Abstract
Soluble amyloid β (Aβ) oligomers cause memory loss and synaptic dysfunction in Alzheimer disease (AD). Despite intensive studies on Aβ assembly in vitro and in vivo, the localization and cellular mechanism of Aβ oligomerization are not fully understood. Previously, we demonstrated that cytoplasmic superoxide radicals contribute to drusen deposition, a hallmark of age-related macular degeneration as well as other geriatric diseases (fatty liver, skin thinning, and osteoporosis). Using a transgenic mouse model of AD, we recently clarified the role of cytoplasmic oxidative stress in cognitive impairment and oligomer formation. Moreover, we also found that these phenomena were associated with neuroinflammation, tau phosphorylation, and synaptic loss. Notably, studies using human brains support the involvement of cytoplasmic superoxide radicals in AD pathology. In this addendum to Murakami et al. (JBC 2011), we discuss and comment on intracellular Aβ oligomer formation and the possible therapeutic effects of intracellular redox state modulators.
Alzheimer disease (AD) is characterized by the presence of senile plaques that mainly contain 40- and 42-mer amyloid β-proteins (Aβ40, Aβ42).Citation1,Citation2 These proteins are generated from amyloid β-protein precursor by β-site hAPP-cleaving enzyme 1 (BACE1), followed by γ-secretase processing (the amyloidogenic pathway). Many studies have demonstrated that soluble Aβ oligomers (50~60 kDa: Aβ-derived diffusible ligands, Aβ*56, and globulomers; > 100 kDa: amylospheroid, protofibrils, and annuli) rather than insoluble fibrils cause memory loss and synaptic dysfunction.Citation3 Oxidative stress contributes to the pathogenesis of several neurodegenerative diseases, such as AD, Parkinson disease, and amyotrophic lateral sclerosis.Citation4,Citation5 In addition, a study that detected the in vitro radicalization of Aβ suggested that the Aβ-induced neurotoxicity observed in AD is associated with oxidative damage.Citation6
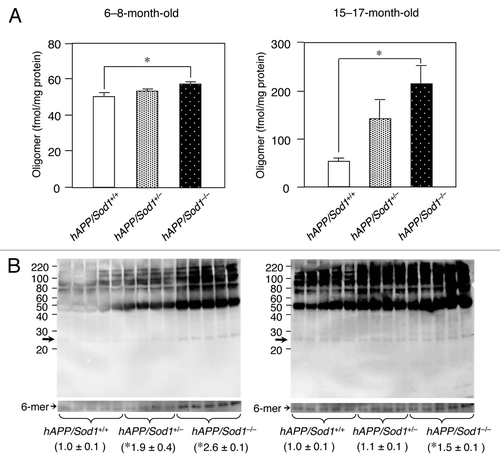
Superoxide dismutase (SOD) is one of the major antioxidant metalloenzymes that converts toxic superoxide radicals (O2-) to H2O2, followed by Fenton reaction.Citation7 SOD consists of three isozymes: CuZn-SOD (SOD1), which is found in the cytosol, nucleus, and intermembrane space of mitochondria; Mn-SOD (SOD2), which is localized in the mitochondrial matrix; and extracellular SOD (SOD3), which is also a complex of Cu and Zn and is found in the blood. We previously reported that Sod1-deficient (Sod1-/-) mice displayed drusen deposition, which is a typical characteristic of age-related macular degeneration,Citation8 fatty liver,Citation9 skin thinning,Citation10 and osteoporosis.Citation11 Some of these phenotypes can be alleviated by vitamin C.Citation10,Citation11 Although mitochondrial oxidative stress has hitherto been believed to be a potent cause of neuronal disorders,Citation12 whether the damage induced by mitochondrial radicals is the main contributor to age-related diseases like AD is questionable because a systemic deficiency in SOD2 causes neonatal lethality in mice.Citation13,Citation14 Our previous study, which involved the use of a human amyloid precursor protein (hAPP) transgenic AD mouse model lacking Sod1 (hAPP/Sod1-/-), shed new light on the importance of cytoplasmic superoxide radicals in the pathogenesis of AD, as the hAPP/Sod1-/- mice accelerated Aβ oligomerization, memory loss, neuroinflammation, tau phosphorylation, and synaptic loss compared with the control AD mouse.Citation15 As shown in , western blotting of anti-Aβ1-16 antibody (6E10) revealed that the formation of Aβ hexamer was significantly increased in hAPP/Sod1-/- rather than hAPP/Sod1+/+. These results are in good agreement with the previous study using anti-N-terminal Aβ antibody (82E1).Citation15 Intriguingly, the levels of SOD1 were significantly lower in human AD patients compared with non-AD individuals, but SOD2 and SOD3 were not.Citation15 Higher levels of BACE1 were also detected in the hAPP/Sod1-/- mice, indicating that the amyloidogenic pathway had been stimulated by the presence of cytoplasmic superoxide radicals.Citation16
Figure 1. An increase of Aβ oligomer by Sod1 deletion in Alzheimer mice. (A) ELISA analysis of 82E1-specific oligomers using the TBS-soluble fraction of brains of mice (n = 5~7 per genotype) of the indicated genotypes and age. In ELISA for Aβ oligomers (Immuno Biochemical Laboratories: IBL, Gunma, Japan), the same N-terminal Aβ antibody (82E1) is used both for antigen capture and detection. In the younger ages (6−8-mo-old), the level of only Aβ oligomers was significantly increased in hAPP/Sod1-/- as compared with the hAPP/Sod1+/+ mice, but not Aβ42 or Aβ40 (ref. Citation15). On the other hand, older (15−17-mo-old) hAPP/Sod1-/- showed a significant elevation of Aβ oligomers as well as Aβ42 and Aβ40 (ref. Citation15) as compared with the hAPP/Sod1+/+ mice. These data are rearrangement of the previous work (ref. Citation15). (B) Distribution of Aβ aggregates by western blotting of mice (n = 4~5 per genotype) of the indicated genotypes and age. The detailed procedure was described previously (ref. Citation15). In brief, Tris buffered saline (TBS)-soluble fractions (2 µg/µL) were subjected to western blotting using 10–20% Tricine gel (Invitrogen) and transferred to a PVDF membrane (0.2 μm pore size, Bio-rad). Anti-Aβ antibody (6E10, 1:1,000, Signet) was used for Aβ detection. Overexposed bands corresponding to the hexamer (arrows: ~30 kDa) were used for relative quantification. left: 6−8-mo-old, right: 15−17-mo-old. *p < 0.05 vs. hAPP/Sod1+/+, mean ± s.e.m.
Intracellular Aβ oligomers have recently become the focus of much AD research. It is indispensible to elucidate where Aβ oligomerizes; i.e., whether Aβ oligomerizes in the cytosol after extracellular secretion and reuptakes into cells, or whether Aβ oligomerizes in the extracellular space and reuptakes into cells. Alternatively, misfolding of Aβ in endoplasmic reticulum-Golgi may lead to oligomerization in the cytosol. Excessive free radicals in the cytosol could directly alter the conformation of intracellular Aβ; e.g., a toxic turn formation of intracellular Aβ42 were proposed by our groups.Citation6,Citation17 Ohyagi and coworkers revealed that intraneuronal Aβ accumulation and memory disturbance occur before extracellular Aβ deposition in mice.Citation18 In the human brain, it was also reported that intracellular Aβ accumulation frequently precedes senile plaque formation.Citation19 Mori, Tomiyama, and coworkers suggested that intraneuronal Aβ oligomers induce neuronal death by activating endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction.Citation20 On the other hand, there have been several reports on the relationship between SOD1 protein and intracellular Aβ. As a result, it has been found that the interaction of intracellular Aβ with SOD1 decreases the activity of SOD1,Citation21 and the deletion of the copper chaperone for SOD1 enhances the cytoplasmic concentration of Aβ.Citation22 These findings strongly suggest the importance of cytoplasmic superoxide radicals in the intracellular Aβ-mediated AD pathologies and support our conclusion that it is an alternative therapeutic target for AD treatments. Further investigation of molecular mechanism will be required because cytoplasmic radicals may contribute to the pathogenesis of other various neurodegenerative diseases.
Regarding therapies based on SOD, several AD phenotypes in mice can be improved by the direct administration of SOD,Citation23 and the dietary intake of Cu stabilizes SOD1 activity.Citation24 However, it should be taken into account that excessive changes in the levels of redox-active metal ions could have adverse effects due to hydroxyl radical generation. On the other hand, a number of synthetic compounds that decrease the concentrations of cellular radicals have been developed. Among them, EUK-8, a salen-manganese complex with SOD and catalase activities,Citation25 and porphyrin-like compound [Mn(III) tetrakis(4-benzoic acid) porphyrin chloride, denoted as Mn-TBAPCitation26] are promising agents. Recently, some orally available compounds that protect against oxidative damage (mito-QCitation27,Citation28 and metalloporphyrin AEOL11207Citation29) were found to rescue the phenotypes of mouse models of AD and Parkinson disease, respectively. On the other hand, unexpectedly, an increase of SOD1 activity may produce H2O2, resulting in generation of hydroxyl radicals. With regard to this context, decreasing H2O2 levels by catalase or glutathione peroxidase can also be valuable to attenuate oxidative stress. Recently, apomorphineCitation30 was reported to reduce the amounts of H2O2, leading to inhibition of intracellular hydroxyl radicals in AD. The combination therapy using various types of anti-oxidative stress drugs can be a promising way to delay AD. In conclusion, our research will thus not only help to advance our understanding of the molecular basis of intracellular Aβ oligomerization, but may eventually provide a starting point for the development of novel redox status modulators.
Acknowledgments
This work was supported in part by the Program for the Promotion of Basic Research Activities for Innovative Biosciences; Grants-in-aid for Scientific Research; and funds for the Promotion of Science for Young Scientists Grant from the Ministry of Education, Culture, Sports, Science, and Technology of the Japanese Government.
Disclosure of Potential Conflicts of Interest
No Potential conflicts of interest were disclosed.
References
- Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984; 120:885 - 90; http://dx.doi.org/10.1016/S0006-291X(84)80190-4; PMID: 6375662
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A 1985; 82:4245 - 9; http://dx.doi.org/10.1073/pnas.82.12.4245; PMID: 3159021
- Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid β-protein assembly and Alzheimer disease. J Biol Chem 2009; 284:4749 - 53; http://dx.doi.org/10.1074/jbc.R800036200; PMID: 18845536
- Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 2004; 3:205 - 14; http://dx.doi.org/10.1038/nrd1330; PMID: 15031734
- Murakami K, Shimizu T, Irie K. Formation of the 42-mer amyloid β radical and the therapeutic role of superoxide dismutase in Alzheimer's disease. J Amino Acids 2011; 2011:654207; http://dx.doi.org/10.4061/2011/654207; PMID: 22332002
- Murakami K, Irie K, Ohigashi H, Hara H, Nagao M, Shimizu T, et al. Formation and stabilization model of the 42-mer Abeta radical: implications for the long-lasting oxidative stress in Alzheimer’s disease. J Am Chem Soc 2005; 127:15168 - 74; http://dx.doi.org/10.1021/ja054041c; PMID: 16248658
- Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem 2001; 276:38388 - 93; http://dx.doi.org/10.1074/jbc.M105395200; PMID: 11507097
- Imamura Y, Noda S, Hashizume K, Shinoda K, Yamaguchi M, Uchiyama S, et al. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: a model of age-related macular degeneration. Proc Natl Acad Sci U S A 2006; 103:11282 - 7; http://dx.doi.org/10.1073/pnas.0602131103; PMID: 16844785
- Uchiyama S, Shimizu T, Shirasawa T. CuZn-SOD deficiency causes ApoB degradation and induces hepatic lipid accumulation by impaired lipoprotein secretion in mice. J Biol Chem 2006; 281:31713 - 9; http://dx.doi.org/10.1074/jbc.M603422200; PMID: 16921198
- Murakami K, Inagaki J, Saito M, Ikeda Y, Tsuda C, Noda Y, et al. Skin atrophy in cytoplasmic SOD-deficient mice and its complete recovery using a vitamin C derivative. Biochem Biophys Res Commun 2009; 382:457 - 61; http://dx.doi.org/10.1016/j.bbrc.2009.03.053; PMID: 19289104
- Nojiri H, Saita Y, Morikawa D, Kobayashi K, Tsuda C, Miyazaki T, et al. Cytoplasmic superoxide causes bone fragility owing to low-turnover osteoporosis and impaired collagen cross-linking. J Bone Miner Res 2011; 26:2682 - 94; http://dx.doi.org/10.1002/jbmr.489; PMID: 22025246
- Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J Neurochem 2005; 93:953 - 62; http://dx.doi.org/10.1111/j.1471-4159.2005.03053.x; PMID: 15857398
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 1995; 11:376 - 81; http://dx.doi.org/10.1038/ng1295-376; PMID: 7493016
- Lebovitz RM, Zhang H, Vogel H, Cartwright J Jr., Dionne L, Lu N, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci U S A 1996; 93:9782 - 7; http://dx.doi.org/10.1073/pnas.93.18.9782; PMID: 8790408
- Murakami K, Murata N, Noda Y, Tahara S, Kaneko T, Kinoshita N, et al. SOD1 deficiency drives amyloid β oligomerization and memory loss in a mouse model of Alzheimer's disease. J Biol Chem 2011; 287:44557 - 68; http://dx.doi.org/10.1074/jbc.M111.279208
- Murakami K, Murata N, Noda Y, Irie K, Shirasawa T, Shimizu T. Stimulation of the amyloidogenic pathway by cytoplasmic superoxide radicals in an Alzheimer's disease mouse model. Biosci Biotechnol Biochem 74:541 - 7; http://dx.doi.org/10.1271/bbb.90729; PMID: 20208365
- Murakami K, Horikoshi-Sakuraba Y, Murata N, Noda Y, Masuda Y, Kinoshita N, et al. Monoclonal antibody against the turn of the 42-residue amyloid β protein at positions 22 and 23. ACS Chem Neurosci 2010; 1:747 - 56; http://dx.doi.org/10.1021/cn100072e
- Himeno E, Ohyagi Y, Ma L, Nakamura N, Miyoshi K, Sakae N, et al. Apomorphine treatment in Alzheimer mice promoting amyloid-β degradation. Ann Neurol 2011; 69:248 - 56; http://dx.doi.org/10.1002/ana.22319; PMID: 21387370
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, et al. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 2000; 156:15 - 20; http://dx.doi.org/10.1016/S0002-9440(10)64700-1; PMID: 10623648
- Umeda T, Tomiyama T, Sakama N, Tanaka S, Lambert MP, Klein WL, et al. Intraneuronal amyloid β oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J Neurosci Res 2011; 89:1031 - 42; http://dx.doi.org/10.1002/jnr.22640; PMID: 21488093
- Yoon EJ, Park HJ, Kim GY, Cho HM, Choi JH, Park HY, et al. Intracellular amyloid β interacts with SOD1 and impairs the enzymatic activity of SOD1: implications for the pathogenesis of amyotrophic lateral sclerosis. Exp Mol Med 2009; 41:611 - 7; http://dx.doi.org/10.3858/emm.2009.41.9.067; PMID: 19478559
- Gray EH, De Vos KJ, Dingwall C, Perkinton MS, Miller CC. Deficiency of the copper chaperone for superoxide dismutase increases amyloid-β production. J Alzheimers Dis 2010; 21:1101 - 5; PMID: 20693630
- Niwa K, Carlson GA, Iadecola C. Exogenous A β1-40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J Cereb Blood Flow Metab 2000; 20:1659 - 68; http://dx.doi.org/10.1097/00004647-200012000-00005; PMID: 11129782
- Bayer TA, Schäfer S, Simons A, Kemmling A, Kamer T, Tepest R, et al. Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Abeta production in APP23 transgenic mice. Proc Natl Acad Sci U S A 2003; 100:14187 - 92; http://dx.doi.org/10.1073/pnas.2332818100; PMID: 14617773
- Baudry M, Etienne S, Bruce A, Palucki M, Jacobsen E, Malfroy B. Salen-manganese complexes are superoxide dismutase-mimics. Biochem Biophys Res Commun 1993; 192:964 - 8; http://dx.doi.org/10.1006/bbrc.1993.1509; PMID: 8484797
- Ferrer-Sueta G, Ruiz-Ramírez L, Radi R. Ternary copper complexes and manganese (III) tetrakis(4-benzoic acid) porphyrin catalyze peroxynitrite-dependent nitration of aromatics. Chem Res Toxicol 1997; 10:1338 - 44; http://dx.doi.org/10.1021/tx970116h; PMID: 9437523
- McManus MJ, Murphy MP, Franklin JL. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci 2011; 31:15703 - 15; http://dx.doi.org/10.1523/JNEUROSCI.0552-11.2011; PMID: 22049413
- Ma T, Hoeffer CA, Wong H, Massaad CA, Zhou P, Iadecola C, et al. Amyloid β-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J Neurosci 2011; 31:5589 - 95; http://dx.doi.org/10.1523/JNEUROSCI.6566-10.2011; PMID: 21490199
- Liang LP, Huang J, Fulton R, Day BJ, Patel M. An orally active catalytic metalloporphyrin protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity in vivo. J Neurosci 2007; 27:4326 - 33; http://dx.doi.org/10.1523/JNEUROSCI.0019-07.2007; PMID: 17442816
- Ma L, Ohyagi Y, Nakamura N, Iinuma KM, Miyoshi K, Himeno E, et al. Activation of glutathione peroxidase and inhibition of p53-related apoptosis by apomorphine. J Alzheimers Dis 2011; 27:225 - 37; PMID: 21799252