 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Synthesis and accumulation of conserved cell cycle regulators such as cyclins are thought to promote G1/S and G2/M transitions in most eukaryotes. 1 When cells at different stages of the cell cycle are fused to form heterokaryons, the shared complement of regulators in the cytoplasm induces the nuclei to become synchronized.2 However, multinucleate fungi often display asynchronous nuclear division cycles, even though the nuclei inhabit a shared cytoplasm. 3 Similarly, checkpoints can induce nuclear asynchrony in multinucleate cells by arresting only the nucleus that receives damage. 4-6 The cell biological basis for nuclear autonomy in a common cytoplasm is not known. Here we show that in the filamentous fungus Ashbya gossypii, sister nuclei born from one mitosis immediately lose synchrony in the subsequent G1 interval. A conserved G1 transcriptional regulatory circuit involving the Rb-analogue Whi5p promotes the asynchronous behavior yetWhi5 protein is uniformly distributed among nuclei throughout the cell cycle. The homologous Whi5p circuit in S. cerevisiae employs positive feedback to promote robust and coherent entry into the cell cycle. We propose that positive feedback in this same circuit generates timing variability in a multinucleate cell. These unexpected findings indicate that a regulatory program whose products (mRNA transcripts) are translated in a common cytoplasm can nevertheless promote variability in the individual behavior of sister nuclei.
Introduction
Among the most influential classic experiments in cell cycle research is a series of elegant cell-fusion studies conducted by Rao and Johnson with cultured HeLa cells. When cells at different stages of the cell cycle were fused together to form heterokaryons, their nuclei rapidly became synchronized. These findings indicated that cytoplasmic signals control the nuclear cycle so that nuclei sharing the same cytoplasm act in unison.Citation7–Citation12 Subsequent work identified a regulatory network centered around cyclins and cyclin-dependent kinases (CDKs) as the key regulators responsible for triggering various cell cycle transitions. The current paradigm for cell cycle control is that regulated synthesis and degradation of cyclins and other regulators manage the timing of cell cycle events. These regulators shuttle between nuclear and cytoplasmic compartments, and when two nuclei share a common cytoplasm, exposure to the same levels of these regulators is thought to lead both nuclei to transit the cell cycle synchronously.
A well-documented counterexample to the rule established by the classic heterokaryon experiments occurs upon localized checkpoint activation. In binucleate sea urchin zygotesCitation4 or binucleate budding yeast cells,Citation5 DNA damage introduced into one nucleus arrests that nucleus in interphase but does not delay mitotic progression of an undamaged nucleus in the same cytoplasm. Even more remarkably, in cultured marsupial cells containing two mitotic spindles, perturbation of chromosome attachment in one spindle delays anaphase only for that spindle, leading to asynchronous metaphase/anaphase transitions in a common cytoplasm.Citation6 These engineered situations reveal that cell cycle transitions are not simply triggered by freely diffusible regulators, but also incorporate local information regarding nuclear or spindle status. The nature of the local regulation of checkpoint activation is unknown.
Other dramatic exceptions to the rule that nuclei sharing a common cytoplasm transit the cell cycle in unison include the naturally occurring multinucleate fungi Neurospora crassa and Ashbya gossypii. In A. gossypii, nuclei divide without nuclear envelope breakdown or cell division, producing large syncytia with many nuclei. Over time, the nuclei migrate, exchange positions and pass within <1 µm of neighboring nuclei.Citation13 Remarkably, the nuclei divide asynchronously despite their residence in a common cytoplasm and pedigree analysis of individual nuclei indicates that the overall nuclear cycle time varies from 40 to over 200 minutes.Citation14 Sustained asynchronous cycling requires that there be some source of timing variability that causes sister nuclei to cycle at different rates. In addition, there must be a nucleus-specific component to cell cycle regulation that allows nuclei to behave autonomously despite inhabiting a common cytosol where regulators are presumably shared. The molecular basis for either the variability or the nuclear autonomy is currently unknown.
Using timelapse microscopy we determined that G1 time is variable between nuclei that share the same cytoplasm in A. gossypii and that even sister nuclei born from one mitosis lose synchrony from one another during G1. Deregulation of the G1/S transition with increased dose or mutation of key regulators including the Rb-analog Whi5p diminishes this variability, leading to more nuclear synchrony than is observed in wild-type cells. Thus variable nuclear division cycle timing in a syncytium can be under genetic control. We speculate that nuclear cycle timing is influenced by molecular noise introduced by limited levels of central regulators.
Results
G1 times vary between nuclei in the same cytoplasm.
Our previous data demonstrated that even genetically identical sister nuclei, born from one mitosis, differ in their subsequent overall nuclear division cycle times.Citation14 To determine when sister nuclei lose synchrony with each other, we filmed cells in which nuclei were labeled with Tub4-mCherry and a nuclear-localized GFP. Tub4p is gamma tubulin and marks spindle pole bodies (SPBs), which are the fungal analogs of centrosomes and are embedded in the nuclear envelope. In S. cerevisiae, SPBs are duplicated near the G1/S transition in response to G1 cyclin/CDK activity.Citation15,Citation16 Given that 95% of the A. gossypii genome is syntentic and contains homologs to S. cerevisiae genes, we use SPB duplication to mark the end of G1 in A. gossypii. The G1 phase (time from birth to SPB duplication) was 41.5 ± 24.4 min and ranged from 10 to over 65 min for a population of nuclei (, n = 40). Several nuclei did not duplicate SPBs within the observation period (and therefore were in G1 indefinitely, grey bars on histogram in ). When pairs of sister nuclei could be followed as in the example pedigree in , it was clear that sisters born of the same mitosis varied substantially in the time it took to duplicate their SPBs. For 11 pairs of sisters that could be tracked, the difference in G1 time between sisters was 22 ± 11 min (). Thus, early stages of the nuclear division cycle are variable even when genetically identical nuclei are bathed in the same cytosol.
An SPB-based synchrony index.
To determine the basis for variability in G1 duration, we wanted to assess synchrony between sister nuclei in mutant strains with perturbed G1 regulators. However, we found that many of these mutants are sensitive to fluorescence microscopy time-lapse imaging conditions and thus it was not possible to collect sufficient data from nuclear pedigrees for statistically meaningful comparisons of times in wild-type and mutant strains. Therefore, we derived a synchrony index (SI) as a way to evaluate synchrony based on static image data, allowing us to analyze hundreds of nuclei per mutant strain. The SI considers the synchrony relationship between neighboring nuclei that is measured and uses statistics to assess whether the observed synchrony is different from the wild-type, even if strains differ from wild-type in their overall nuclear cycle profiles. Synchrony relationships between neighbors will be impacted by variable nuclear division timing but may also be sensitive to changes in nuclear migration dynamics.
The first step in calculating an SI is to determine the observed synchrony in a given strain. To do this, we imaged large numbers of nuclei and used the SPB status to assign each nucleus to a discrete cell cycle interval (, left side). Nuclei assigned to G1 had a single SPB and nuclei in M had 2 SPBs more than 0.5 µm apart on opposite sides of the nucleus. Nuclei assigned to S/G2 either had a single SPB that was twice the intensity and size as a G1 SPB or had two adjacent SPBs that were less than 0.5 µm apart. Then we focused on pairs of neighboring nuclei in the images and determined if they were in the same (synchronous) or in different (asynchronous) cell cycle stages (). The frequency with which two neighbors were in the same stage of the cell cycle was determined for each strain (the “synchronous pairs” in , right side).
We next calculated the synchrony that would be expected by chance in each strain as a means to normalize the synchrony score so that it is comparable between strains that have different nuclear cycle distributions. The chance synchrony shown in is the probability that any two randomly chosen nuclei would be in the same stage of the cell cycle given the observed distribution of nuclei in various stages of the cell cycle (using the proportions in under SPB category, calculations described in Materials and Methods). The SI is then calculated as the observed synchrony divided by the chance synchrony. Because the SI is a relative, rather than absolute, measure, it provides an indicator of synchrony for each strain that is independent of the overall proportions of nuclei in each stage of the division cycle. This is important as it allows us to separate the effect of mutations on nuclear synchrony from the effect on overall cell cycle progression. This enables us to make pair-wise comparisons of SIs between strains that have different proportions of cell cycle stages using an unequal variance t-test (see Materials and Methods for further explanation of statistics).
Increased levels of G1 cyclin promote synchrony.
We hypothesize that asynchrony in wild-type cells arises due to the action of G1 regulators given the variable G1 times we observed in nuclear pedigrees (). Therefore, we predict that we would see a change in synchrony levels in mutants and this would be evidenced by a change in the SI in mutants compared to wild-type.
We first tested whether increases in G1 cyclin abundance could alter synchrony. In S. cerevisiae, G1 times vary considerably from cell to cell for at least two reasons. First, cell size is variable and progression past Start is delayed until cells reach a critical size.Citation17,Citation18 This is unlikely to be relevant to A. gossypii, where the nuclei coexist within a single cell. Second, molecular noise in the abundance of the G1 cyclins Cln1p and Cln2p contributes a cell-size-independent variability in S. cerevisiae, as revealed by the reduced variability in G1 duration observed upon increased G1 cyclin expression.Citation19 Molecular noise arises in part from the intrinsically noisy nature of transcription, which produces variation in the levels of non-abundant proteins. AgCLN1/2 is the single, syntenic homolog of ScCLN1 and ScCLN2 in the A. gossypii genome and it is an essential gene.Citation20 The AgCln1/2 protein is difficult to detect, perhaps because it is present at very low levels.Citation14,Citation21 To assess whether AgCln1/2p abundance contributes to the observed asynchrony ( and ), we supplied an extra copy of CLN1/2 expressed either from its own promoter or from a constitutive promoter (the ScHIS3 promoter). In both cases, the nuclei were significantly more synchronous than wild-type cells (SI = 1.24 compared 0.96 in wt, p < 0.02, 1-tailed t-test, ). Thus, additional CLN1/2 is sufficient to increase synchrony between neighbors indicating that limitation in the abundance of Cln1/2p is one possible source of variability between nuclei in the time spent in G1.
Deletion of G1 transcription factors increases synchrony.
In S. cerevisiae, Cln1p and Cln2p operate as part of a positive feedback loop that enhances the sharpness and coherence of the G1/S transition.Citation22 CLN1/2 transcription is driven by the activating factors SBF (Swi4/Swi6) and MBF (Mbp1/Swi6), which in turn are targets of the Cln1/2/3p-associated CDK.Citation23–Citation27 A side-effect of positive feedback is that molecular noise becomes particularly important in determining when the positive feedback loop is triggered in a given cell, because the transition can be triggered by a relatively small initial change in Cln1/2p abundance. Deletion of SBF or MBF components in S. cerevisiae reduces positive feedback and makes cell cycle transitions less coherent, but does not arrest the cell cycle because cells transcribe intermediate levels of CLN1/2 sufficient for cell cycle progression.Citation28,Citation29 If a similar situation applies in A. gossypii, we would predict that SBF and MBF mutants would reduce positive feedback and hence make molecular noise less effective at introducing variability. This would be reflected in increased SIs in SBF and MBF mutants compared to wild-type cells. Both swi4 and mbp1 mutants were viable in A. gossypii and had similar proportions of G1 nuclei as wild-type cells (). Notably, the nuclei in these strains were considerably more synchronous than wild type in their nuclear division cycles, with an SI of 1.93 (swi4) or 1.63 (mbp1) (p < 0.001, 1-tailed t-test, ). While we presently cannot rule out some contribution of dampened nuclear dynamics on increased synchrony in these mutants, the spacing between nuclei in the strains was comparable to wild type. Therefore we envision that much of this increase in synchrony originates from the alterations to cell cycle regulation.
SBF-mediated CLN1/2 transcription in yeast is regulated by Whi5p, a transcriptional repressor analogous to Rb in animal cells. Phosphorylation of Whi5p by Cln3/CDK and Cln1/2p-CDK relieves repression and causes Whi5p nuclear export at Start.Citation30,Citation31 There is a CLN3 homolog in A. gossypii; however, the mutant is pleiotropic with a complex phenotype indicating that its function may have expanded since these species diverged. Therefore analysis of Cln3p will be presented in detail elsewhere. Instead we have focused here on the action of the Whi5 repressor. We predicted that deletion of AgWHI5 would increase synchrony in A. gossypii because: (1) loss of Whi5p should lead to increased CLN1/2 transcription, directly reducing molecular noise and (2) it should reduce the variability introduced as a result of positive feedback within individual nuclei, making the G1/S transition less sensitive to molecular noise. Indeed, the whi5 mutant had the largest observed effect on nuclear asynchrony, increasing the SI to 2.18 (p < 0.001, 1-tailed t-test, ). Unlike the other mutant strains, we were able to film whi5Δ cells in several short movies to capture a small number of G1 times and we could see that the length of G1 was shorter in these cells and in fact less variable in the population. The time from birth to SPB duplication for a whi5Δ nucleus was 18.0 ± 7.0 min (n = 22, compared to 41 ± 24 min in the wt).
Our findings indicate that sister nuclei in A. gossypii exhibit considerable variability in the duration of G1 and that altering levels of the components of a conserved G1 regulatory circuit can significantly diminish this variability. We hypothesized that the G1 regulators that we altered could be partitioned asymmetrically at nuclear division, leading to inherited differences between sister nuclei that promote variability in the subsequent cycle. As Whi5p had the greatest effect on timing variability, we examined Whi5p localization during the nuclear division cycle.
AgWhi5p is constitutively nuclear and equally partitioned during mitosis.
In S. cerevisiae, overall cellular Whi5p levels are constant but the protein is swiftly exported from G1 nuclei upon phosphorylation by G1-cyclin/CDK1 complexes at Start.Citation30–Citation32 ScWhi5p does not re-accumulate in nuclei until low CDK levels are reached in late mitosis. Additionally, it has been shown in animal cells that Rb localization can be regulated by CDK activity.Citation33 To examine subcellular localization of Whi5p, we visualized in living cells a functional Whi5-GFP and Tub4-mCherry, each expressed from their endogenous loci and promoters. Unexpectedly, we observed that Whi5p localized to all nuclei, regardless of nuclear division cycle stage with the same mean fluorescence intensity in all stages ( and B and data not shown). To determine whether there is transient asymmetry between sister nuclei in the inheritance of Whi5p, cells expressing Whi5-GFP were imaged by time-lapse fluorescence microscopy (Supp. Vid. 1). Sister nuclei measured immediately after mitosis have nearly equivalent mean fluorescence signals for Whi5-GFP that vary in most cases by <5.0% in the signal intensity ( and D and Supp. Vid. 1). These data indicate that Whi5p is present in nuclei throughout the cell cycle and is symmetrically inherited at mitosis. Therefore, differences in the levels of Whi5p between sisters are not a likely source of variability. Instead, we propose that Whi5p may be inactivated at different times in sisters due to variable rates at which G1 cyclins accumulate in nuclei. These timing differences may in part arise due to low levels of G1 cyclins that make G1 times sensitive to effects of molecular noise.
Discussion
Cell-to-cell variability in the timing of the cell division cycle is widespread throughout prokaryotes and eukaryotes. In the 1960s and 1970s, a number of researchers noticed that genetically identical cells, cultured in the same environment, do not take the same amount of time to pass through the division cycle.Citation17,Citation34–Citation38 “Non-genetic individuality” (coined by Spudich and Koshland in 1976) was noted for the division cycle times of bacteria, algae, yeast and mammalian cells.Citation39 Time-lapse analyses of S. cerevisiae revealed that much of the variability in division cycle times was generated in G1. While some of the timing differences are accounted for by cell size differences (with small cells taking longer in G1), it was also appreciated that there is also substantial size-independent variability in the length of G1.Citation40–Citation43
Study of G1-phase variability has experienced a recent renaissance. The use of more precise protein markers of the G1/S transition supported the presence of size-dependent and sizeindependent features that modulate the length of G1 in individual cells.Citation19,Citation32 These studies indicate that molecular noise, some of which arises from fluctuations in low abundance regulatory proteins, does contribute substantial kinetic variability within populations of cells. Similarly, analyses of single mammalian cells support the presence of a considerable and under-appreciated reservoir of variability in the mammalian cell cycle in a population of seemingly identical cells.Citation44–Citation45 In A. gossypii, we have shown that a similar variability exists between nuclei sharing a common cytoplasm. Because the shared cytoplasm eliminates the cell-size component of variability that is a major contributor in uninucleate cells, A. gossypii is particularly well-suited to discover other sources of variability in the cell cycle.
Here we show that asynchronous division of genetically identical nuclei sharing a common cytoplasm depends upon an intact G1/S control pathway. Mutants lacking any component of the pathway become more synchronous, indicating that variability in this system is under genetic control (). It is intriguing that perturbation of this transcriptional program should have such a profound impact on nuclear variability within a syncytium. As transcripts generated in one nucleus are presumably translated in a common cytoplasm, how is it that their effects are not homogenized?
One possibility is that transcripts stay close to their originating nuclei and that the protein products preferentially re-enter those nuclei. However, previous work in A. gossypii heterokaryons has shown that B-type cyclin transcripts encoded in one nucleus generate proteins that can diffuse and enter nearby nuclei that do not themselves encode that cyclin.Citation14 Thus, there do not appear to be general diffusion barriers between nuclei. Nevertheless, transcript- or protein-specific barriers may exist. Alternatively, it is also possible that while proteins or transcripts can diffuse, they do not accumulate to a functional level in adjacent nuclei.
We predict that individual nuclei are creating “neighborhoods” of cytoplasm that are functionally distinct despite being adjacent to one another. The biophysical properties of these zones can then influence the concentration of transcripts and proteins regulating the G1/S transition. These territories of cytoplasm may restrain the transcriptional output of SBF/MBF within the vicinity of one nucleus so that key transcripts/proteins do not influence timing of neighboring nuclei (). The cytoskeleton or nuclear-associated ER may establish these regions and dynamic nuclei may actually carry with them a functional shell as they navigate the cytoplasm.
The organization of distinct zones of cytoplasm around nuclei may generate functional compartments in the syncytia of many organisms. For example, nuclei in Drosophila syncytial embryos have been shown to have organized, compartmentalized islands of cytoplasm and nearby plasma membrane prior to cellularization.Citation46–Citation48 Similarly binucleate sea urchins,Citation4 yeast,Citation5 and marsupial cellsCitation6 in which checkpoints have been triggered in only one of the nuclei limit diffusion of checkpoint signals to the unaffected nucleus. Compartments without continuous membrane borders may allow for spatially and temporally distinct behaviors of nuclei sharing a common cytoplasm in multinucleate cells. How cells can establish islands of cytoplasm with distinct properties in a single cell is unknown but work from diverse systems suggests that they exist. It is likely that some level of cytoplasmic compartmentalization can occur even in uninucleate cells to generate gradients of cytoplasm with distinct properties. A. gossypii, a simple filamentous fungus that is amenable to molecular genetics and cytology, will be a powerful system to study the molecular basis of this compartmentalization.
Materials and Methods
Growth conditions and strain construction.
A. gossypii was cultured and transformed using previously established protocols.Citation49 DNA manipulations were carried out as described previously using DH5α.Citation50 Taq polymerase from Roche (Indianapolis, IN) was used in PCR reactions performed using standard methods. All oligonucleotides used for tagging proteins or creating deletions were synthesized by IDT (Coralville, IA) or Invitrogen (Carlsbad, CA) and all restriction enzymes were obtained from New England Biolabs (Beverly, MA). The strains generated and used in this study are listed under Supplemental Table 1 and ≥2 independent transformants were analyzed for each strain. Plasmids used here are listed in Supplemental Table 2. Oligonucleotide primers used are listed in Supplemental Table 3. Plasmid isolation from yeast was performed as described previouslyCitation51 using DHD5 as a yeast host strain. All sequencing was carried out by Dartmouth College Core Facilities (Hanover, NH).
PCR-mediated gene targeting with dominant selection markers was used to create A. gossypii deletion mutantsCitation9,Citation52 pAGT140 served as the template for amplifying the G418 resistance cassette using “gene name”-D1/D2 (or Fwd del/Rev del) primer pairs which had ∼45 bp homology to the termini of the gene of interest and ∼20 bp homology to the termini of a selection marker. pAGT100 was used for amplifying the NATPS resistance cassette for Nourseothricin (CloNAT, obtained from Werner Bioagents, Germany) resistance. Verification of deletion mutants and correct integration of selection markers into the genome was verified by three independent analytical PCR reactions for each strain using combinations of oligonucleotides “gene name”-V1, V2, Int (gene/locus-specific sequences), VG3, VG5 (G418-specific sequences) V2 NAT1 and V3 NAT1 (NAT1-specific sequences). Transformation of the multinucleate mycelium with PCR products resulted in heterokaryotic mycelia that contained a mixture of both transformed and wild-type nuclei. In order to obtain homokaryotic mycelia, single uninucleate spores from heterokaryotic mycelia were picked and grown on selective plates. At least two independent transformants were characterized for each strain.
Tagging of WHI5 and TUB4 was done via co-transformation in yeast. For Whi5-GFP, the primer pair AGO1 and AGO38 was used to generate PCR product, using pAGT141 as a template.Citation53 The 3′ ends of these primers have sequence homology to the GFP-GEN3 cassette in plasmid pAGT141. The 5′ ends of the oligos have sequence homology to the end of WHI5 immediately upstream and several nucleotides downstream of the stop codon, respectively. The PCR product containing the GFP-GEN3 cassette was co-transformed with the plasmid pAGB002 into the yeast DHD5 strain. Yeast in which the PCR product recombined with the plasmid were selected on plates containing 200 µg/ml G418 (Calbiochem). The resulting recombinant plasmid recovered from yeast, pAGB006, was then verified by diagnostic enzymatic digestion and sequencing. The plasmid pAGB006 was digested with NotI and XhoI and the 5,165 bp fragment was gel purified and transformed into A. gossypii to generate the strain AG98.
In order to create AG290 (WHI5-GFP TUB4-mCherry), the mCHERRY-NAT1 cassette was first amplified from pAGT211 and co-transformed into yeast with pAGB144. The resulting recombined plasmid was verified by digestion and sequencing. This plasmid was then digested with SacII and the 5,040 bp product was gel-purified and transformed into AG98 to obtain AG285, which was sporulated and individual spores were dissected to make the AG290 homokaryon.
To replace the endogenous AgCLN1/2 promoter with the ScHIS3 promoter, ScHIS3pr was amplified from AGB096,Citation54 using primer pair AGO438 and 439 and the PCR product was co-transformed into yeast with pAGB188. The plasmid recovered from yeast, AGB192, was verified by digest, sequenced and transformed into WT.
Statistical methods for synchrony index.
All raw data and calculations can be found in the Supplemental Excel file. Statistical analysis involved assessing whether the SI for each mutant was significantly different from that for the WT. Hypothesis testing was performed using “join count” statistics for spatial autocorrelation.Citation55,Citation56 The first step of this test requires the determination of the chance synchrony. This involves first using the measured proportion of nuclei in the population that are in each cell cycle stage to calculate the number of each type of neighbor relationship (i.e., “join”) expected to result from chance, according to the expressions:
where
is the expected number of matching neighbors,
is the expected number of non-matching neighbors, k is the total number of observed neighbor relations, pA is the proportion of nuclei in any one stage, and pA, is the proportion in a different stage (=1 − pA). The uncertainty in the number of each type of neighbor relation expected to result from chance has been shown to follow a normal distribution with standard errors calculated as:
where n is the number of nuclei observed.
To determine whether the SI for each mutant was significantly different from that for WT, we used an unequal variance t-test. Standard errors, sAA and sAA, calculated for each strain from the join count statistics given above were translated into standard errors for the corresponding SI values using first-order error analysis. This leads to the following expression for the t-statistic:
where
with
and
where m and wt indicate the appropriate values for the mutant and wild-type strains, respectively. The number of degrees of freedom for the t-statistic is determined from:
Microscope setup, image acquisition and image processing.
All images were acquired on a Zeiss AxioImager-M1 upright light microscope equipped with the following Zeiss oil immersion objectives: EC Plan-Neofluar 40x/1.3NA, Plan-Neofluar 100x/1.3NA, Plan-Fluar 100x/1.45NA and PlanApochromat 63X/1.4NA. Chroma filter set 41025 and Zeiss filter set 38HE were used for visualization of GFP. Chroma filter sets 41002B and 41043 were used for visualizing AlexaFluor 568 and mCherry respectively. Hoechst staining was visualized using a Zeiss filter set 49. An Exfo X-Cite 120 lamp was employed as the fluorescence light source. A Hamamatsu Orca-ER (C4742-80-12AG) CCD camera driven by either OpenLab 5 (Improvision-PerkinElmer) or Volocity 4 (Improvision-PerkinElmer) was used for acquisition of images. The resulting images were processed by fast or iterative deconvolution using calculated point spread functions in Volocity 4 to generate movies. All still images were linearly contrast enhanced in Volocity 4 and Photoshop CS2 (Adobe). All images and movies presented are extended projections of >10 × 0.5 µm step images acquired along the z axis to ensure that the nuclei and SPBs were imaged completely.
For determining neighbor relationships, nuclei were evenly spaced apart in most cells but in cases where nuclei were clumped, the relationship was assessed based on the nearest neighbor to form a pair.
Cells used in time-lapse experiments were grown in liquid AFM with continued shaking for ∼13 hours at 30°C. The low-fluor minimal medium (Sunrise Science products) to limit background fluorescence. An agar pad was made by dropping 1 ml of molten 2X CSM low fluor minimal medium +1% agarose on to a glass slide and dropping a cover slip on it to make a flat surface. The cells were imaged in a humid chamber at 30°C after incubating 30 min on the gel pad. For data, to enrich for division and observe the behavior of sister pairs, after the overnight growth in AFM, nocodazole (Sigma) was added at a final concentration of 8–10 µg/mL and allowed to incubate at 30°C for another hour (this is half the nocodazole concentration and a quarter of the time needed to synchronize nuclei). The cells were then spun down gently at 500 rpm and washed three times with 1/4xAFM. 10 µl of cells were placed on the solidified agarose pad and a coverslip was gently placed on top of the cell suspension. Data derived from time-lapse movies () were acquired using the PlanApochromat 63X/1.4NA objective at 10–20% fluorescence light transmission, exposed for 100 ms per z-step, through ten 0.5 µm steps in the z direction with 2X binning. Images were taken every minute for the GFP signal and every two minutes for the mCherry signal and focusing was performed using the auto-focus function in Openlab 5. For data in , strains were processed for anti-tubulin immunofluorescence as a second measure of SPBs in addition to Tub4-mCherry as previously described.Citation14 Supplemental Video 1 was obtained using a Quorum Wave FX Spinning Disc Confocal System with which images were taken every 30 seconds to visualize Whi5-GFP and every 5 min to visualize Tub4-mCherry. There was no post-acquisition processing and the relative intensity of sister nuclei was determined by Volocity 4 using the measurements modules which summed the total fluorescence for each pixel in the nucleus to generate a mean fluorescence for the nucleus and subtracted the cytosolic background. Each sister's corrected mean fluorescence was then divided by the sum of the mean fluorescence signal of the sisters combined and the absolute value of the differences between sisters is reported.
Figures and Tables
Figure 1 G1 is variable between nuclei in the same cell. (A) Cells with GFP-localized to nuclei and SPBs visualized with mCherry were filmed using 4D fluorescence microscopy (AG290, Whi5-GFP, Tub4-mCherry). Histogram represents the distribution of times observed from birth to SPB duplication for 40 nuclei. Grey bars represent nuclei that were born but never duplicated their SPBs during the observation period and therefore ultimate G1 times are unknown but are greater than the movie length (65–100 min). The average G1 duration includes the minimal G1 lengths of these nuclei that did not transition during observation. (B) Example pedigree showing variable G1 lengths between sister nuclei. (C) Overlapping barplot of G1 times for 11 pairs of sister nuclei. The G1 time for the slow sister is in black and the G1 time for the fast sister is overlapping in grey. (D) Schematic summarizing how pairs of neighboring nuclei are considered for scoring in .
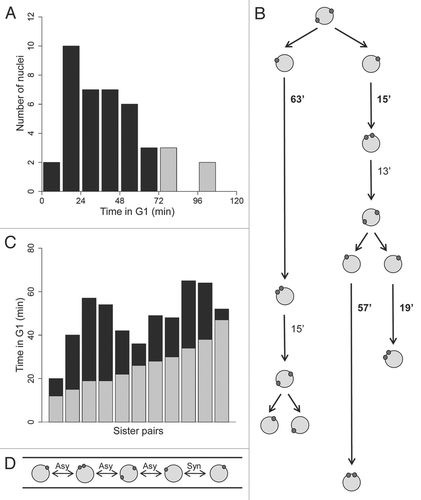
Figure 2 Whi5p is nuclear in all the stages of the cell cycle. (A) A young mycelium expressing Whi5-GFP and Tub4-mCherry shows that all nuclei have Whi5-GFP (AG290). Bar, 5 µm. (B) Individual nuclei in different stages of the nuclear division cycle containing Whi5-GFP. Bar, 5 µm. (C) Images from a time series showing Whi5-GFP appears equally segregated in mitosis between sister nuclei. The nucleus undergoing mitosis is indicated by an asterisk and the resulting sister nuclei are indicated by arrowheads. Bar, 5 µm. (D) Histogram depicting the percent difference in Whi5-GFP mean fluorescence intensity between sister nuclei born from one mitosis. This difference in intensity is calculated by dividing the mean fluorescence intensity of each sister nucleus by the total intensity of the two sister nuclei and taking the absolute value of the difference between the values (n = 19 sister pairs).
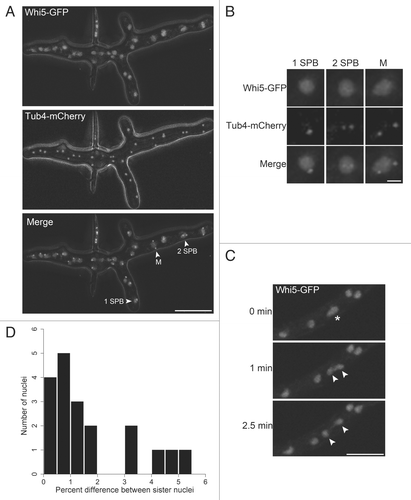
Figure 3 Model for asynchrony arising in G1. Neighboring nuclei divide asynchronously and newborn sister nuclei spend different amounts of time in G1. In mutants with altered regulation of the G1/S transition, nuclei become more synchronous.
Table 1 Observed frequency distributions of nuclear cycle stages and neighbor relations
Table 2 Synchrony index statistics
Additional material
Download Zip (676.4 KB)Acknowledgements
We are grateful for the stimulating discussions from the Gladfelter lab and to Sharon Bickel, Daniel Lew and Roger Sloboda for reading the manuscript. This work was supported by American Cancer Society grant #IRG-82-003-24 (A.G.), a Basil O'Connor award from the March of Dimes (A.G.), a Howard Hughes Medical Institute post-doctoral fellowship (D.N.) and NIH award T32GM008704 (C.D.).
References
- Bloom J, Cross FR. Multiple levels of cyclin specificity in cell cycle control. Nat Rev Mol Cell Biol 2007; 8:149 - 160
- Rao PN, Johnson RT. Mammalian cell fusion: studies on the regulation of DNA synthesis and mitosis. Nature 1970; 225:159 - 164
- Gladfelter AS. Nuclear anarchy: Mitosis in multinucleated cells. Current Opinion in Microbiology 2006;
- Hinchcliffe EH, Thompson EA, Miller FJ, Yang J, Sluder G. Nucleo-cytoplasmic interactions that control nuclear envelope breakdown and entry into mitosis in the sea urchin zygote. J Cell Sci 1999; 112:1139 - 1148
- Demeter J, Lee SE, Haber JE, Stearns T. The DNA damage checkpoint signal in budding yeast is nuclear limited. Mol Cell 2000; 6:487 - 492
- Rieder CL, Khodjakov A, Paliulis LV, Fortier TM, Cole RW, Sluder G. Mitosis in vertebrate somatic cells with two spindles: implications for the metaphase/anaphase transition checkpoint and cleavage. Proc Natl Acad Sci USA 1997; 94:5107 - 5112
- Rao PN, Johnson RT. Mammalian cell fusion: studies on the regulation of DNA synthesis and mitosis. Nature 1970; 225:159 - 162
- Johnson RT, Rao PN. Mammalian cell fusion: induction of premature chromosome condensation in interphase nuclei. Nature 1970; 226:717 - 722
- Johnson RT, Rao PN. Nucleo-cytoplasmic interactions in the acheivement of nuclear synchrony in DNA synthesis and mitosis in multinucleate cells. Biol Rev Camb Philos Soc 1971; 46:97 - 155
- Johnson RT, Rao PN, Hughes HD. Mammalian cell fusion. 3. A HeLa cell inducer of premature chromosome condensation active in cells from a variety of animal species. J Cell Physiol 1970; 76:151 - 157
- Rao PN, Johnson RT. Mammalian cell fusion. IV. Regulation of chromosome formation from interphase nuclei by various chemical compounds. J Cell Physiol 1971; 78:217 - 223
- Rao PN, Johnson RT. Premature chromosome condensation: a mechanism for the elimination of chromosomes in virus-fused cells. J Cell Sci 1972; 10:495 - 513
- Alberti-Segui C, Dietrich F, Altmann-Johl R, Hoepfner D, Philippsen P. Cytoplasmic dynein is required to oppose the force that moves nuclei towards the hyphal tip in the filamentous ascomycete Ashbya gossypii. PG. J Cell Sci 2001; 114
- Gladfelter AS, Hungerbuehler AK, Philippsen P. Asynchronous nuclear division cycles in multinucleated cells. J Cell Biol 2006; 172:347 - 362
- Simmons Kovacs LA, Nelson CL, Haase SB. Intrinsic and cyclin-dependent kinase-dependent control of spindle pole body duplication in budding yeast. Mol Biol Cell 2008; 19:3243 - 3253
- Pringle JR, Hartwell LH. Strathern JD, Jones EW, Broach JR. The Saccharomyces cerevisiae cell cycle. The Molecular Biology of the Yeast Saccharomyces 1981; Cold Spring Harbor NY Cold Spring Harbor Laboratory 97 - 142
- Hartwell L, Unger M. Unequal division in Saccharomyces cerevisiae and its implications for the control of cell division. J Cell Biol 1977; 75:422 - 435
- Johnston GC, Pringle JP, Hartwell LH. Coordination of growth with cell division in the yeast S. cerevisiae. Exp Cell Res 1977; 105:75 - 98
- Di Talia S, Skotheim JM, Bean JM, Siggia ED, Cross FR. The effects of molecular noise and size control on variability in the budding yeast cell cycle. Nature 2007; 448:947 - 951
- Hungerbuehler AK, Philippsen P, Gladfelter AS. Limited functional redundancy and oscillation of cyclins in multinucleated A. gossypii fungal cells. Eukaryot Cell 2007; 6:473 - 486
- Hungerbuehler AK, Philippsen P, Gladfelter AS. Limited functional redundancy and oscillation of cyclins in multinucleated Ashbya gossypii fungal cells. Eukaryot Cell 2007; 6:473 - 486
- Skotheim JM, Di Talia S, Siggia ED, Cross FR. Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature 2008; 454:291 - 296
- Wijnen H, Landman A, Futcher B. The G(1) cyclin Cln3 promotes cell cycle entry via the transcription factor Swi6. Mol Cell Biol 2002; 22:4402 - 4418
- Nasmyth K, Dirick L. The role of SWI4 and SWI6 in the activity of the G1 cyclins in yeast. Cell 1991; 66:995 - 1013
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, et al. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell 1998; 9:3273 - 3297
- Wittenberg C, Reed SI. Cell cycle-dependent transcription in yeast: promoters, transcription factors and transcriptomes. Oncogene 2005; 24:2746 - 2755
- Lowndes NF, Johnson AL, Breeden L, Johnston LH. SWI6 protein is required for transcription of the periodically expressed DNA synthesis genes in budding yeast. Nature 1992; 357:505 - 508
- Stuart D, Wittenberg C. Cell cycle-dependent transcription of CLN2 is conferred by multiple distinct cis-acting regulatory elements. Mol Cell Biol 1994; 14:4788 - 4801
- Breeden L, Mikesell G. Three independent forms of regulation affect expression of HO, CLN1 and CLN2 during the cell cycle of Saccharomyces cerevisiae. Genetics 1994; 138:1015 - 1024
- de Bruin RA, McDonald WH, Kalashnikova TI, Yates J 3rd, Wittenberg C. Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 2004; 117:887 - 898
- Costanzo M, Nishikawa JL, Tang X, Millman JS, Schub O, Breitkreuz K, et al. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 2004; 117:899 - 913
- Bean JM, Siggia ED, Cross FR. Coherence and timing of cell cycle start examined at single-cell resolution. Mol Cell 2006; 21:3 - 14
- Jiao W, Datta J, Lin HM, Dundr M, Rane SG. Nucleocytoplasmic shuttling of the retinoblastoma tumor suppressor protein via Cdk phosphorylation-dependent nuclear export. J Biol Chem 2006; 281:38098 - 38108
- Sennerstam R, Stromberg JO. A comparative study of the cell cycles of nullipotent and multipotent embryonal carcinoma cell lines during exponential growth. Dev Biol 1984; 103:221 - 229
- Zetterberg A, Larsson O. Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of Swiss 3T3 cells. Proc Natl Acad Sci USA 1985; 82:5365 - 5369
- Fox TO, Pardee AB. Animal cells: noncorrelation of length of G1 phase with size after mitosis. Science 1970; 167:80 - 82
- Fantes P, Nurse P. Control of cell size at division in fission yeast by a growth-modulated size control over nuclear division. Exp Cell Res 1977; 107:377 - 386
- Koch AL, Schaechter M. A model for statistics of the cell division process. J Gen Microbiol 1962; 29:435 - 454
- Spudich JL, Koshland DE Jr. Non-genetic individuality: chance in the single cell. Nature 1976; 262:467 - 471
- Tyson CB, Lord PG, Wheals AE. Dependency of size of Saccharomyces cerevisiae cells on growth rate. J Bacteriol 1979; 138:92 - 98
- Lord PG, Wheals AE. Asymmetrical division of Saccharomyces cerevisiae. J Bacteriol 1980; 142:808 - 818
- Lord PG, Wheals AE. Variability in individual cell cycles of Saccharomyces cerevisiae. J Cell Sci 1981; 50:361 - 376
- Lord PG, Wheals AE. Rate of cell cycle initiation of yeast cells when cell size is not a rate-determining factor. J Cell Sci 1983; 59:183 - 201
- Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008; 14:111 - 122
- Holland AJ, Cleveland DW. Beyond genetics: surprising determinants of cell fate in antitumor drugs. Cancer Cell 2008; 14:103 - 105
- Frescas D, Mavrakis M, Lorenz H, Delotto R, Lippincott-Schwartz J. The secretory membrane system in the Drosophila syncytial blastoderm embryo exists as functionally compartmentalized units around individual nuclei. J Cell Biol 2006; 173:219 - 230
- Mavrakis M, Rikhy R, Lippincott-Schwartz J. Cells within a cell: Insights into cellular architecture and polarization from the organization of the early fly embryo. Commun Integr Biol 2009; 2:313 - 314
- Mavrakis M, Rikhy R, Lippincott-Schwartz J. Plasma membrane polarity and compartmentalization are established before cellularization in the fly embryo. Dev Cell 2009; 16:93 - 104
- Wendland J, Ayad-Durieux Y, Knechtle P, Rebischung C, Philippsen P. PCR-based gene targeting in the filamentous fungus Ashbya gossypii. Gene 2000; 242:381 - 391
- Sambrook J. Molecular Cloning: A Laboratory Manual 2001; Cold Spring Harbor Cold Spring Harbor Laboratory Press
- Schmitz HP, Kaufmann A, Kohli M, Laissue PP, Philippsen P. From function to shape: a novel role of a formin in morphogenesis of the fungus Ashbya gossypii. Mol Biol Cell 2006; 17:130 - 145
- Wach A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 1996; 12:259 - 265
- Kaufmann A. A plasmid collection for PCR-based gene targeting in the filamentous ascomycete Ashbya gossypii. Fungal Genet Biol 2009; 46:595 - 603
- Helfer H, Gladfelter AS. AgSwe1p regulates mitosis in response to morphogenesis and nutrients in multinucleated Ashbya gossypii cells. Mol Biol Cell 2006; 17:4494 - 4512
- Cliff AD, Ord JK. Spatial Autocorrelation 1973; London Pion Press
- Moran PA. The interpretation of statistical maps. J R Stat Soc Series B Stat Methodol 1948; 10:243 - 251
- Altmann-Johl R, Philippsen P. AgTHR4, a new selection marker for transformation of the filamentous fungus Ashbya gossypii, maps in a four-gene cluster that is conserved between A. gossypii and Saccharomyces cerevisiae. Mol Gen Genet 1996; 250:69 - 80
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 1989; 122:19 - 27
- Dietrich FS, Voegeli S, Brachat S, Lerch A, Gates K, Steiner S, et al. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science 2004; 304:304 - 307