Abstract
Metastable epialleles (MEs) are mammalian genomic loci where epigenetic patterning occurs before gastrulation in a stochastic fashion leading to systematic interindividual variation within one species. Importantly, periconceptual nutritional influences may modulate the establishment of epigenetic changes, such as DNA methylation at MEs. Based on these characteristics, we exploited Infinium HumanMethylation450 BeadChip kits in a 2-tissue parallel screen on peripheral blood leukocyte and colonic mucosal DNA from 10 children without identifiable large intestinal disease. This approach led to the delineation of 1776 CpG sites meeting our criteria for MEs, which associated with 1013 genes. The list of ME candidates exhibited overlaps with recently identified human genes (including CYP2E1 and MGMT, where methylation has been associated with Parkinson disease and glioblastoma, respectively) in which perinatal DNA methylation levels where linked to maternal periconceptual nutrition. One hundred 18 (11.6%) of the ME candidates overlapped with genes where DNA methylation correlated (r > 0.871; p < 0.055) with expression in the colon mucosa of 5 independent control children. Genes involved in homophilic cell adhesion (including cadherin-associated genes) and developmental processes were significantly overrepresented in association with MEs. Additional filtering of gene expression-correlated MEs defined 35 genes, associated with 2 or more CpG sites within a 10 kb genomic region, fulfilling the ME criteria. DNA methylation changes at a number of these genes have been linked to various forms of human disease, including cancers, such as asthma and acute myeloid leukemia (ALOX12), gastric cancer (EBF3), breast cancer (NAV1), colon cancer and acute lymphoid leukemia (KCNK15), Wilms tumor (protocadherin gene cluster; PCDHAs) and colorectal cancer (TCERG1L), suggesting a potential etiologic role for MEs in tumorigenesis and underscoring the possible developmental origins of these malignancies. The presented compendium of ME candidates may accelerate our understanding of the epigenetic origins of common human disorders.
Introduction
Epigenetic changes, including DNA methylation and histone modification, can influence gene expression independently from the genetic code. These tightly regulated mechanisms play an essential role in mammalian differentiation and phenotype development. As an exception to tight regulation, epigenetic modifications at metastable epialleles (MEs) occur stochastically during early embryonic development, leading to systematic (affecting all tissues of the body) and prominent interindividual variation. DNA methylation at MEs can be nutritionally responsive, which implicates MEs in the developmental origins of common human disorders.Citation1,Citation2 Therefore, MEs are prime examples of environmental epigenetics.Citation3
A 2-tissue parallel epigenomic screen on peripheral blood leukocyte (PBL) and hair follicle DNA indicated the existence of MEs in humans.Citation1 However, the microarrays used in that study were of low coverage and did not allow for the quantitative estimation of DNA methylation at ME candidates. Potential associations between the putative MEs and transcription were not examined either. Here, we exploited Infinium HumanMethylation450 BeadChip kits in a 2-tissue parallel screen. These arrays can support an accurate, quantitative determination of DNA methylation at select CpG sites both from PBL (mesodermal origin)Citation4,Citation5 and colonic mucosal DNA (endodermal origin).Citation6 Association between the novel ME candidates and gene expression was examined in an independent cohort.
Results
ME candidates are relevant for homotypic cell adhesion
Microarray data was analyzed from 10 control children (pediatric patients with gastrointestinal complaints, but in whom the endoscopic—histologically normal intestinal mucosa—and clinico-laboratory investigations did not reveal abnormal organic etiology for their complaints; see Materials and Methods and Table S1A). These patients had both PBLCitation4 and colonic mucosal DNA samples examined (data: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE32148). Our earlier publication on the PBL samples showed significant correlation between DNA methylation levels and β values by bisulfite pyrosequencing validation of the arrays.Citation4 Similarly, a significant correlation was found between microarray and pyrosequencing results in the colonic mucosa of 8 controls at 5 independent loci in this work (p = 0.0002; Fig. S1; see Table S2 for primer sequences).
A total of 1776 CpG sites met our criteria for MEs, which associated with 1013 genes (Table S3). Importantly, ZFYVE28, one of the five earlier validated human MEs,Citation1 was among this list, in spite of the different tissues studied and the dissimilar molecular methods employed. For ontology analyses, the ME associated genes were compared with the rest of the genome, since the array covers 99% of known human genes. Remarkably, Interpro (http://www.ebi.ac.uk/interpro) comparisons revealed that cadherin, N-terminal (IPR:013164, 8.7 fold) and cadherin (IPR:002126, 5.12 fold), genes in which DNA methylation has been linked to metastasis in multiple forms of cancer,Citation7,Citation8 were significantly (p < 0.05) overrepresented, while the conserved protein category of olfactory receptor (IPR:000725, 0.14 fold) was underrepresented in connection with MEs (Table S4A). Cadherins play an important role in homotypic cell adhesion.Citation9 Our finding that MEs are overrepresented in association with these genes indicates that periconceptual environmental influences may be important in modifying cell adhesion in humans. On the contrary, epigenetic development at olfactory receptors may be protected against early prenatal environmental changes.
MEs overlap with genes epigenetically responsive to periconceptual nutrition
A recent prospective human study found that periconceptual maternal micronutrient supplementation induced gender-specific DNA methylation changes that could be detected from cord blood.Citation10 Twenty-one female genes and 14 male genes were observed to have associations between DNA methylation and periconceptual dietary supplementation. Importantly, 2 of the male genes (14.3%) and 2 of the female genes (9.5%) had overlap with our candidate ME list. In addition, there were no such associations with the control list of 1399 genes, in which DNA methylation variation did not correlate (r < 0.5; p > 0.14) between PBL and colonic mucosa (Chi square p = 0.04). Namely, C18orf22 (RBFA, ) and PTPN20B () overlapped between the ME list of this report and the male-specific epigenetically responsive genes to maternal periconceptual nutrition, while MGMT () and CYP2E1 () matched between the female-specific list with our ME candidates.
Figure 1. Metastable epiallele (ME) candidates, where epigenetic responsiveness to periconceptual maternal micronutrient supplementation was observed in the cord blood of male (A–B) and female (C) newborns (see text for details). DNA methylation from peripheral blood (mesodermal origin) DNA, and colonic mucosa (colon, endodermal origin) showed significant intraindividual correlation, but > 10% interindividual methylation differences. n = 10. C18orf22: r = 0.66, p = 0.038; PTPN20B: r = 0.97, p < 0.001; MGMT: r = 0.98, p < 0.001
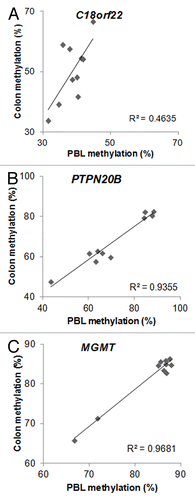
Figure 2. Examples for metastable epiallele (ME) candidates with 3 or more CpG sites meeting ME criteria in close vicinity (within 10 kb) of each other. DNA methylation from peripheral blood leukocyte (mesodermal origin) DNA, and colonic mucosa (colon, endodermal origin) showed significant intraindividual correlation, but > 10% interindividual methylation differences. A. CYP2E1, where gene expression associated with DNA methylation in Parkinson disease brain. Epigenetic responsiveness to periconceptual maternal micronutrient supplementation was also observed in the cord blood of female newborns at this gene. B. HCG9, where DNA methylation associated with bipolar disorder in three different [brain, white blood cells, sperms (germ line)] tissues (see text for references and details). n = 10. CYP2E1: r = 0.8, p = 0.005; HCG9: r = 0.87, p = 0.001
![Figure 2. Examples for metastable epiallele (ME) candidates with 3 or more CpG sites meeting ME criteria in close vicinity (within 10 kb) of each other. DNA methylation from peripheral blood leukocyte (mesodermal origin) DNA, and colonic mucosa (colon, endodermal origin) showed significant intraindividual correlation, but > 10% interindividual methylation differences. A. CYP2E1, where gene expression associated with DNA methylation in Parkinson disease brain. Epigenetic responsiveness to periconceptual maternal micronutrient supplementation was also observed in the cord blood of female newborns at this gene. B. HCG9, where DNA methylation associated with bipolar disorder in three different [brain, white blood cells, sperms (germ line)] tissues (see text for references and details). n = 10. CYP2E1: r = 0.8, p = 0.005; HCG9: r = 0.87, p = 0.001](/cms/asset/fe902d34-5051-4d7e-b871-32d90778795f/kepi_a_10923438_f0002.gif)
MEs link to common human diseases
Further filtering of MEs—where 2 or more CpG sites met criteria within a 10 kb distance from each other—provided 401 loci (22.6% of the ME list), which associated with 140 genes (Table S5). DNA methylation at several of the MEs in which 3 or more CpG sites were found within 10 kb from each other has already been linked to common human disorders, including glioblastoma (GSTM5Citation11), bladder cancer (AQP11Citation12), breast cancer (NAV1Citation13), colorectal cancer (APC2Citation14), gastric cancer (EBF3Citation15) and other cancers (KCNK15Citation16), ileal Crohn disease (S100A13Citation17), Parkinson disease (CYP2E1Citation18) and bipolar disorder (HCG9Citation19) (Table S5; and B).
MEs overlap with epigenetic discordance between monozygotic twins
Interindividual genetic variation can associate with epigenetic variation.Citation20,Citation21 However, epigenetic variation in genetically identical monozygotic (MZ) twins is devoid of such bias. Therefore, we matched the ME loci with CpGs in which DNA methylation differences within at least 1 out of 6 MZ twin pairs exceeded 10% [MZ PBL DNA samples were obtained form the Danish twin registry (see ref. Citation4); for raw data see: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE32148]. Two hundred and forty-eight (14%) of the ME CpGs matched with the MZ twin list of CpGs in which at least 1 MZ pair showed methylation differences greater than 10% in PBL DNA (Table S6), while there was significantly fewer CpGs matching that criteria (2.6%; Fischer exact p < 0.0001) in a control list of CpGs in which DNA methylation variation did not correlate between PBL and colonic mucosa (r < 0.5; p > 0.14). This result underscores that interindividual DNA methylation variation at a significant number of ME candidates can occur independently from genetic differences.
MEs overlap with genes in which DNA methylation correlates with gene expression
Direct correlation between MEs and gene expression at nearby genomic regions is of major functional significance. However, it should be noted that epigenetic changes can influence rather distant areas of the genome compared with where the changes take place. Therefore, any area of the genome behaving as an ME may bear potential physiologic importance. Nevertheless, we examined how the ME candidates identified in this study compare with genes whose expression correlated with DNA methylation variation in the colonic mucosa of control children. Since further biopsies from the discovery cohort of this study were not available, we examined DNA methylation and gene expression associations in the colonic mucosa of 5 other control patients (Table S1B).
There were 1181 CpG sites in which DNA methylation correlated (r > 0.871; p < 0.055) with the expression of 904 genes in the colonic mucosa of children, as determined by associating Infinium HumanMethylation450 v1.1 probe gene annotations to Affymetrix GeneChip PrimeView Human Gene Expression Array probe annotations (Table S7). One hundred and eighteen (11.6%) of the ME genes overlapped with this independent list. This overlap was significantly higher (Yates Chi square p < 0.0001) than expected by chance (2.8%). Additionally, the ME-associated, transcriptionally correlating genes had functional overrepresentation in biological processes involved in cell adhesion and development of anatomical structures in multicellular organisms (Table S4B). Furthermore, the transcriptionally relevant ME candidates maintained the significant association with biological function (Fatigo) and proteins (Interpro) at the level of homophilic cell adhesion and cadherin, respectively, similar to our observations in the list of MEs based only on methylation characteristics (Tables S4A and B).
NetworkMiner (Babelomics4.3) revealed a functional link between 26 of the 118 transcriptionally relevant MEs (Fig. S2) that was significantly higher (p < 0.034) than in a randomly selected list of 2000 genes.
Importantly, 35 (25%) out of the 140 filtered (with 2 or more candidate CpG sites within 10 kb, see above) ME associated genes overlapped with the epigenetically linked gene expression in the colonic mucosa of children from the independent cohort. This association was significantly higher (Fischer exact p < 0.0001) than with the original list, indicating that the clustering of ME candidate CpGs predicts the transcriptional relevance of the methylation changes identified. NAV1, EBF and KCNK15 (see above) still remained among these 35 genes, further implicating the role of MEs in human tumorigenesis.
Discussion
The term “metastable epiallele” was introduced by Emma Whitelaw and colleagues to describe mammalian alleles in which variable expression associated with epigenetic differences instead of genetic heterogeneity. The murine agouti viable yellow (Avy) is an outstanding example of such an allele.Citation22 Similarly behaving loci, even in imprinted genomic regions, have been observed in humans as well.Citation23
The seminal work of Cooney and colleagues demonstrated that maternal micronutrient supplementation can shift DNA methylation distribution and the corresponding fur phenotype at the population level in Avy mice, thereby implicating MEs in the nutritional (or environmental) origins of common human diseases.Citation24 Further studies confirmed the nutritionally driven establishment of interindividually variable, but intraindividually systematic DNA methylation to occur before gastrulation (i.e., in the periconceptual period)Citation25 and showed similar behavior at other mammalian MEs.Citation26 Post-gastrulation tissue-specific epigenetic modification can also occur at MEs, resulting in differing absolute levels of DNA methylation between the tissues of one individual, but the persistence of the similar magnitude of interindividual methylation variation when the same tissues are compared between different organisms.Citation27 This could explain the behavior of DNA methylation at C18orf22, where methylation was lower in PBL DNA than in the colonic mucosa (). In addition, the significant and similar interindividual variation between the select tissues persisted, implicating these loci as MEs.
The existence of human MEs was recently supported by the examination of PBL DNA from children in The Gambia.Citation1 In this geographic region, a bimodal annual variation of nutritional abundance (i.e., abundance and paucity of food within the same year) allowed for testing the effects of periconceptual maternal diet on offspring DNA methylation at a handful of putative MEs. A consecutive prospective study in the same genetically homogeneous population revealed a number of genomic loci in which infantile DNA methylation was modified as a result of maternal micronutrient supplementation in the periconceptual period.Citation10 Importantly, none of the 107 putative MEs from our original publication (see Table S2A from ref. Citation1) showed overlaps with the list of genes, which had DNA methylation associations with periconceptual nutrition from the cord blood of Gambian infants (see Supplementary Data set Table 2 and Gene list 1 and 3 from ref. Citation10). However, the candidate MEs identified in this work matched with 9.5% () and 14.3% () of these latter lists. Additionally, 8 (21.1%) genes (AKAP12, CCDC102A, ITPKB, PANX2, PARD6G, SPNS2, SPTBN4 and ZFYVE28) from the former filtered putative human ME list (38 genes after filtering for interfering single nucleotide polymorphisms, copy number variations and segmental duplications: see Table S2C from ref. Citation1) had matches in our current compendium. Also, we observed significantly lower (2.6%; p = 0.028) association between the former putative MEs and our control list. This is a remarkable result, considering the different populations and tissues studied and the vastly dissimilar microarray methods employed between the two works.
The key feature of MEs is similar intraindividual methylation within all tissues of the body. Therefore, we specifically allowed either whole PBL or isolated PBMC DNA to be included on the arrays as DNA of mesodermal origin (see Materials and Methods) from the patients. Additionally, the diverse and clinically poorly defined (e.g., abdominal pain) proband inclusion allowed for the identification of common human ME candidates. With lack of whole genome sequencing we cannot rule out that interindividual genetic variation is the initiator of the epigenetic variation observed at many of the ME candidates identified. To address this issue, we filtered out CpGs from our candidate lists in which single nucleotide polymorphisms (SNPs) have been found to either overlap or to be near the loci. Nevertheless, trimodal or bimodal clustering of methylation values (such as at PTN20B; ) do raise the possibility for genetic (i.e., allelic) variation-stimulated DNA methylation differences. In addition, the homogeneous dispersion of interindividual systemic DNA methylation (i.e., significant correlation between DNA methylation from two tissues of different germ layer origin, leukocytes and colonic epithelium, within the same individual, such as at C18orf22, CYPE1 and HCG9; and ) is more supportive of the ME status. Similarly, the overlaps with genes epigenetically sensitive to periconceptual micronutrient supplementation in a genetically homogeneous populationCitation10 (see above) argue for the non-genetic origins of DNA methylation differences at a number of our ME candidates. Furthermore, the significant overlap between inter-twin DNA methylation differences in MZ twin pairs and the ME list of this report indicates that a large number of the loci identified are true MEs. It should also be noted that the studied children were not healthy but had gastrointestinal complaints. Also, they provided a non-homogeneous population in respect to sex and age. They can only be considered as controls because anatomical abnormalities were not detected by colonoscopy or other examination of the intestines. Sex and age-dependent DNA methylation variation was excluded during the ME selection. However, it is possible that the clustering pattern of DNA methylation at select ME candidates may represent functional disease associations, such as subtypes of irritable bowel syndrome/recurrent abdominal pain, for example. In the meantime, no obvious discriminating phenotypic characteristics could be determined in association with methylation variation in the examples of and .
Within the highlighted MEs of this report (Figs. 1 and 2), CYP2E1 in respect to Parkinson disease,Citation18 HCG9 in bipolar disorder,Citation19 and MGMT in glioblastoma,Citation28 merit further consideration. The high monozygotic twin discordance ratesCitation29 and lower prevalence of Parkinson disease (PD) in developing countries, such as Tanzania,Citation30 indicate the potential importance of environmentally-driven epigenetic changes, especially in developed countries, in the etiology of the disease. The increased cerebral expression of CYP2E1 in association with decreased DNA methylation, its periconceptual epigenetic responsiveness,Citation10 and our findings suggesting it as an ME point toward CYP2E1 as an outstanding candidate in respect to the developmental origins of PD. Similarly, the epidemiology of bipolar disorder (BPD) supports epigenetic factors playing a pathogenic role.Citation31 The association between BPD and HCG9 methylation in 3 different tissues,Citation19 and our results indicating it as an ME, underscore the possible etiologic importance of HCG9 in BPD. Furthermore, MGMT methylation within the tumor links the gene to earlier presentation with glioblastoma and predicts increased survival.Citation28 Epidemiologic observations show a tendency for increased incidence of gliomas in industrialized countriesCitation32 pointing to the importance of environmental changes in this malignancy. The epigenetic responsiveness of MGMT to periconceptual intrauterine nutritionCitation10 and its potential ME nature outlined here indicate its role in the developmental origins of glioblastoma. Secondary to their ME nature, in addition to contributing to the pathology of human diseases, DNA methylation at CYP2E1, HCG9 and MGMT may aid in the diagnostics (e.g., by determining relevant DNA methylation in the critically affected tissues based on PBL DNA methylation), prevention (by identifying and eliminating critical environmental influences responsible for the periconceptual DNA methylation changes), prognostics (MGMT methylation in PBL DNA may predict survival in glioblastoma, for instance) and even treatment of PD, BPD and glioblastoma in the future.
In order to determine the potential transcriptional relevance of the expanded ME compendium of this report, we examined the correlation between DNA methylation and gene expression in the colonic mucosa of an independent group of control children. We could delineate 118 ME associated genes in which DNA methylation changes correlated with gene expression modification. Remarkably, the functional (homophilic cell adhesion) and protein (cadherin) based separation persisted between the methylation characteristics, and the further gene expression association based ME compendiums (Table S4A and B). Additional filtering of gene expression-correlated MEs defined 35 genes in association with 2 or more CpG sites within a 10 kb genomic region fulfilling the ME criteria. DNA methylation changes at a number of these genes have been associated with various forms of human disease, including cancers, such as asthma and acute myeloid leukemia (ALOX12), gastric cancer (EBF3), breast cancer (NAV1), colon cancer and acute lymphoid leukemia (KCNK15), Wilms tumor (protocadherin gene cluster; PCDHAs) and colorectal cancer (TCERG1L), suggesting a potential etiologic role for MEs in tumorigenesis and underscoring the possible developmental origins of these malignancies.
This is the first study to examine the potential transcriptional relevance of human MEs. Additionally, DNA methylation-associated common gene expression variation in histologically normal colonic mucosa of children was delineated. This latter list of genes will likely prove significant for future studies into pediatric gastrointestinal disorders. Based on the discussion above, the presented compendium of ME candidates may accelerate our understanding of the epigenetic developmental origins of multiple common human disorders.
Materials and Methods
Patients and PBL DNA isolation
Control patients (Table S1A) were recruited prior to endoscopy following informed consent through the institutional review board (IRB) approved tissue banks of the Charles University, Prague, Czech Republic (EK-1796/08) and the Pediatric Inflammatory Bowel Disease Consortium Registry at the Baylor College of Medicine (BCM; H-17654). The patients and data were included and described in a previous publication.Citation4 Further control patients examined by both DNA methylation and gene expression microarrays were recruited solely at BCM (Table S1B). Only patients with grossly and histologically normal mucosa at colonoscopy were designated as controls. For the peripheral blood leukocyte (PBL) DNA either Gentra Puregene (Qiagen, Valencia, CA) for whole blood DNA isolation was used according to manufacturer’s recommendation on the samples from the Charles University, or the QuiAmp DNA mini kit (Qiagen, Valencia, CA) was utilized on isolated peripheral blood mononuclear cells (PBMCs) form the cohort of the Baylor College of Medicine. BD Vacutainer® CPT™ Cell Preparation Tube with Sodium Citrate was used in the latter case to isolate peripheral blood mononuclear cells (PBMCs) from PBL.
Tissues and nucleic acid processing for microarray
Colonic mucosa was obtained by pinch biopsies during endoscopy. Samples were flash frozen on dry ice and stored at -80°C until further processing. After thawing, the colonic mucosal biopsies were centrifuged at 14,000 rpm for 30 sec and resuspended in 500 µl RLT buffer (Qiagen, Valencia, CA) (with β-mercaptoethanol). Sterile 5mm steel beads (Qiagen) and 500µl sterile 0.1mm glass beads (Scientific Industries, Inc., NY, USA) were added for complete cellular lysis in a Qiagen TissueLyser (Qiagen), and run at 30Hz for 5min. Samples were centrifuged briefly, 350 µl of RTL and 200 µl of 100% ethanol were added to a 100 µl aliquot of the sample supernatant. This mixture was added to a DNA spin column, and DNA recovery protocols were followed as instructed in the QIAamp DNA Mini Kit (Qiagen) starting at step 5 of the Tissue Protocol. DNA was eluted from the column with 30 µl water and samples were diluted accordingly to a final concentration of 20 ng/µl. DNA samples were quantified using a Nanodrop spectrophotometer (Nyxor Biotech, Paris, France).
The PBL and colonic mucosal DNA samples following quality control with PicoGreen (http://probes.invitrogen.com/media/pis/mp07581.pdf) were processed by Infinium HumanMethylation450 BeadChip Kits (Illumina San Diego, CA, USA; http://www.illumina.com/products/methylation_450_beadchip_kits.ilmn) according to the manufacturer’s recommendations through automated processes in the Core Laboratory for Translational Genomics of the Baylor College of Medicine. Arrays were imaged with BeadArray Reader using standard recommended Illumina scanner settings. GenomeStudio software version 2010.3.0.30128 was used to generate β values normalized to internal control probes. Internal controls determined the array processing to be of good quality. Only the 482,421 CpG probes on the array were used for subsequent analysis of the independent control samples (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=pvojjiqcqiuqori&acc=GSE42921). Bisulfite pyrosequencing validation of the arrays showed good correlation between DNA methylation levels and β values (ref. Citation4 and Fig. S1).
Colonic mucosal RNA was isolated by Qiangen-Qiazol - miRNA Isolation Kit. cDNA amplification and labeling was performed with Ovation Pico WTA System V2 and Encore Biotin Module (NuGEN), respectively. Array hybridization was performed according to Affymetrix FS450_0002 Hybridization Protocol. The Affymetrix GeneChip® PrimeView™ Human Gene Expression Arrays were scanned with Affymetrix Genechip Scanner 7G. The R Bioconductor affy package rma function was used to compute expression. The CEL files and RMA normalized expression values can be downloaded from http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=pzyffweqggmsgnk&acc=GSE42911.
Metastable epiallele selection
MEs were selected by identifying CpG sites where the highest and the lowest methylation (based on β values) was in the same patients both from the peripheral blood leukocyte (PBL) and the colonic mucosal DNA. Only those CpG sites were selected where the average methylation difference (from PBL and colon) between individuals with the highest and the lowest methylation was ≥ 10% and ≤ 60% (to eliminate genetic bias arising from un-anticipated C > T polymorphisms), correlation (r) between PBL and colonic methylation was ≥ 0.64 or ≤ -0.64 (two tailed Pearson p ≤ 0.046), and methylation was independent from age (r ≥ -0.6 or ≤ 0.6; p ≥ 0.067) and gender (male vs. female methylation two tailed unpaired T test p > 0.05). Finally, Infinium 450K probes which contain SNPs at or near the target CpG site were removed based on the information here: http://ima.r-forge.r-project.org/.Gene associations were based on the Infinium HumanMethylation450 v1.1 manifest.
Methylation correlation with expression
Probe gene annotations in the Infinium HumanMethylation450 v1.1 manifest were used to identify associated probe gene annotations in the Affymetrix GeneChip PrimeView Human Gene Expression Array manifest based on matching gene annotations. HumanMethylation450 probes with a range of β values > 0.1 across the 5 samples were compared with PrimeView RMA normalized expression values using Pearson’s correlation. Genes with β values and RMA normalized expression values correlated at r > 0.871, the p < 0.055 significance level for a sample size of 5, were used for subsequent analysis.
Additional material
Download Zip (586.6 KB)Acknowledgments
The authors would like to acknowledge the patients for providing samples, and all of our colleagues who contributed to the successful banking of the tissues. This study utilized the NIDDK supported Pediatric Inflammatory Bowel Disease Consortium Registry of the Baylor College of Medicine (DK56338; Texas Medical Center Digestive Disease Center (PI: Estes, MK).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Waterland RA, Kellermayer R, Laritsky E, Rayco-Solon P, Harris RA, Travisano M, et al. Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet 2010; 6:e1001252; http://dx.doi.org/10.1371/journal.pgen.1001252; PMID: 21203497
- Barouki R, Gluckman PD, Grandjean P, Hanson M, Heindel JJ. Developmental origins of non-communicable disease: implications for research and public health. Environ Health 2012; 11:42; http://dx.doi.org/10.1186/1476-069X-11-42; PMID: 22715989
- Cortessis VK, Thomas DC, Levine AJ, Breton CV, Mack TM, Siegmund KD, et al. Environmental epigenetics: prospects for studying epigenetic mediation of exposure-response relationships. Hum Genet 2012; 131:1565 - 89; http://dx.doi.org/10.1007/s00439-012-1189-8; PMID: 22740325
- Harris RA, Nagy-Szakal D, Pedersen N, Opekun A, Bronsky J, Munkholm P, et al. Genome-wide peripheral blood leukocyte DNA methylation microarrays identified a single association with inflammatory bowel diseases. Inflamm Bowel Dis 2012; 18:2334 - 41; http://dx.doi.org/10.1002/ibd.22956; PMID: 22467598
- Pan H, Chen L, Dogra S, Teh AL, Tan JH, Lim YI, et al. Measuring the methylome in clinical samples: improved processing of the Infinium Human Methylation450 BeadChip Array. Epigenetics 2012; 7:1173 - 87; http://dx.doi.org/10.4161/epi.22102; PMID: 22964528
- Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011; 6:692 - 702; http://dx.doi.org/10.4161/epi.6.6.16196; PMID: 21593595
- DesRochers TM, Shamis Y, Alt-Holland A, Kudo Y, Takata T, Wang G, et al. The 3D tissue microenvironment modulates DNA methylation and E-cadherin expression in squamous cell carcinoma. Epigenetics 2012; 7:34 - 46; http://dx.doi.org/10.4161/epi.7.1.18546; PMID: 22207358
- Wang Z, Liu L, Wang M, Shen M, Li J, Liu J, et al. NOTCH1 regulates migration and invasion of skin cancer cells by E-cadherin repression. Mol Cell Biochem 2012; 362:35 - 41; http://dx.doi.org/10.1007/s11010-011-1125-6; PMID: 22038626
- Katsamba P, Carroll K, Ahlsen G, Bahna F, Vendome J, Posy S, et al. Linking molecular affinity and cellular specificity in cadherin-mediated adhesion. Proc Natl Acad Sci U S A 2009; 106:11594 - 9; http://dx.doi.org/10.1073/pnas.0905349106; PMID: 19553217
- Khulan B, Cooper WN, Skinner BM, Bauer J, Owens S, Prentice AM, et al. Periconceptional maternal micronutrient supplementation is associated with widespread gender related changes in the epigenome: a study of a unique resource in the Gambia. Hum Mol Genet 2012; 21:2086 - 101; http://dx.doi.org/10.1093/hmg/dds026; PMID: 22307237
- Etcheverry A, Aubry M, de Tayrac M, Vauleon E, Boniface R, Guenot F, et al. DNA methylation in glioblastoma: impact on gene expression and clinical outcome. BMC Genomics 2010; 11:701; http://dx.doi.org/10.1186/1471-2164-11-701; PMID: 21156036
- Veerla S, Panagopoulos I, Jin Y, Lindgren D, Höglund M. Promoter analysis of epigenetically controlled genes in bladder cancer. Genes Chromosomes Cancer 2008; 47:368 - 78; http://dx.doi.org/10.1002/gcc.20542; PMID: 18196590
- Li L, Lee KM, Han W, Choi JY, Lee JY, Kang GH, et al. Estrogen and progesterone receptor status affect genome-wide DNA methylation profile in breast cancer. Hum Mol Genet 2010; 19:4273 - 7; http://dx.doi.org/10.1093/hmg/ddq351; PMID: 20724461
- Naghibalhossaini F, Hosseini HM, Mokarram P, Zamani M. High frequency of genes’ promoter methylation, but lack of BRAF V600E mutation among Iranian colorectal cancer patients. Pathol Oncol Res 2011; 17:819 - 25; http://dx.doi.org/10.1007/s12253-011-9388-5; PMID: 21455633
- Kim J, Min SY, Lee HE, Kim WH. Aberrant DNA methylation and tumor suppressive activity of the EBF3 gene in gastric carcinoma. Int J Cancer 2012; 130:817 - 26; http://dx.doi.org/10.1002/ijc.26038; PMID: 21387304
- Shu J, Jelinek J, Chang H, Shen L, Qin T, Chung W, et al. Silencing of bidirectional promoters by DNA methylation in tumorigenesis. Cancer Res 2006; 66:5077 - 84; http://dx.doi.org/10.1158/0008-5472.CAN-05-2629; PMID: 16707430
- Nimmo ER, Prendergast JG, Aldhous MC, Kennedy NA, Henderson P, Drummond HE, et al. Genome-wide methylation profiling in Crohn’s disease identifies altered epigenetic regulation of key host defense mechanisms including the Th17 pathway. Inflamm Bowel Dis 2012; 18:889 - 99; http://dx.doi.org/10.1002/ibd.21912; PMID: 22021194
- Kaut O, Schmitt I, Wüllner U. Genome-scale methylation analysis of Parkinson’s disease patients’ brains reveals DNA hypomethylation and increased mRNA expression of cytochrome P450 2E1. Neurogenetics 2012; 13:87 - 91; http://dx.doi.org/10.1007/s10048-011-0308-3; PMID: 22238121
- Kaminsky Z, Tochigi M, Jia P, Pal M, Mill J, Kwan A, et al. A multi-tissue analysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol Psychiatry 2012; 17:728 - 40; http://dx.doi.org/10.1038/mp.2011.64; PMID: 21647149
- Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet 2009; 41:112 - 7; http://dx.doi.org/10.1038/ng.283; PMID: 19098912
- Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E, et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet 2008; 40:904 - 8; http://dx.doi.org/10.1038/ng.174; PMID: 18568024
- Rakyan VK, Blewitt ME, Druker R, Preis JI, Whitelaw E. Metastable epialleles in mammals. Trends Genet 2002; 18:348 - 51; http://dx.doi.org/10.1016/S0168-9525(02)02709-9; PMID: 12127774
- Weksberg R, Shuman C, Caluseriu O, Smith AC, Fei YL, Nishikawa J, et al. Discordant KCNQ1OT1 imprinting in sets of monozygotic twins discordant for Beckwith-Wiedemann syndrome. Hum Mol Genet 2002; 11:1317 - 25; http://dx.doi.org/10.1093/hmg/11.11.1317; PMID: 12019213
- Cooney CA, Dave AA, Wolff GL. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr 2002; 132:Suppl 2393S - 400S; PMID: 12163699
- Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 2003; 23:5293 - 300; http://dx.doi.org/10.1128/MCB.23.15.5293-5300.2003; PMID: 12861015
- Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A 2007; 104:13056 - 61; http://dx.doi.org/10.1073/pnas.0703739104; PMID: 17670942
- Waterland RA, Dolinoy DC, Lin JR, Smith CA, Shi X, Tahiliani KG. Maternal methyl supplements increase offspring DNA methylation at Axin Fused. Genesis 2006; 44:401 - 6; http://dx.doi.org/10.1002/dvg.20230; PMID: 16868943
- Skiriute D, Vaitkiene P, Saferis V, Asmoniene V, Skauminas K, Deltuva VP, et al. MGMT, GATA6, CD81, DR4, and CASP8 gene promoter methylation in glioblastoma. BMC Cancer 2012; 12:218; http://dx.doi.org/10.1186/1471-2407-12-218; PMID: 22672670
- Wirdefeldt K, Gatz M, Reynolds CA, Prescott CA, Pedersen NL. Heritability of Parkinson disease in Swedish twins: a longitudinal study. Neurobiol Aging 2011; 32:1923 - , e1-8; http://dx.doi.org/10.1016/j.neurobiolaging.2011.02.017; PMID: 21482443
- Winkler AS, Tütüncü E, Trendafilova A, Meindl M, Kaaya J, Schmutzhard E, et al. Parkinsonism in a population of northern Tanzania: a community-based door-to-door study in combination with a prospective hospital-based evaluation. J Neurol 2010; 257:799 - 805; http://dx.doi.org/10.1007/s00415-009-5420-z; PMID: 20024574
- Petronis A. Epigenetics and bipolar disorder: new opportunities and challenges. Am J Med Genet C Semin Med Genet 2003; 123C:65 - 75; http://dx.doi.org/10.1002/ajmg.c.20015; PMID: 14601038
- Ohgaki H. Epidemiology of brain tumors. Methods Mol Biol 2009; 472:323 - 42; http://dx.doi.org/10.1007/978-1-60327-492-0_14; PMID: 19107440