
Abstract
Cognitive dysfunction has been reported in patients with spinal cord injury (SCI), but it has been questioned whether such changes may reflect concurrent head injury, and the issue has not been addressed mechanistically or in a well-controlled experimental model. Our recent rodent studies examining SCI-induced hyperesthesia revealed neuroinflammatory changes not only in supratentorial pain-regulatory sites, but also in other brain regions, suggesting that additional brain functions may be impacted following SCI. Here we examined effects of isolated thoracic SCI in rats on cognition, brain inflammation, and neurodegeneration. We show for the first time that SCI causes widespread microglial activation in the brain, with increased expression of markers for activated microglia/macrophages, including translocator protein and chemokine ligand 21 (C–C motif). Stereological analysis demonstrated significant neuronal loss in the cortex, thalamus, and hippocampus. SCI caused chronic impairment in spatial, retention, contextual, and fear-related emotional memory—evidenced by poor performance in the Morris water maze, novel objective recognition, and passive avoidance tests. Based on our prior work implicating cell cycle activation (CCA) in chronic neuroinflammation after SCI or traumatic brain injury, we evaluated whether CCA contributed to the observed changes. Increased expression of cell cycle-related genes and proteins was found in hippocampus and cortex after SCI. Posttraumatic brain inflammation, neuronal loss, and cognitive changes were attenuated by systemic post-injury administration of a selective cyclin-dependent kinase inhibitor. These studies demonstrate that chronic brain neurodegeneration occurs after isolated SCI, likely related to sustained microglial activation mediated by cell cycle activation.
Introduction
The impact of spinal cord injury (SCI) has long focused on motor deficits or such secondary complications as neuropathic pain, compromised bladder/bowel function, loss of sexual function, or emotional distress. Cognitive deficits have been reported after SCI and its prevalence has likely been underestimated.Citation1 It has also been questioned as to whether such changes may reflect unappreciated concurrent head injury. Studies that evaluated SCI patients using a battery of neuropsychological tests have identified performance impairments including span memory, executive functioning, attention, processing speed, and learning ability.Citation1-Citation8 Lazzaro et al.Citation5 focused on the impact of SCI on central psychophysiological indices of information processing using scalp-recorded late component event-related potentials (ERPs) and speculated that ERP alterations in SCI reflect impairments in distributed integrative cortical/subcortical networks that are engaged in stimulus detection, evaluation, and executive functioning. It has also been documented that chronic hypotension in persons with SCI is associated with deficits in memory and possibly attention and processing speed.Citation9,Citation10 However, this issue has not been addressed experimentally with regard to mechanism. Only one prior experimental study has suggested cognitive decline after SCI- a projectile injury in pigs wearing body armor.Citation11 But the model, species and outcome used (conditioned feeding behavior) makes both interpretation and relevant comparison difficult.
The pathophysiological changes following experimental SCI have been extensively examined, but most prior work has focused predominantly on the spinal cord and its afferent/efferent pathways. Chang et al.,Citation12 showed that SCI produced a significant apoptotic change and cell death not only in the spinal cord but also in the supraventricular cortex and hippocampal cornu ammonis 1 (CA1) region in the rat brains. The presence of hippocampal pathology after SCI was also suggested by Felix et al.,Citation13 who described long-term reduction of neurogenesis in the hippocampus after subchronic cervical SCI. In our own recent studies examining mechanisms of post-SCI-induced hyperpathia in rats, we observed sustained neuroinflammation not only in brain regions regulating pain sensation,Citation14 but also more diffusely in cortex, thalamus and hippocampus. As the degree and pattern of changes was similar to those associated with the chronic neurodegeneration and cognitive deficits observed after traumatic brain injury (TBI) in rodents,Citation15 we initiated an independent study to examine whether brain neuroinflammation after isolated thoracic SCI, is associated with brain neurodegeneration and related functional changes. Clinically, half of SCI cases result from contusion injuries that produce similar pathophysiological changes to those observed in rat spinal cord impact models. Here we utilized a well characterized rat contusion model.Citation14
Microglia/macrophages have multiple phenotypes: a classically activated, proinflammatory (M1) state that appears to contribute to neurotoxicity; and an alternatively activated (M2) state that may promote repair.Citation16,Citation17 M1/M2 phenotypic switch likely depends on the local signals in the microenvironment. Emerging evidence indicates that increased expression of the 18 kDa translocator protein (TSPO) is a feature of microglial/macrophage activation.Citation18 It has been shown that TSPO is a much more sensitive indicator in detecting brain damage than standard histological markers, as significant elevations in TSPO levels are measured prior to the onset of pathological changes. In addition, induction of the chemokine ligand 21 (C-C motif) (CCL21) following SCI was found in the thalamus,Citation14,Citation19 which induces microglial activation associated with chronic hyperesthesia. Therefore, in the present study, we also examined the effects of SCI on these factors.
Cell cycle related genes and proteins are upregulated in the spinal cord after SCI, as well as in the brain after TBI, and contribute to secondary injury.Citation20,Citation21 More recently, we observed chronic cell cycle activation (CCA) in the posterior thalamic nucleus after rat thoracic contusion SCI, which was associated with hyperesthesia, increased neuronal excitability and microglial activation.Citation14 Therefore, in the current study we examined expression levels of CCA in selected brain regions following SCI, as well as the effects of inhibiting CCA on behavioral and histological outcomes. SCI caused neurodegeneration in selected brain regions, including the cerebral cortex, thalamus, and hippocampus, and cognitive impairment which were associated with chronic neuroinflammation and sustained CCA. Treatment with a selective cyclin-dependent kinase (CDK) inhibitor CR8 significantly reduced both the behavioral and inflammatory changes, suggesting a role for CCA in the brain pathophysiology following SCI.
Results
Thoracic contusion SCI alters microglial activation in the brain
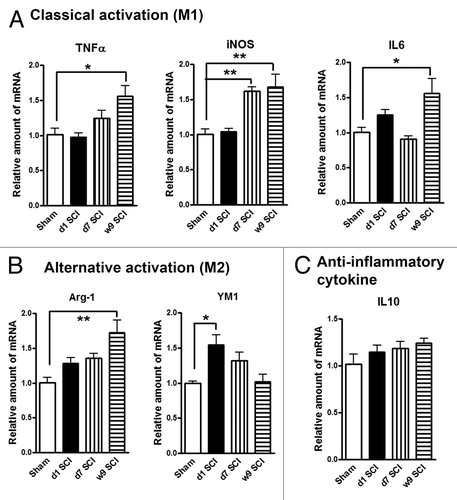
In our recent studies examining mechanisms of post-SCI-induced hyperpathia in rats, we observed sustained inflammation not only in the thalamus,Citation14 but also more diffusely in cortex and hippocampus. Here we expanded examination in separate experiments of the effects of isolated thoracic SCI in rats on microglia activation. We collected hippocampal tissue at day 1, days 7 and week 9 after SCI to determine the expression of markers of M1/M2 microglia phenotypes. showed the relative expression of several M1genes: tumor necrosis factor-α (TNFα), inducible nitric oxide synthase (iNOS), and interleukin 6 (IL6). SCI significantly increased the mRNA levels of these M1 type genes in the hippocampus when compared with sham-injured controls (). showed the relative expression of 2 key M2 genes: arginase-1 (Arg-1) and YM1. While Arg-1 increase toward the 9 wk time point is similar to the consistent gradual elevation in the expression of M1 markers, YM1 is transiently increased at day 1 followed by a decline. We observed no significantly changes in the expression of the, anti-inflammatory cytokine interleukin 10 (IL10) (). These results indicate that lesion to the spinal cord changes the expression patterns of M1 and M2 microglia phenotypes markers in the hippocampus.
Figure 1. SCI alters expression of M1 (classical)/M2 (alternative) microglia genes in the hippocampus. (A) The mRNA levels of M1 type genes (TNFα, iNOS, IL6) were significantly increased in SCI rats at 7 d or 9 wk post-injury. (B) SCI induced M2a gene YM1 expression transiently at day 1, whereas Arg-1 elevation at 9 wk post-injury. (C) Anti-inflammatory cytokine IL10 was found no significantly changes at all-time points. *P < 0.05, **P < 0.01 vs. sham group. n = 4 (Sham), 6 (d1 SCI), 6 (d7 SCI), 5 (9 wk SCI).
Resting microglia display ramified cellular morphologies, whereas activated forms display cellular hypertrophic or bushy morphologies. Unbiased stereological assessment was used to examine microglial cell numbers and phenotype in the hippocampus and cerebral cortex following SCI. presents representative images and reconstructions (Neurolucida) of resting microglia (ramified, small cell body with elongated and thin projections) and activated microglia (hypertrophic, large cell body with shorter and thicker projections; bushy, enlarged cell body with multiple short processes that form thick bundles). Seven days after SCI there was a significant increase in the number of activated microglia and a decrease in the number of ramified phenotypes in hippocampus/cortex (). At 10 wk posttrauma, there were significantly increased numbers of microglia displaying the highly activated (bushy) phenotype and reduced ramified phenotypes in both hippocampus and cerebral cortex. Treatment with the CDK inhibitor CR8 resulted in a significant attenuation in numbers of total and activated microglia at 7 d and bushy microglia at 10 wk post-injury (). These data demonstrate that microglial activation occurs in the brain after SCI, in part relating to CCA.
Figure 2. SCI increases activated microglial phenotypes in the brain at 7 d and 10 wk post-lesion. (A and B) Representative Iba-1 immunohistochemical images displaying resting (rami□ed morphology) or activated (hypertrophic or bushy morphology) microglial phenotypes and the corresponding Neurolucida reconstructions. (C and D) Unbiased stereological quantitative assessment in the hippocampus (C) and cerebral cortex (D) revealed increased numbers of highly activated microglia displaying a hypertrophic and bushy cellular morphology and reduced numbers of resting microglia displaying the ramified cellular morphology in SCI-brain when compared with Sham/Vehicle rats. CR8 treatment reversed these changes. *P < 0.05, **P < 0.01, ***P < 0.001, SCI/Vehicle vs. Sham/Vehicle groups; #P < 0.05, 7d SCI/CR8 vs. 7d SCI/Vehicle groups. $P < 0.05, $$$P < 0.001, 10w SCI/CR8 vs. 10w SCI/Vehicle groups. n = 4 (Sham/Vehicle), 5 (Sham/CR8), 4 (7d SCI/Vehicle), 6 (7d SCI/CR8), 5 (10w SCI/Vehicle), and 5 (10w SCI/CR8). (E) In vitro [125I]iodo-DPA-713 autoradiography in rat brain sections at 7 d post-SCI. All rats were sacrificed 7 d after sham or injury. Their brains were collected, sectioned on a cryomicrotome, and then probed with the indicated radiotracer. Blue bars indicate regional binding of [125I]iodo-DPA-713 in nmol/mg of wet tissue for sham treated rats while red bars indicate tracer binding in injured rats. Radioracer binding in injured rats was significantly higher across all measured regions as compared with sham-treated rats. To determine the extent of TSPO radiotracer differences, a two-tailed t test was performed. *P < 0.05; **P < 0.005; ***P < 0.0005 vs. Sham group. n = 5 for both groups. Cpu = caudate/putamen; CWM = cerebellar white matter; CGM = cerebellar gray matter.
![Figure 2. SCI increases activated microglial phenotypes in the brain at 7 d and 10 wk post-lesion. (A and B) Representative Iba-1 immunohistochemical images displaying resting (rami□ed morphology) or activated (hypertrophic or bushy morphology) microglial phenotypes and the corresponding Neurolucida reconstructions. (C and D) Unbiased stereological quantitative assessment in the hippocampus (C) and cerebral cortex (D) revealed increased numbers of highly activated microglia displaying a hypertrophic and bushy cellular morphology and reduced numbers of resting microglia displaying the ramified cellular morphology in SCI-brain when compared with Sham/Vehicle rats. CR8 treatment reversed these changes. *P < 0.05, **P < 0.01, ***P < 0.001, SCI/Vehicle vs. Sham/Vehicle groups; #P < 0.05, 7d SCI/CR8 vs. 7d SCI/Vehicle groups. $P < 0.05, $$$P < 0.001, 10w SCI/CR8 vs. 10w SCI/Vehicle groups. n = 4 (Sham/Vehicle), 5 (Sham/CR8), 4 (7d SCI/Vehicle), 6 (7d SCI/CR8), 5 (10w SCI/Vehicle), and 5 (10w SCI/CR8). (E) In vitro [125I]iodo-DPA-713 autoradiography in rat brain sections at 7 d post-SCI. All rats were sacrificed 7 d after sham or injury. Their brains were collected, sectioned on a cryomicrotome, and then probed with the indicated radiotracer. Blue bars indicate regional binding of [125I]iodo-DPA-713 in nmol/mg of wet tissue for sham treated rats while red bars indicate tracer binding in injured rats. Radioracer binding in injured rats was significantly higher across all measured regions as compared with sham-treated rats. To determine the extent of TSPO radiotracer differences, a two-tailed t test was performed. *P < 0.05; **P < 0.005; ***P < 0.0005 vs. Sham group. n = 5 for both groups. Cpu = caudate/putamen; CWM = cerebellar white matter; CGM = cerebellar gray matter.](/cms/asset/5b2f64a5-e70a-4c49-8aff-0166714dafd0/kccy_a_10929420_f0002.gif)
Furthermore, [125I] iodo-DPA-713 in vitro autoradiography was performed in fresh frozen brain sections at 7 d post-trauma and regions of interest were chosen to reflect major brain structures. All 5 regions showed significantly elevated radiotracer binding in the injured rat brains (), with binding in the thalamus robustly increased (P ≤ 0.0005). These data corroborate the microscopy data showing significant accumulation of microglia, particularly in the thalamus, at 7 d post-injury but not in sham-treated rats. The cortex, hippocampus and cerebellum also showed significantly increased (each P ≤ 0.005) radiotracer uptake in injured rats.
CCL21 is upregulated in the hippocampus and cerebral cortex after SCI
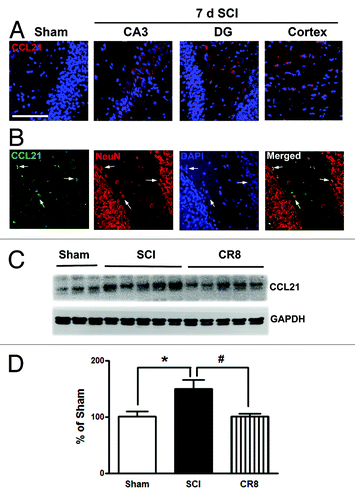
The neuroimmune modulator CCL21 is upregulated in the thalamus after SCI and may serve to trigger thalamic microglial activation in association with hyperesthesia.Citation14,Citation19 In uninjured animals, CCL21 immunoreactivity is weak in the hippocampus (). At 7 d after SCI, CCL21 immunolabeling was increased in neuronal cell bodies and parenchyma within both the hippocampus and cortex (). Western blot analysis confirmed that SCI increased CCL21 protein expression levels in the cortex. Notably, there was a reduction of CCL21 expression at 7 d post-injury in CR8-treated animals ().
Figure 3. CCL21 is upregulated in the hippocampus and cerebral cortex after SCI. (A) In the intact animal, immunoreactivity of CCL21 (red) was weakly detected in the hippocampus. At 7 d after SCI, CCL21 immunolabeling showed a clear increase in both the hippocampus and cortex. (B) CCL21 (green) was detected in neuronal cell bodies (NeuN, red) and parenchyma in the hippocampus. Scale bars = 100 μm. (C) western blot analysis showed that SCI upregulated CCL21 protein expression levels in the cortex. Notably, there was a reduction of CCL21 expression at 7 d post-injury in CR8-treated animals. *P < 0.05, vs. sham group; #P < 0.05, vs. SCI group. n = 5 (Sham), 8 (SCI), and 7 (CR8).
SCI mediates chronic neuronal loss in the cortex, thalamus, and hippocampus
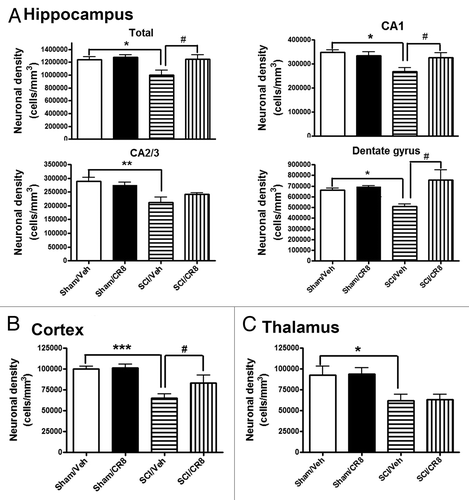
Stereological assessment of surviving neurons was performed in CA1, cornu ammonis 2/3 (CA2/3), and dentate gyrus (DG) hippocampal sub-regions as well as the cerebral cortex and thalamus. At 7 d after SCI, no differences were observed in the number of neurons in the hippocampus across the groups (data not shown). At 10 wk post-injury, there was significant neuronal cell loss in the hippocampus (; P < 0.05, vs. Sham/Vehicle group), cortex (), and thalamus (). Systemic administration of CR8 significantly improved neuronal survival in CA1 and DG sub-regions and cortex when compared with SCI/Vehicle samples (P < 0.05). Thus, SCI causes chronic neurodegeneration in the brain, in part through CCA.
Figure 4. SCI causes reduction of surviving neurons chronically in the hippocampus, cortex, and thalamus. SCI resulted in significant neuronal cell loss in the hippocampus (A), cortex (B), and thalamus (C) at 10 wk post-injury. Systemic administration of CR8 significantly improved neuronal survival in the CA1 and DG sub-regions of the hippocampus as well as cortex, compared with SCI/Vehicle samples. *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham/Vehicle mice; #P < 0.05, vs. SCI/CR8. n = 5 (Sham/Vehicle), 6 (Sham/CR8), 6 (SCI/Vehicle), 5 (SCI/CR8).
Cognitive and motor functions are impaired in rats after SCI
To investigate whether SCI rats show cognitive impairment, animals were evaluated using well-established cognitive tests through 2 mo post-injury. The Morris water maze (MWM) test was employed to evaluate hippocampus-dependent spatial learning and memory at 8 wk after injury. Initial pilot studies demonstrated that the modified protocol applied for the MWM test is feasible in experimental SCI, indicating that notwithstanding the motor deficits, injured rats retain swimming capability. SCI rats showed significantly increased latency to locate the hidden platform during the acquisition phase when compared with uninjured sham animals () (P < 0.001). There were no significant differences between the Sham/Vehicle- and Sham/CR8-groups, SCI/Vehicle- and SCI/CR8-groups across each of the training days. In the probe test, SCI rats spent significantly less time in the target quadrant compared with Sham/Vehicle animals (P < 0.01, ). Notably, a significant difference was also observed between the SCI/Vehicle- and SCI/CR8- groups (P < 0.05). No differences were observed between the 2 sham groups. However, injured rats showed significantly reduced swim speeds and distance in the MWM and CR8 did not reverse these changes in SCI animals (data now shown). To limit a perceived confounding effect between swim speed and latency time, a search strategy analysis was performed to evaluate the efficiency of locating the platform, an outcome that is less influenced by locomotion deficits. Based on previously published criteria,Citation22 3 search strategies were evaluated for each of the 4 trials on training day 4. Occasionally, rats changed search strategies during a trial. When this happened, the strategy that best described the major swimming path was assigned. Search strategy in the MWM showed good group separation (chi-square = 16.73, P = 0.01; ). SCI rats exhibited more reliance on systematic or looping search strategies than spatial search strategy compared with Sham animals. Specifically, the use of spatial search strategies ranged from 68% in the Sham/Vehicle group to 25% in the SCI/Vehicle group. CR8 increased the use of this type of swim path to 36%. shows different representative search strategy pathways between Sham and SCI rats.
Figure 5. Cognitive functions are impaired in rats after SCI. (A) Acquisition trials in Morris water maze (MWM) test. A significant difference was detected between the Sham/Vehicle (n = 11) and SCI/Vehicle (n = 17) groups at training day 4 (***P < 0.001). No differences were found between the Sham/Vehicle (n = 11) and Sham/CR8 (n = 11) groups, SCI/Vehicle (n = 17) and SCI/CR8 (n = 14) groups. (B) Probe trial in MWM test. SCI/Vehicle rats spent significantly less time in the target quadrant compared with Sham/Vehicle group (**P < 0.01) and to SCI/CR8 group (#P < 0.05). (C) Search strategy. Systematic type is predominant in SCI/Vehicle group (64%) compared with Sham/Vehicle group (27%), whereas SCI rats with CR8 treatment displayed increased spatial type (36% vs. 25% from SCI/Vehicle rats) and reduced looping type (6% vs. 11% from SCI/Vehicle rats). (D) Representative types of search strategies. SCI rats showed less traveled distance than that in uninjured sham rats. (E) Assessment of the novel object recognition test. During the test phase, sham-injured rats showed the predicted preference for the novel object. In contrast, SCI/Veh rats had reduced preference to the novel object, and the discrimination index was 69.1 ± 5.5% for Sham/Veh (n = 11) and 28.0 ± 4.9% for SCI/Veh (n = 17, P < 0.001). CR8 treatment caused significant increase in the discrimination index (P < 0.05 vs SCI/Veh rats). (F) The passive avoidance (PA) test. There were no significant differences in the latency to enter the dark compartment in the training phase between the sham and the injured groups. Twenty-four hours after training, SCI/Vehicle rats (n = 10) spent significant less time to enter to the dark compartment compared with Sham animals (n = 11, *P < 0.05). CR8 treatment (n = 9) reversed these reduced latency (#P < 0.05, vs. SCI/Vehicle group).
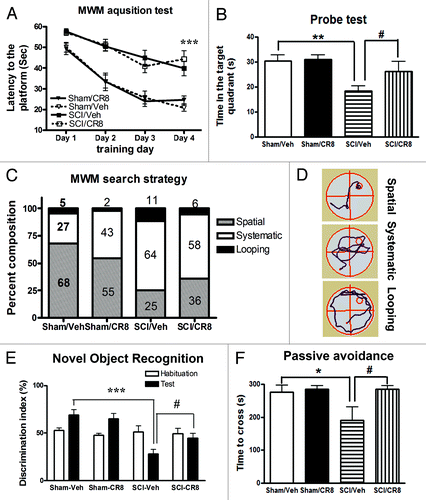
Retention memory was assessed by the novel object recognition (NOR) test that is less dependent on locomotion. During the test phase, sham-injured rats showed the predicted preference for the novel object, indicating intact memory. In contrast, SCI/Veh rats had reduced preference to the novel object, and the discrimination index was 69.1 ± 5.5% for Sham/Veh (n = 11) and 28.0 ± 4.9% for SCI/Veh (n = 17) (). Notably, CR8 treatment significantly increased the discrimination index, indicating improvement in retention memory performance.
Non-spatial, contextual and emotional memory was determined by the passive avoidance test, which is also less dependent on locomotor activity. There were no significant differences in the latency to enter the dark compartment in the training phase between the sham and the injured groups. Twenty-four hours (24 h) after training, SCI/vehicle rats spent significantly less time to enter to the dark compartment compared with sham animals (P < 0.05, (). CR8 treatment significantly limited these changes (P < 0.05, vs. SCI/Vehicle group). Collectively, these data show that SCI rats performed poorly on cognitive tests, indicating spatial and non-spatial learning and memory deficits and such changes were limited by cell cycle inhibition.
The open-field test was performed at 9 wk after SCI to examine spontaneous locomotor activity (). Sham animals displayed similar locomotor activity (distance traveled and walking speed) during the 5 min of testing. SCI rats showed significantly reduced traveled distance (1.94 ± 0.15 min, P < 0.001) when compared with Sham/Vehicle mice (3.21 ± 0.34 min). Decreased traveled distance in SCI rats was paralleled by reduced walking speed. We did not observe any improvement of spontaneous locomotor activity in SCI/CR8 group, consistent with the result of swim speed in MWM test.
Figure 6. Determination of spontaneous locomotor activity and hind limb functions using the open-field test. (A and B) Traveled distance and speed. SCI resulted in a significant reduced distance traveled and walking speed compared with Sham/Vehicle rats. CR8 treatment did not improve spontaneous locomotor activity. **P < 0.01, ***P < 0.001, vs. SCI/Vehicle. n = 5 (Sham/CR8), 5 (Sham/Vehicle), 17 (SCI/Vehicle), and 19 (SCI/CR8). (C and D) Hind limb sensory-motor deficits were estimated in these rats on day 1 after injury and weekly thereafter for up to 8 wk using the BBB and CBS scores. Significantly improved functional recovery in CR8-treated rats was observed compared with saline-treated rats. *P < 0.05, **P < 0.01, **P < 0.001, vs. saline-treated group. n = 17 (SCI/Vehicle) and 12 (SCI/CR8).
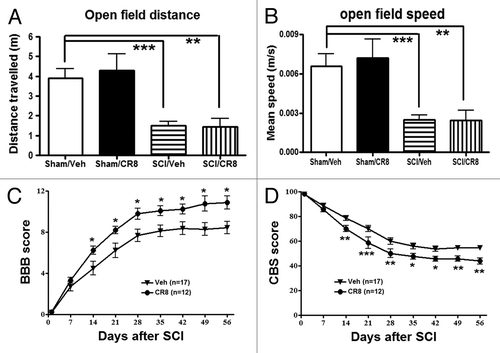
Hind limb function was evaluated in the open-field test on day 1 after injury and weekly thereafter for up to 8 wk using the Basso, Beattie and Bresnahan (BBB) scores.Citation23 One day after SCI, all animals had a BBB score of 0 or 1, indicating nearly complete loss of motor function (). In addition, overall hind limb sensory-motor deficits were estimated using a battery of behavioral tests (the combined behavioral score, CBS).Citation24 By day 1, rats with nearly complete injury had a score of 100 (). Three hours post-injury and once daily for 6 consecutive days after the injury, the rats received systemically intraperitoneal (IP) injection of CR8 (1 mg/kg) or saline. CR8-treated SCI rats (n = 12) had significantly better BBB and CBS scores than the saline-treated SCI rats (n = 17) by day 14, and this effect persisted through day 56 after SCI. Therefore, inhibiting cell cycle activation improves the recovery of hind limb function in SCI rats, without affecting spontaneous locomotor activity.
SCI causes upregulation of cell cycle genes and proteins in the hippocampus and cerebral cortex
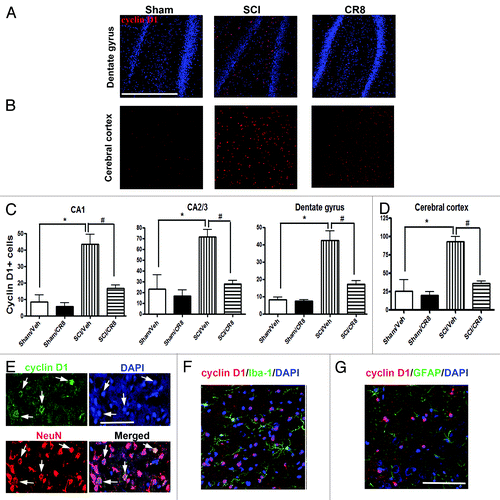
To further investigate the involvement of the cell cycle pathway in SCI-induced brain abnormalities, we first examined whether upregulation of cell cycle genes occurs in the brain following SCI. Quantitative real-time PCR analysis showed an upregulation of multiple cell cycle related genes (cyclins A1, A2 and D1 as well as proliferating cell nuclear antigen [PCNA]) in the hippocampus at 9 wk after SCI (). The E2F1 transcription factor showed a rapid, transient elevation at 24 h after injury (; expression of cyclin A1, A2, D1, and PCNA were significantly increased at 10 wk post-injury ( and ), whereas cyclin B1 remained unchanged at all time-points (). In the intact hippocampus and cerebral cortex, immunoreactivity of cyclin D1 was weakly detected in some neurons. At 10 wk post-injury, numerous cyclin D1-positive cells were observed in both of these brain regions (). Most cyclin D1+ cells co-localized with NeuN+ neurons, but not ionized calcium binding adaptor molecule 1 (Iba-1)+ microglia or glial fibrillary acidic protein (GFAP)+ astrocytes (). Systemic administration of CR8 significantly suppressed SCI-mediated upregulation of cell cycle genes and protein expression in the brain.
Figure 7. Impact SCI causes upregulation on the expression of a cluster of cell cycle genes in the hippocampus. Quantitative real-time PCR analysis was used to quantify mRNA levels in the hippocampal tissues from sham or SCI mice at 1 d, 7 d or 9 wk post-injury. The E2F1 transcription factor (A) showed a rapid, transient elevation at 24 h after injury. The expression of cyclin A1, A2, D1, and PCNA genes (B and C, E and F) were significantly increased in the SCI rats at 9 wk post-injury, whereas cyclin B1 remained unchanged at all time-points (D). *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham group. n = 4 (Sham), 6 (d1 SCI), 6 (d7 SCI), 5 (9 wk SCI).
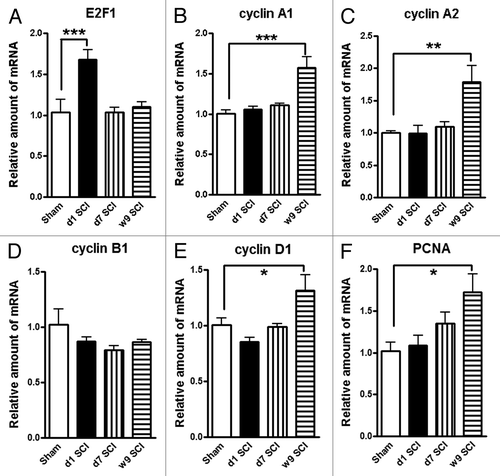
Figure 8. SCI increases numbers of cyclin D1+ cells in the hippocampus and cerebral cortex at 10 wk post-injury. (A and B) Representative cyclin D1 immunofluorecent images in both hippocampus (DG) and cerebral cortex. Scale bars = 100 μm. (C and D) Quantification showed that SCI increased total numbers of cyclin D1+ cells in both of these brain regions, such changes were limited by CR8 treatment. *P < 0.05 vs. Sham/Vehicle group; #P < 0.05, vs. SCI/Vehicle group. n = 4 (Sham/Vehicle), 4 (sham/CR8), 6 (SCI/Vehicle), and 6 (SCI/CR8). (E–G) Double staining showed that most cyclin D1+ cells were co-labeled with NeuN+ neurons, but not Iba-1+ microglia or GFAP+ astrocytes. Scale bars = 100 μm.
Discussion
This study provides the first experimental evidence that isolated rat thoracic spinal cord contusion causes chronic diffuse inflammation in the brain associated with neurodegeneration and cognitive loss. Injury increased numbers of reactive microglia, caused a microglial M1/M2 balance switch, and elevated expression levels for markers of activated microglia/macrophages—including radioiodinated DPA-713 and CCL21. These changes were associated with neuronal loss in the hippocampus, cortex, and thalamus. In addition, increased expression of cell cycle-related genes and proteins was observed in these regions after SCI. All changes were attenuated by inhibition of cell cycle pathways, suggesting that CCA may contribute to these supratentorial pathophysiological events.
TSPO has been studied as a biomarker of reactive gliosis and inflammation associated with a variety of neuropathological conditions.Citation18 Under normal physiological conditions, TSPO levels are low in the brain but they are markedly increased at sites of brain injury, making it useful for assessing active inflammation and gliosis. We and others have used TSPO as a molecular target for imaging neuroinflammation.Citation25 Iodo-DPA-713 is a synthetic low-molecular-weight pyrazolopyrimidine ligand for TSPO and can be labeled with either γ (I-123/5) or positron-emitting (I-124) radioisotopes. In the present study, we applied [125I]iodo-DPA-713 in the fresh-frozen rat brain sections to detect radiotracer uptake. Our data demonstrate that all 5 regions of interest showed significantly elevated radiotracer binding in the SCI-brains, consistent with the microglial activation observed histologically. However, increased TPSO may also reflect involvement of macrophages and astrocyte activation.Citation26
Central nervous system resident microglia, like peripheral macrophages, have multiple activation phenotypes. Classical M1 microglia are characterized as reactive phenotype that produce pro-inflammatory cytokines, reactive oxygen species, and nitric oxide, contributing to tissue inflammation and damage. In contrast, alternative M2 phenotype microglia secrete anti-inflammatory cytokines and neurotrophic factors, contributing to wound healing and tissue-remodeling. We found that the expression levels of M1 genes (TNFα, iNOS) gradualy increased with time in the hippocampus following SCI when compared with sham rats. There was additionally an gradual increase in the proinflammatory cytokine IL6, which has been previously shown to be elevated in the thalamus in response to SCI.Citation19 Furthermore, IL6 has a dual role as a microglial activator as well as a neuromodulator. More recently, Guerrero et al. reported that blocking IL6 signaling promotes functional recovery by inhibiting M1 and promoting M2 macrophage activation after SCI.Citation27 SCI also induced changes in the expression of alternatively activated M2a genes (Arg-1 and YM1) in the hippocampus although the overall pattern was not consistent. Arg-1, which competes with iNOS for arginine to produce L-ornithine and urea rather than nitric oxide increased in a time-dependent manner similar to that of M1 markers. In contrast, YM1 expression was only transiently increased at day 1 post-injury; despite being expressed in the injured brain and spinal cord there is limited information on its expression and function under physiological and pathophysiological conditions.Citation17 Our data based on tissue homogenate cannot definitively demonstrate the cellular origin of the gene expression changes. The precise interpretation of our findings is also made difficult by the complex patterns observed. Nonetheless, they suggest that SCI results in significant modulation of expression of well-known M1/M2 markers and future studies should continue to evaluate their consequences on microglia phenotypes.
Stereological analysis of the hippocampus and cerebral cortex in injured animals showed an enhanced microglial activation response, with increased numbers of activated microglia displaying a bushy and hypertrophic cellular morphology and reduced numbers of resting microglia displaying the ramified cellular morphology. In addition, an increase in the chemokine CCL21 signal, a potent microglial activator, was observed in various brain regions within neuronal cell bodies and parenchyma. Microglial activation and neuroinflammation have been linked to changes in cognitive function through both direct and indirect effects on neurons.Citation15 Two recent clinical studies—one using positron emission tomography (PET) scanning to delineate microglial activity, and the other pathological—have further underscored the important potential role of microglial activation in chronic neurodegeneration after TBI.Citation28,Citation29 After SCI, assessments of neurons in the brain have been focused on the motor cortex and medullary pyramid. These results are conflicted with reports ranging from no cell death to extensive retrograde degeneration.Citation12,Citation13,Citation30-Citation50 One of the limitations of these prior SCI studies is that they were performed over poorly defined areas of the cerebral cortex and/or did not involve rigorous quantitative assessment techniques. In contrast, our study utilizes stereology techniques with computer-driven, random, systematic sampling to provide unbiased and quantitative data for cell counting. We show here that SCI results in neuronal cell loss in hippocampus, cortex, and thalamus at 10 wk, but not early (7 d) after injury. Thus, these data demonstrate chronic neurodegeneration in the brain after SCI that is associated with microglial activation.
We employed a battery of cognitive behavioral tests to determine whether these tests are feasible in experimental SCI rats and whether the use of multiple behavioral outcomes can overcome the potential confounding effects of motor dysfunction in the animals. The MWM has been commonly used to examine hippocampus-dependent spatial learning and memory impairments in rodents. However, despite a certain level of endogenous motor recovery, injured rats showed significantly reduced swim speeds and distance in the MWM and decreased spontaneous activity in the open-field test. Thus, significantly delayed escape latencies and less time spent in target quadrant following SCI in the MWM test may not reflect pure cognitive deficits as for other models. Therefore, we also examined the search strategy used by the injured rats to locate the hidden platform, paralleling the type of search strategy analysis initially proposed for the Barnes maze; this method has been detailed by us for both the Barnes maze and MWM tests after TBI and provides effective and complementary outcome measures that help to discriminate cognitive changes. Sham-injured rats primarily employed a spatial strategy to locate the platform and use of this strategy decreased after SCI. In contrast, injured rats more prominently used systematic or looping strategies to locate the platform. Moreover, whereas sham animals swam in the center of the maze to find the hidden platform, injured animals spent most of their time swimming around the entrance quadrant of the maze. The NOR and PA tests are also less dependent on locomotor activity reflecting non-spatial, contextual and fear-related emotional memory. These tests therefore provide good complement to spatial memory assessment by the MWM test. Injured rats also spent significantly less time with the novel object and had reduced transfer latency compared with sham rats. These results provide strong evidence that contusion SCI in rats causes learning and memory impairment, despite the potentially confounding effects of reduced motor function.
The role of CCA in pathophysiology of SCI has been extensively studied by us and others, with most work focusing on the primary lesion site.Citation14,Citation51-Citation60 Here we report, for the first time, an upregulation of a cluster of cell cycle related genes and proteins in the brain over the course of several weeks after SCI. Among them, the E2F1 transcription factor shows a rapid, transient elevation at 24 h following SCI. Immunohistochemistry at 10 wk after SCI revealed increased numbers of cyclin D1 neurons (most of them co-labeled with NeuN) in both hippocampus and cerebral cortex. CR8, administrated systemically after SCI, significantly attenuated CCA, as well as reducing microglial activation and chronic neurodegeneration in the brain.
The mechanisms underlying SCI-mediated effects in the brain remain speculative. Endo et al. found,Citation33 using in situ hybridization that the Nogo receptor and its co-receptor LINGO-1 are downregulated specifically in cortical areas deprived of sensory input and in adjacent cortex from 1 d after complete transection of the spinal cord at the thoracic level, whereas BDNF is upregulated. These observations suggest an involvement of Nogo signaling in cortical activity-dependent plasticity after SCI. Further, synapsin expression and neurite sprouting are increased in lamprey brain after spinal cord transections.Citation43 By contrast, acute SCI in rats reduces brain-derived neurotrophic factor (BDNF) levels in the hippocampus in conjunction with the activated forms of synapsin 1, cyclic AMP-responsive element-binding protein (CREB), and calcium/calmodulin-dependent protein kinase II (CaMKII).Citation35,Citation61 It has been well established that SCI can produce extensive long-term reorganization of the cerebral cortex, as well as thalamic changes, through anterograde and retrograde mechanisms.Citation62 Induction of CCL21 at distal brain regions, synthesized by damaged neurons in spinal cord after injury, may be, at least in part, an important source that may trigger microglial activation at more distant sites.Citation19,Citation63 Whether this process can occur across multiple synapses has not been examined; however, SCI can markedly alter systemic immune functionsCitation64 which can secondarily affect the brain. Although it is also possible that factors are released that may have access to the brain through the cerebrospinal fluid, flow considerations argue against this potential explanation.
In summary, our data indicate that in a clinically relevant, well-characterized rat SCI model, isolated thoracic contusion induces a chronic inflammatory response and neurodegeneration in the brain associated with cognitive impairment. These findings provide the first experimental confirmation of clinical evidence suggesting SCI-related cognitive deficits considerably revising concepts about the nature of SCI as a focal acute neurodegenerative disorder. Moreover, the use of TSPO-specific ligands may serve as a biomarker of active brain inflammation using in vivo imaging modalities such as positron emission tomography for future human studies. Additionally, the finding that CCA continues chronically after SCI in each of the regions associated with long-term progressive changes, may permit treatment with a far greater therapeutic window than previously considered.
Materials and Methods
Spinal cord injury and drug administration
All surgical procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by NIH (DHEW publication NIH 85-23-2985), and the protocols were approved by the University of Maryland School of Medicine Institutional Animal Care and Use Committee. Male Sprague-Dawley rats (275–325 g) were anesthetized with sodium pentobarbital (65 mg/kg IP). Body temperature was kept by maintaining the animal on a heating pad (37 °C) throughout the procedure. Contusion SCI was produced at spinal segment T8 by dropping a 10 g weight from 2.5 cm onto an impounder positioned on the exposed spinal cord without disrupting the dura.Citation14 Sham rats were subjected to anesthesia and a laminectomy but were not injured. Three hours post-injury, rats received 1 mg/kg CR8 (Tocris Bioscience, dissolved in sterile saline) or equal volume saline IP, once daily continuing for 7 d. Dosages were based upon prior investigations in TBI and SCI models.Citation14,Citation65
Quantitative real-time-polymerase chain reaction (qRT-PCR)
Total RNA was isolated from the hippocampus at 1 d, 7 d, and 9 wk after SCI or sham rats by using TRIzol reagent (Invitrogen) following the manufacturer's instructions. Verso™ cDNA Kit (Thermo Fisher, Waltham, MA USA was used to synthesize cDNA from purified total RNA. Quantitative real-time PCR amplification was performed by using cDNA TaqMan® Universal Master Mix II (Applied Biosystems). Gene Expression assays for following genes were used: TNFα (Rn01525859_g1), iNOS (Rn00561646_m1), IL6 (Rn01410330_m1), IL10 (Rn 00563409_m1), Arg-1 (Rn00691090_m1), YM1 (Rn01523660_g1), cyclin A1 (Rn 01761351_m1), cyclin A2 (Rn01493715_m1), cyclin B1 (Rn01494177_m1), cyclin D1 (Rn 00432360_m1), E2F1 (Rn01536222_m1), PCNA (Rn01514538_g1). Gene expression was normalized to GAPDH (Rn99999915_g1), and the relative quantity of mRNAs was calculated based on the comparative Ct method.
Stereological quantification of microglial phenotypes and surviving neurons
At the indicated time points, rats were perfused and coronal sections were cut and serially collected (3 × 60 μm followed by 3x20 μm sections) throughout the brain. Every fourth 60-μm section was processed for Iba-1 or cresyl violet staining beginning from a random start point, and analyzed using a Leica DM4000B microscope (Leica, Leica Microsystems Inc) and Stereoinvestigator software (MBF Biosciences). Microglial phenotypic classification was based on the length and thickness of the projections, the number of branches, and the size of the cell body as described previously.Citation14 Cresylviolet neuronal cell bodies were counted for each subfield of the hippocampus as well as cortex and thalamus. The estimated number of microglia in each phenotypic class or surviving neurons in each field was divided by the volume of the region of interest to obtain cellular density expressed in cells/mm3. Neurolucida software (MBF Biosciences) was used to trace the cell bodies and dendrites of microglia at resting or activated stage following injury as described previously.Citation14 The cell body was outlined using the contour tool followed by the tracing of the individual dendrites, using the dendrite line tool.
Autoradiography
Fresh-frozen rat brains at 7 d post injury/sham were stored at −80 °C until being sectioned to 20 μm onto charged glass slides using a Microm HM 550 cryotome (Thermo-Fisher). Slides containing regions of interest were thawed immediately prior to use and were briefly washed in PBS, pH7.5 prior to addition of 344 pM [125I]iodo-DPA-713 in PBS, pH 7.5. The radiotracer solution remained for 1 h at room temperature before washing twice for 2 min each with ice-cold PBS followed by a brief dip in ice-cold water. The slides were then dried and apposed to Kodak Biomax XAR film (Fisher Scientific) for 1 h prior to development and fixation using a Mini Medical film processor (AFP Imaging). The film was then digitized using a QICAM QIC-F-M-12 camera and Northern Light Illuminator and analyzed using the MCID Core densitometry software (MCID). Regions of interest (caudate-putamen, hypothalamus, cortex, cerebellar white and gray matter, and thalamus) were identified with the aid of a rat brain atlas (Paxinos and Watson “The Rat Brain, 6th Ed.” Academic Press, 2009) and compared with calibrated standards. The values are expressed as nmol/mg of wet tissue and reflect at least 3 regions-of-interest per brain region.
Immunohistochemistry and quantification
Frozen 20-μm brain sections were incubated overnight with primary antibodies followed by appropriate secondary antibodies. The following primary antibodies were used: rabbit anti-CCL21 (1:200, Abcam), mouse anti-NeuN (1:500, Millipore), mouse anti-cyclin D1 (1:500, Neomarker), rabbit anti-Iba-1 (1:1000, Wako Chemicals), and rabbit anti-GFAP (1:500, Millipore). Fluorescent-conjugated secondary antibodies (Alexa 488-conjugated goat anti-mouse or rabbit, 1:1000, Molecular Probes) were incubated with tissue sections for 1 h at room temperature. Cell nuclei were labeled with 4’, 6-diamidino-2-phenylindole (DAPI, 1 μg/mL, Sigma). Finally, slides were washed and mounted with an anti-fading medium (Invitrogen). Immunofluorescence microscopy was performed using a Leica TCS SP5 II Tunable Spectral Confocal microscope system (Leica Microsystems Inc). The images were processed using Adobe Photoshop 7.0 software (Adobe Systems). All immunohistological staining experiments were performed with appropriate positive control tissue as well as primary/secondary-only negative controls. For quantitative image analysis, digital images at 20× magnification were captured from the cerebral cortex, CA1, CA2/3, and DG sub-regions of the hippocampus, based on atlas boundaries and using a confocal laser-scanning microscope (n = 3 sections/location/rat for 6 rats/SCI groups or 3 rats/Sham groups). The number of cyclin D1-expressing cells was analyzed at predefined areas.
Western blots
The tissue was homogenized and sonicated in radioimmunoprecipitation assay (RIPA) buffer (Sigma), and then centrifuged at 20 600 × g for 20 min at 4 °C. The supernatant was removed and protein concentration was determined using the Pierce BCA Protein Assay kit (Thermo Scientific) with a bovine serum albumin standard. Each sample contained proteins from one animal. Equal amounts of protein were electrophoretically separated on 4–12% NuPAGE Novex Bis-Tris gradient gels (Invitrogen) and transferred to nitrocellulose membranes (Invitrogen). After blocking in 5% nonfat milk for 1 h at room temperature, membranes were incubated with antibody against CCL21 (polyclonal, 1:500, Abcam) overnight at 4 °C followed by horseradish peroxidase-conjugated secondary antibody (GE Healthcare) for 1.5 h at room temperature. The immunoreactivity was detected using SuperSignal West Dura Extended Duration Substrate (Thermo Scientific), and quantified by band densitometry of scanned films using the Gel-Pro Analyzer program (Media Cybernetics, Inc). Some blots were further stripped in a stripping buffer (Thermo Scientific) for 45 min at 55 °C. The loading and blotting of equal amounts of protein were verified by re-probing the membrane with anti-GAPDH (monoclonal, 1:1000, Chemicon).
Motor function evaluation
The open field test was used to measure locomotor activity at 9 wk post-SCI. Rats were individually placed in a corner facing the wall of the open-field chamber (40 cm × 80 cm) and allowed to freely explore the chamber for 10 min. The distance and speed traveled was recorded by computer-based Any-Maze automated video tracking system (Stoelting Co). Hind limb function was assessed on day 1 post-injury and weekly thereafter for up to 8 wk, using the Basso, Beattie and Bresnahan (BBB) open field expanded locomotor score.Citation23 Neurological functional deficits were also estimated with a combined behavioral score (CBS), which included open field locomotion (motor score); withdrawal reflex to hind limb extension, pain and pressure; foot placing, toe spread and righting reflexes; maintenance of position on an inclined plane and swimming tests.Citation24
Morris water maze test
Spatial learning and memory was evaluated by using the standard Morris water maze (MWM) at 8 wk post-injury, as described previously with some modifications.Citation22 In brief, each rat was allowed a maximum of 60 s to find the hidden submerged platform, and rats that failed to find the platform within this time were placed onto the platform and allowed to remain on the platform for 10 s on subsequent training days. Rats were trained to find the platform located in the North East (NE) quadrant of tank for 4 consecutive days. The swim path, latency to platform, time spent in each zone and velocity were recorded by computer-based Any-Maze automated video tracking system (Stoelting Co). Reference memory was assessed by a probe test performed on 5th day. The platform was removed and rats were released from the South West (SW) position and the time spent in each quadrant was recorded out of a maximum of 60 s. Water maze search strategy analysis was performed as previously described.Citation22
Novel object recognition test
Novel object recognition (NOR) evaluated retention or intact memory at 9 wk post-injury, as described previously.Citation65
Passive avoidance test
The passive avoidance (PA) test was used to evaluate nonspatial fear-based amygdala-dependent contextual and emotional memory at 10 wk post-injury, as previously described using one-trial step-through PACS-30 passive avoidance apparatus (Columbus Instruments) adjusted to an electric foot shock (0.5 mA for 3 s) with a 24-h lag between training and testing.Citation65
Statistical analysis
Quantitative data are plotted as mean ± standard error of the mean. For the acquisition trials of the Morris Water Maze test, BBB and CBS scores repeated measures 2-way analyses of variance (ANOVAs) were conducted, followed by Bonferroni post-hoc test to compare the differences between each group. Statistical significance was evaluated between 2 individual samples using Student unpaired t tests. For multiple comparisons, one-way analysis of variance (ANOVA) followed by Student Newman-Keuls-post-hoc test. Statistical analysis was performed using SigmaPlot Program, Version 12 (Systat Software) or GraphPad Prism software, version 4.00 for windows (GraphPad Software, Inc). For non-parametric data, the Wilcoxon Exact test was used. SAS 9.3 (Statistical Analysis System) was used for this test. Statistical significance was set at P value ≤ 0.05.
| Abbreviations: | ||
| BBB | = | Basso, Beattie and Bresnahan locomotor rating scale |
| CDK | = | cyclin-dependent kinase |
| CCA | = | cell cycle activation |
| CCL21 | = | chemokine ligand 21 (C-C motif) |
| CBS | = | Combination Behaviral Scores |
| ERPs | = | event-related potentials |
| MWM | = | morris water maze |
| NOR | = | novel objective recognition |
| PT | = | positron emission tomography |
| SCI | = | spinal cord injury |
| TSPO | = | translocator protein |
| TBI | = | traumatic brain injury |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Katherine Cardiff and Angela Pan for expert technical support, Dr Soren M Bentzen and Ms Shari Kronsberg for expert statistical consultation and analysis. This study was supported by the National Institutes of Health Grants R01 NS054221 (AIF), R01 NR013601 (AIF), R21 NR014053 (JW).
Author contributions
J.W. and A.I.F. designed research; J.W., T.L., B.S., Z.Z., K.G., S.K.N., and C.A.F. performed research; J.W., T.L., B.S., Z.Z., S.K.N., and C.A.F. analyzed data; J.W., B.A.S., M.G.P., and A.I.F. wrote the paper.
References
- Davidoff GN, Roth EJ, Richards JS. Cognitive deficits in spinal cord injury: epidemiology and outcome. Arch Phys Med Rehabil 1992; 73:275 - 84; PMID: 1543433
- Roth E, Davidoff G, Thomas P, Doljanac R, Dijkers M, Berent S, Morris J, Yarkony G. A controlled study of neuropsychological deficits in acute spinal cord injury patients. Paraplegia 1989; 27:480 - 9; http://dx.doi.org/10.1038/sc.1989.75; PMID: 2608301
- Dowler RN, O’Brien SA, Haaland KY, Harrington DL, Feel F, Fiedler K. Neuropsychological functioning following a spinal cord injury. Appl Neuropsychol 1995; 2:124 - 9; http://dx.doi.org/10.1080/09084282.1995.9645349; PMID: 16318515
- Dowler RN, Harrington DL, Haaland KY, Swanda RM, Fee F, Fiedler K. Profiles of cognitive functioning in chronic spinal cord injury and the role of moderating variables. J Int Neuropsychol Soc 1997; 3:464 - 72; PMID: 9322406
- Lazzaro I, Tran Y, Wijesuriya N, Craig A. Central correlates of impaired information processing in people with spinal cord injury. J Clin Neurophysiol 2013; 30:59 - 65; http://dx.doi.org/10.1097/WNP.0b013e31827edb0c; PMID: 23377444
- Jensen MP, Kuehn CM, Amtmann D, Cardenas DD. Symptom burden in persons with spinal cord injury. Arch Phys Med Rehabil 2007; 88:638 - 45; http://dx.doi.org/10.1016/j.apmr.2007.02.002; PMID: 17466734
- Murray RF, Asghari A, Egorov DD, Rutkowski SB, Siddall PJ, Soden RJ, Ruff R. Impact of spinal cord injury on self-perceived pre- and postmorbid cognitive, emotional and physical functioning. Spinal Cord 2007; 45:429 - 36; http://dx.doi.org/10.1038/sj.sc.3102022; PMID: 17228355
- Strubreither W, Hackbusch B, Hermann-Gruber M, Stahr G, Jonas HP. Neuropsychological aspects of the rehabilitation of patients with paralysis from a spinal injury who also have a brain injury. Spinal Cord 1997; 35:487 - 92; http://dx.doi.org/10.1038/sj.sc.3100495; PMID: 9267911
- Jegede AB, Rosado-Rivera D, Bauman WA, Cardozo CP, Sano M, Moyer JM, Brooks M, Wecht JM. Cognitive performance in hypotensive persons with spinal cord injury. Clin Auton Res 2010; 20:3 - 9; http://dx.doi.org/10.1007/s10286-009-0036-z; PMID: 19842013
- Wecht JM, Bauman WA. Decentralized cardiovascular autonomic control and cognitive deficits in persons with spinal cord injury. J Spinal Cord Med 2013; 36:74 - 81; http://dx.doi.org/10.1179/2045772312Y.0000000056; PMID: 23809520
- Zhang B, Huang Y, Su Z, Wang S, Wang S, Wang J, Wang A, Lai X. Neurological, functional, and biomechanical characteristics after high-velocity behind armor blunt trauma of the spine. J Trauma 2011; 71:1680 - 8; http://dx.doi.org/10.1097/TA.0b013e318231bce7; PMID: 22182875
- Chang CM, Lee MH, Wang TC, Weng HH, Chung CY, Yang JT. Brain protection by methylprednisolone in rats with spinal cord injury. Neuroreport 2009; 20:968 - 72; http://dx.doi.org/10.1097/WNR.0b013e32832d0a28; PMID: 19525878
- Felix MS, Popa N, Djelloul M, Boucraut J, Gauthier P, Bauer S, Matarazzo VA. Alteration of forebrain neurogenesis after cervical spinal cord injury in the adult rat. Front Neurosci 2012; 6:45; http://dx.doi.org/10.3389/fnins.2012.00045; PMID: 22509147
- Wu J, Raver C, Piao C, Keller A, Faden AI. Cell cycle activation contributes to increased neuronal activity in the posterior thalamic nucleus and associated chronic hyperesthesia after rat spinal cord contusion. Neurotherapeutics 2013; 10:520 - 38; http://dx.doi.org/10.1007/s13311-013-0198-1; PMID: 23775067
- Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun 2012; 26:1191 - 201; http://dx.doi.org/10.1016/j.bbi.2012.06.008; PMID: 22728326
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol 2003; 3:23 - 35; http://dx.doi.org/10.1038/nri978; PMID: 12511873
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol 2009; 4:399 - 418; http://dx.doi.org/10.1007/s11481-009-9164-4; PMID: 19655259
- Chen MK, Guilarte TR. Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther 2008; 118:1 - 17; http://dx.doi.org/10.1016/j.pharmthera.2007.12.004; PMID: 18374421
- Zhao P, Waxman SG, Hains BC. Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine cysteine chemokine ligand 21. J Neurosci 2007; 27:8893 - 902; http://dx.doi.org/10.1523/JNEUROSCI.2209-07.2007; PMID: 17699671
- Wu J, Stoica BA, Faden AI. Cell cycle activation and spinal cord injury. Neurotherapeutics 2011; 8:221 - 8; http://dx.doi.org/10.1007/s13311-011-0028-2; PMID: 21373950
- Byrnes KR, Faden AI. Role of cell cycle proteins in CNS injury. Neurochem Res 2007; 32:1799 - 807; http://dx.doi.org/10.1007/s11064-007-9312-2; PMID: 17404835
- Zhao Z, Loane DJ, Murray MG 2nd, Stoica BA, Faden AI. Comparing the predictive value of multiple cognitive, affective, and motor tasks after rodent traumatic brain injury. J Neurotrauma 2012; 29:2475 - 89; http://dx.doi.org/10.1089/neu.2012.2511; PMID: 22924665
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 1995; 12:1 - 21; http://dx.doi.org/10.1089/neu.1995.12.1; PMID: 7783230
- Gale K, Kerasidis H, Wrathall JR. Spinal cord contusion in the rat: behavioral analysis of functional neurologic impairment. Exp Neurol 1985; 88:123 - 34; http://dx.doi.org/10.1016/0014-4886(85)90118-9; PMID: 3979506
- Endres CJ, Pomper MG, James M, Uzuner O, Hammoud DA, Watkins CC, Reynolds A, Hilton J, Dannals RF, Kassiou M. Initial evaluation of 11C-DPA-713, a novel TSPO PET ligand, in humans. J Nucl Med 2009; 50:1276 - 82; http://dx.doi.org/10.2967/jnumed.109.062265; PMID: 19617321
- Maeda J, Higuchi M, Inaji M, Ji B, Haneda E, Okauchi T, Zhang MR, Suzuki K, Suhara T. Phase-dependent roles of reactive microglia and astrocytes in nervous system injury as delineated by imaging of peripheral benzodiazepine receptor. Brain Res 2007; 1157:100 - 11; http://dx.doi.org/10.1016/j.brainres.2007.04.054; PMID: 17540348
- Guerrero AR, Uchida K, Nakajima H, Watanabe S, Nakamura M, Johnson WE, Baba H. Blockade of interleukin-6 signaling inhibits the classic pathway and promotes an alternative pathway of macrophage activation after spinal cord injury in mice. J Neuroinflammation 2012; 9:40; http://dx.doi.org/10.1186/1742-2094-9-40; PMID: 22369693
- Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013; 136:28 - 42; http://dx.doi.org/10.1093/brain/aws322; PMID: 23365092
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol 2011; 70:374 - 83; http://dx.doi.org/10.1002/ana.22455; PMID: 21710619
- Nielson JL, Sears-Kraxberger I, Strong MK, Wong JK, Willenberg R, Steward O. Unexpected survival of neurons of origin of the pyramidal tract after spinal cord injury. J Neurosci 2010; 30:11516 - 28; http://dx.doi.org/10.1523/JNEUROSCI.1433-10.2010; PMID: 20739574
- Bonatz H, Röhrig S, Mestres P, Meyer M, Giehl KM. An axotomy model for the induction of death of rat and mouse corticospinal neurons in vivo. J Neurosci Methods 2000; 100:105 - 15; http://dx.doi.org/10.1016/S0165-0270(00)00238-7; PMID: 11040372
- Crawley AP, Jurkiewicz MT, Yim A, Heyn S, Verrier MC, Fehlings MG, Mikulis DJ. Absence of localized grey matter volume changes in the motor cortex following spinal cord injury. Brain Res 2004; 1028:19 - 25; http://dx.doi.org/10.1016/j.brainres.2004.08.060; PMID: 15518637
- Endo T, Spenger C, Tominaga T, Brené S, Olson L. Cortical sensory map rearrangement after spinal cord injury: fMRI responses linked to Nogo signalling. Brain 2007; 130:2951 - 61; http://dx.doi.org/10.1093/brain/awm237; PMID: 17913768
- Feringa ER, Vahlsing HL. Labeled corticospinal neurons one year after spinal cord transection. Neurosci Lett 1985; 58:283 - 6; http://dx.doi.org/10.1016/0304-3940(85)90067-9; PMID: 4047489
- Fumagalli F, Madaschi L, Caffino L, Marfia G, Di Giulio AM, Racagni G, Gorio A. Acute spinal cord injury reduces brain derived neurotrohic factor expression in rat hippocampus. Neuroscience 2009; 159:936 - 9; http://dx.doi.org/10.1016/j.neuroscience.2009.01.030; PMID: 19344636
- Ganchrow D, Bernstein JJ. Thoracic dorsal funicular lesions affect the bouton patterns on, and diameters of, layer VB pyramidal cell somata in rat hindlimb cortex. J Neurosci Res 1985; 14:71 - 81; http://dx.doi.org/10.1002/jnr.490140107; PMID: 4020899
- Giehl KM, Tetzlaff W. BDNF and NT-3, but not NGF, prevent axotomy-induced death of rat corticospinal neurons in vivo. Eur J Neurosci 1996; 8:1167 - 75; http://dx.doi.org/10.1111/j.1460-9568.1996.tb01284.x; PMID: 8752586
- Hains BC, Black JA, Waxman SG. Primary cortical motor neurons undergo apoptosis after axotomizing spinal cord injury. J Comp Neurol 2003; 462:328 - 41; http://dx.doi.org/10.1002/cne.10733; PMID: 12794736
- Hammond EN, Tetzlaff W, Mestres P, Giehl KM. BDNF, but not NT-3, promotes long-term survival of axotomized adult rat corticospinal neurons in vivo. Neuroreport 1999; 10:2671 - 5; http://dx.doi.org/10.1097/00001756-199908200-00043; PMID: 10574390
- Kalil K, Schneider GE. Retrograde cortical aand axonal changes following lesions of the pyramidal tract. Brain Res 1975; 89:15 - 27; http://dx.doi.org/10.1016/0006-8993(75)90130-4; PMID: 1148840
- Klapka N, Hermanns S, Straten G, Masanneck C, Duis S, Hamers FP, Müller D, Zuschratter W, Müller HW. Suppression of fibrous scarring in spinal cord injury of rat promotes long-distance regeneration of corticospinal tract axons, rescue of primary motoneurons in somatosensory cortex and significant functional recovery. Eur J Neurosci 2005; 22:3047 - 58; http://dx.doi.org/10.1111/j.1460-9568.2005.04495.x; PMID: 16367771
- Kost SA, Oblinger MM. Immature corticospinal neurons respond to axotomy with changes in tubulin gene expression. Brain Res Bull 1993; 30:469 - 75; http://dx.doi.org/10.1016/0361-9230(93)90280-O; PMID: 8457896
- Lau BY, Foldes AE, Alieva NO, Oliphint PA, Busch DJ, Morgan JR. Increased synapsin expression and neurite sprouting in lamprey brain after spinal cord injury. Exp Neurol 2011; 228:283 - 93; http://dx.doi.org/10.1016/j.expneurol.2011.02.003; PMID: 21316361
- Lee BH, Lee KH, Kim UJ, Yoon DH, Sohn JH, Choi SS, Yi IG, Park YG. Injury in the spinal cord may produce cell death in the brain. Brain Res 2004; 1020:37 - 44; http://dx.doi.org/10.1016/j.brainres.2004.05.113; PMID: 15312785
- Mason MR, Lieberman AR, Anderson PN. Corticospinal neurons up-regulate a range of growth-associated genes following intracortical, but not spinal, axotomy. Eur J Neurosci 2003; 18:789 - 802; http://dx.doi.org/10.1046/j.1460-9568.2003.02809.x; PMID: 12925005
- Merline M, Kalil K. Cell death of corticospinal neurons is induced by axotomy before but not after innervation of spinal targets. J Comp Neurol 1990; 296:506 - 16; http://dx.doi.org/10.1002/cne.902960313; PMID: 2358550
- Mikucki SA, Oblinger MM. Corticospinal neurons exhibit a novel pattern of cytoskeletal gene expression after injury. J Neurosci Res 1991; 30:213 - 25; http://dx.doi.org/10.1002/jnr.490300122; PMID: 1724469
- Nielson JL, Strong MK, Steward O. A reassessment of whether cortical motor neurons die following spinal cord injury. J Comp Neurol 2011; 519:2852 - 69; http://dx.doi.org/10.1002/cne.22661; PMID: 21618218
- Wannier T, Schmidlin E, Bloch J, Rouiller EM. A unilateral section of the corticospinal tract at cervical level in primate does not lead to measurable cell loss in motor cortex. J Neurotrauma 2005; 22:703 - 17; http://dx.doi.org/10.1089/neu.2005.22.703; PMID: 15941378
- Kaas JH, Qi HX, Burish MJ, Gharbawie OA, Onifer SM, Massey JM. Cortical and subcortical plasticity in the brains of humans, primates, and rats after damage to sensory afferents in the dorsal columns of the spinal cord. Exp Neurol 2008; 209:407 - 16; http://dx.doi.org/10.1016/j.expneurol.2007.06.014; PMID: 17692844
- Wu J, Stoica BA, Faden AI. Cell cycle activation and spinal cord injury. Neurotherapeutics 2011; 8:221 - 8; http://dx.doi.org/10.1007/s13311-011-0028-2; PMID: 21373950
- Byrnes KR, Stoica BA, Fricke S, Di Giovanni S, Faden AI. Cell cycle activation contributes to post-mitotic cell death and secondary damage after spinal cord injury. Brain 2007; 130:2977 - 92; http://dx.doi.org/10.1093/brain/awm179; PMID: 17690131
- Di Giovanni S, Knoblach SM, Brandoli C, Aden SA, Hoffman EP, Faden AI. Gene profiling in spinal cord injury shows role of cell cycle in neuronal death. Ann Neurol 2003; 53:454 - 68; http://dx.doi.org/10.1002/ana.10472; PMID: 12666113
- Tian DS, Dong Q, Pan DJ, He Y, Yu ZY, Xie MJ, Wang W. Attenuation of astrogliosis by suppressing of microglial proliferation with the cell cycle inhibitor olomoucine in rat spinal cord injury model. Brain Res 2007; 1154:206 - 14; http://dx.doi.org/10.1016/j.brainres.2007.04.005; PMID: 17482149
- Tian DS, Xie MJ, Yu ZY, Zhang Q, Wang YH, Chen B, Chen C, Wang W. Cell cycle inhibition attenuates microglia induced inflammatory response and alleviates neuronal cell death after spinal cord injury in rats. Brain Res 2007; 1135:177 - 85; http://dx.doi.org/10.1016/j.brainres.2006.11.085; PMID: 17188663
- Tian DS, Yu ZY, Xie MJ, Bu BT, Witte OW, Wang W. Suppression of astroglial scar formation and enhanced axonal regeneration associated with functional recovery in a spinal cord injury rat model by the cell cycle inhibitor olomoucine. J Neurosci Res 2006; 84:1053 - 63; http://dx.doi.org/10.1002/jnr.20999; PMID: 16862564
- Wu J, Stoica BA, Dinizo M, Pajoohesh-Ganji A, Piao C, Faden AI. Delayed cell cycle pathway modulation facilitates recovery after spinal cord injury. Cell Cycle 2012; 11:1782 - 95; http://dx.doi.org/10.4161/cc.20153; PMID: 22510563
- Wu J, Pajoohesh-Ganji A, Stoica BA, Dinizo M, Guanciale K, Faden AI. Delayed expression of cell cycle proteins contributes to astroglial scar formation and chronic inflammation after rat spinal cord contusion. J Neuroinflammation 2012; 9:169; http://dx.doi.org/10.1186/1742-2094-9-169; PMID: 22784881
- Wu J, Kharebava G, Piao C, Stoica BA, Dinizo M, Sabirzhanov B, Hanscom M, Guanciale K, Faden AI. Inhibition of E2F1/CDK1 pathway attenuates neuronal apoptosis in vitro and confers neuroprotection after spinal cord injury in vivo. PLoS One 2012; 7:e42129; http://dx.doi.org/10.1371/journal.pone.0042129; PMID: 22848730
- Wu J, Renn CL, Faden AI, Dorsey SG. TrkB.T1 contributes to neuropathic pain after spinal cord injury through regulation of cell cycle pathways. J Neurosci 2013; 33:12447 - 63; http://dx.doi.org/10.1523/JNEUROSCI.0846-13.2013; PMID: 23884949
- Gomez-Pinilla F, Ying Z, Zhuang Y. Brain and spinal cord interaction: protective effects of exercise prior to spinal cord injury. PLoS One 2012; 7:e32298; http://dx.doi.org/10.1371/journal.pone.0032298; PMID: 22384207
- Nardone R, Höller Y, Brigo F, Seidl M, Christova M, Bergmann J, Golaszewski S, Trinka E. Functional brain reorganization after spinal cord injury: systematic review of animal and human studies. Brain Res 2013; 1504:58 - 73; http://dx.doi.org/10.1016/j.brainres.2012.12.034; PMID: 23396112
- Hulsebosch CE, Hains BC, Crown ED, Carlton SM. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev 2009; 60:202 - 13; http://dx.doi.org/10.1016/j.brainresrev.2008.12.010; PMID: 19154757
- Ankeny DP, Popovich PG. Mechanisms and implications of adaptive immune responses after traumatic spinal cord injury. Neuroscience 2009; 158:1112 - 21; http://dx.doi.org/10.1016/j.neuroscience.2008.07.001; PMID: 18674593
- Kabadi SV, Stoica BA, Loane DJ, Luo T, Faden AI. CR8, a novel inhibitor of CDK, limits microglial activation, astrocytosis, neuronal loss, and neurologic dysfunction after experimental traumatic brain injury. J Cereb Blood Flow Metab 2014; 34:502 - 13; http://dx.doi.org/10.1038/jcbfm.2013.228; PMID: 24398934