregulation. Genes Dev 2012; 26:2009 - 14; http://dx.doi.org/10.1101/gad.197640.112; PMID: 22987635
- Wilhelm MT, Rufini A, Wetzel MK, Tsuchihara K, Inoue S, Tomasini R, et al. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev 2010; 24:549 - 60; http://dx.doi.org/10.1101/gad.1873910; PMID: 20194434
- Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000; 404:99 - 103; http://dx.doi.org/10.1038/35003607; PMID: 10716451
- Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 2000; 289:304 - 6; http://dx.doi.org/10.1126/science.289.5477.304; PMID: 10894779
- Billon N, Terrinoni A, Jolicoeur C, McCarthy A, Richardson WD, Melino G, et al. Roles for p53 and p73 during oligodendrocyte development. Development 2004; 131:1211 - 20; http://dx.doi.org/10.1242/dev.01035; PMID: 14960496
- Agostini M, Tucci P, Killick R, Candi E, Sayan BS, Rivetti di Val Cervo P, et al. Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proc Natl Acad Sci U S A 2011; 108:21093 - 8; http://dx.doi.org/10.1073/pnas.1112061109; PMID: 22160687
- Agostini M, Tucci P, Steinert JR, Shalom-Feuerstein R, Rouleau M, Aberdam D, et al. microRNA-34a regulates neurite outgrowth, spinal morphology, and function. Proc Natl Acad Sci U S A 2011; 108:21099 - 104; http://dx.doi.org/10.1073/pnas.1112063108; PMID: 22160706
- Tomasini R, Tsuchihara K, Tsuda C, Lau SK, Wilhelm M, Ruffini A, et al. TAp73 regulates the spindle assembly checkpoint by modulating BubR1 activity. Proc Natl Acad Sci U S A 2009; 106:797 - 802; http://dx.doi.org/10.1073/pnas.0812096106; PMID: 19139399
- Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer 2004; 4:335 - 48; http://dx.doi.org/10.1038/nrc1362; PMID: 15122205
- Tucci P, Porta G, Agostini M, Dinsdale D, Iavicoli I, Cain K, et al. Metabolic effects of TiO2 nanoparticles, a common component of sunscreens and cosmetics, on human keratinocytes. Cell Death Dis 2013; 4e:549; http://dx.doi.org/10.1038/cddis.2013.76; PMID: 23519118
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell 2006; 124:471 - 84; http://dx.doi.org/10.1016/j.cell.2006.01.016; PMID: 16469695
- Hands SL, Proud CG, Wyttenbach A. mTOR’s role in ageing: protein synthesis or autophagy?. Aging (Albany NY) 2009; 1:586 - 97; PMID: 20157541
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Newbold A, Vervoort SJ, Martin BP, Bots M, Johnstone RW. Induction of autophagy does not alter the anti-tumor effects of HDAC inhibitors. Cell Death Dis 2012; 3:e387; http://dx.doi.org/10.1038/cddis.2012.128; PMID: 22951984
- Melino G, Knight RA, Nicotera P. How many ways to die? How many different models of cell death?. Cell Death Differ 2005; 12:Suppl 2 1457 - 62; http://dx.doi.org/10.1038/sj.cdd.4401781; PMID: 16247490
- Rosenbluth JM, Mays DJ, Pino MF, Tang LJ, Pietenpol JA. A gene signature-based approach identifies mTOR as a regulator of p73. Mol Cell Biol 2008; 28:5951 - 64; http://dx.doi.org/10.1128/MCB.00305-08; PMID: 18678646
- Rosenbluth JM, Mays DJ, Jiang A, Shyr Y, Pietenpol JA. Differential regulation of the p73 cistrome by mammalian target of rapamycin reveals transcriptional programs of mesenchymal differentiation and tumorigenesis. Proc Natl Acad Sci U S A 2011; 108:2076 - 81; http://dx.doi.org/10.1073/pnas.1011936108; PMID: 21245298
- Hu W, Zhang C, Wu R, Sun Y, Levine AJ, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A 2010; 107:7455 - 60; http://dx.doi.org/10.1073/pnas.1001006107; PMID: 20378837
- Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A 2010; 107:7461 - 6; http://dx.doi.org/10.1073/pnas.1002459107; PMID: 20351271
- Ye J, Mancuso A, Tong X, Ward PS, Fan J, Rabinowitz JD, et al. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci U S A 2012; 109:6904 - 9; http://dx.doi.org/10.1073/pnas.1204176109; PMID: 22509023
- Maddocks ODK, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013; 493:542 - 6; http://dx.doi.org/10.1038/nature11743; PMID: 23242140
- Leontieva OV, Gudkov AV, Blagosklonny MV. Weak p53 permits senescence during cell cycle arrest. Cell Cycle 2010; 9:4323 - 7; http://dx.doi.org/10.4161/cc.9.21.13584; PMID: 21051933
- Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010; 2:344 - 52; PMID: 20606252
- Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A 2010; 107:9660 - 4; http://dx.doi.org/10.1073/pnas.1002298107; PMID: 20457898